
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Examination of the short-range structure of molten salts: ThF4, UF4, and related alkali actinide fluoride systems†
J. A.
Ocádiz-Flores
a,
A. E.
Gheribi
 b,
J.
Vlieland
a,
D.
de Haas
a,
K.
Dardenne
c,
J.
Rothe
c,
R. J. M.
Konings
ad and
A. L.
Smith
*a
b,
J.
Vlieland
a,
D.
de Haas
a,
K.
Dardenne
c,
J.
Rothe
c,
R. J. M.
Konings
ad and
A. L.
Smith
*a
aDelft University of Technology, Faculty of Applied Sciences, Radiation Science & Technology Department, Mekelweg 15, 2629 JB Delft, The Netherlands
bCentre for Research in Computational Thermochemistry, Department of Chemical Engineering, École Polytechnique, C.P, 6079, Succursale “Downtown”, Montreal (Quebec), Canada H3C 3A7
cKarlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), Radionuclide Speciation Department, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dEuropean Commission, Joint Research Centre, P.O. Box 2340, D-76125 Karlsruhe, Germany. E-mail: A.L.Smith@tudelft.nl
First published on 29th April 2021
Abstract
The short-range structures of LiF–ThF4, NaF–AnF4, KF–AnF4, and Cs–AnF4 (An = Th, U), were probed using in situ high temperature Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy. Signally, the EXAFS spectra of pure molten ThF4 and UF4 were measured for the first time. The data were interpreted with the aid of Molecular Dynamics (MD) and standard fitting of the EXAFS equation. As in related studies, a speciation distribution dominated by [AnFx]4−x (x = 7, 8, 9) coordination complexes was observed. The average coordination number was found to decrease with the increasing size of the alkali cation, and increase with AnF4 content. An average coordination number close to 6, which had not been detected before in melts of alkali actinide fluorides, was seen when CsF was used as solvent.
1 Introduction
Molten salts are a class of ionic liquids which have in recent years been the focus of extensive fundamental research. This can be explained by the fact that they are a versatile class of reaction media, e.g. carbonates, nitrates, fluorides, chlorides, with a variety of melting points, heat capacities, vapor pressures, densities, and other thermophysical properties suited for a variety of industrial applications. The most well-known is perhaps the production of materials as important as aluminum and sulfuric acid, yet thermal energy storage is also a notable application. In the future, molten salts may be used for processes as important as the production of hydrogen1 and ammonia,2 carbon capture and storage,3 nuclear fuel pyroprocessing,4 and as nuclear fuels.5,6Understanding the speciation and structure of the molten salts, which are closely related to their thermodynamic and transport properties, is crucial for the design and operation of any intended application. Salt systems which have been subject of this type of analysis in the literature include AF–ZrF4 (A = Li, Na, K),7 KF–ThF4,8 LiF–ThF49 and LiF–ThF4–UF4 systems.10 In these studies, the experimental technique used is in situ high temperature Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy. This element-specific, non-destructive, oxidation state-sensitive technique requires very small masses of sample, making it very powerful and ideally suited for radioactive, corrosive salts at high temperature. It provides detailed information about the local structure such as interatomic distances among nearest neighbors and second-nearest neighbor elements.
However, EXAFS spectroscopy of liquids is less straightforward than that of solids because the standard EXAFS equation11 may be inaccurate to describe the measured experimental data since it relies on a Gaussian distribution of interatomic distances, which liquids do not have. Another approach, known as cumulant expansion, is also inadequate because of a strong correlation between the fitting parameters.8,12,13 Hence, this work relied on configurations generated from Molecular Dynamics (MD) simulations as the underlying structural models. MD has demonstrated already in numerous studies to be a strikingly valuable tool in its own right for calculating the thermo-physical and thermo-chemical properties of molten salts.14–17 In other works, our group has presented EXAFS data of the LiF–ThF4,9 LiF–UF4 (submitted), and NaF–ThF49 systems. In order to arrive at a comprehensive picture of the local structure of alkali actinide fluoride binary systems as a function of the alkali and actinide ions, this work extends to NaF–UF4 and focuses on larger alkali ions: KF–ThF4, KF–UF4, CsF–UF4, and CsF–ThF4. Notably, EXAFS data for pure molten ThF4 and UF4 were collected for the first time.
2 Experimental method
EXAFS measurements were performed at the INE beamline18 of the KARA (Karlsruhe Research Accelerator) synchrotron facility (Karlsruhe, Germany), with 2.5 GeV and 150–170 mA as operating conditions in the storage ring. The beamline uses a Ge(422) double-crystal monochromator (DCM). Rh-coated mirrors before (flat, cylindrically bent) and after (toroidal) the DCM are used to collimate and focus the synchrotron beam, respectively, producing a spot size of 300 mm × 500 mm at the sample surface. Transmission and fluorescence geometries could be measured in unison, and samples were probed at the L3 edges of Th (16.300 keV) and U (17.166 keV), scanning from ∼16.1 to ∼17.05 keV and ∼17.14 to ∼17.77 keV, respectively.A dedicated experimental set-up, previously described in detail by Smith et al.9 and built to operate at the INE beamline, was used for the measurements. The set-up consists of a specifically designed furnace inside a custom-made glovebox. The salts themselves were sealed in pre-dried boron nitride containment cells loaded into the furnace chamber, which was evacuated under operation down to ∼2 × 10−5 mbar to avoid reaction of the salts with residual oxygen or water. Once under vacuum, the samples were heated up to (50 ± 10) K above their liquidus temperatures. Quick scans were made during the heating ramp to detect the melting of the material. If the measurement temperature of the sample was less than the melting point of the pure alkali fluoride end-member, the temperature was ramped up to the melting point of the end-member and held for about 15 minutes to ensure complete melting and homogenization. The temperature was subsequently adjusted to a set value (50 ± 20) K above liquidus. In addition, an equilibration time of ∼15–30 min was employed before collecting the X-ray Absorption Spectroscopy (XAS) data to ensure the signal had stabilized. EXAFS data were collected up to ∼12.5 Å, and were Fourier transformed using the Hanning window over the k-range 3–12 Å−1 (dk = 2) for solids, and 3–9 Å−1 (dk = 2) for liquids. A step size of 0.8 eV was used in the X-ray absorption near edge structure (XANES) region.
The samples (8–25 mg) were prepared by mixing and grinding stoichiometric amounts of metal fluorides in the desired stoichiometric ratio, and then pressed in the form of pellets of thickness less than 100 mm by applying a pressure of 10 tons cm−2. Sodium, potassium, and cesium fluorides were sourced from Alfa Aesar (99.99%, metals basis) and dried at 673 K for 4 h in an open nickel boat under Ar flow. The thorium fluoride was supplied by the Joint Research Center, Karlsruhe, Germany, (0.995 ± 0.005), synthesized as described in ref. 19. The UF4 was obtained from International Bio-Analytical Industries (0.9999 ± 0.0001 metals basis). All the samples in this work were handled in the inert and dry atmosphere of an argon-filled glovebox (H2O and O2 contents below 5 ppm), and in a nitrogen atmosphere when handled in the self-designed glovebox containing the furnace set-up at the KARA-INE beamline.
Each liquid-state scan took close to 30 minutes, and three to four scans were accumulated to be averaged. The energy E0 of the edge absorption threshold position was identified as the first x-intercept of the second derivative of the signal, which also corresponds to the first inflexion point of the signal. Before averaging, the spectra were aligned with the XANES spectrum of an appropriate reference: a ThO2–BN pellet in the case of AF–ThF4 (A = Li, Na, K, Cs) and ThF4, a metallic yttrium (K edge = 17.0384 keV) plate in the case of AF–UF4 (A = Na, K, Cs). A UO2–BN pellet was used as reference for the UF4 sample at room temperature. The references were located between the second and third ionization chambers and measured simultaneously with the sample. Data treatment of the raw XAS data was done with the ATHENA software,20 version 9.25. The XANES data were analyzed to confirm that the actinide cations remained tetravalent (refer to ESI†).
Fitting of the treated data with the standard EXAFS equation was done with Artemis software,20 version 0.8.012. No third-order cumulant parameter was used. The use of the standard EXAFS equation on liquids has limitations, since liquids are highly disordered and display strong anharmonicity. Moreover, the coordination number and distance to nearest neighbor distributions are lost, replaced by averages. To complement the information obtained with fitting, this work has also relied on MD simulations.
3 Molecular dynamics simulations
MD simulations were performed using the PIMAIM code14 for all compositions measured by EXAFS at the corresponding experimental temperature: 50 K above the liquidus line. The form of the potential used for the study of these molten salt systems is the Polarizable Ion Model (PIM).21 It has been chosen because it has already shown its usefulness in the study of several molten systems such as alkali fluoride mixtures,21 LiF–BeF2,17,22 AF–ZrF4 (A = Li, Na, K),7 LiF–UF4,23 and LiF–ThF4.16 The potential has four contributions with functional forms given in eqn (1)–(5): charge–charge (eqn (1)), dispersion (eqn (2)), overlap repulsion (eqn (4)) and polarization (eqn (5)).• Charge–charge:
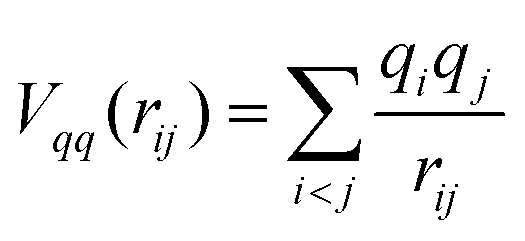 | (1) |
• Dispersion:
 | (2) |
 | (3) |
This work only consider dipoles and quadrupoles.
• Overlap repulsion
 | (4) |
Here Aij and aij are fitting parameters.
• Polarization
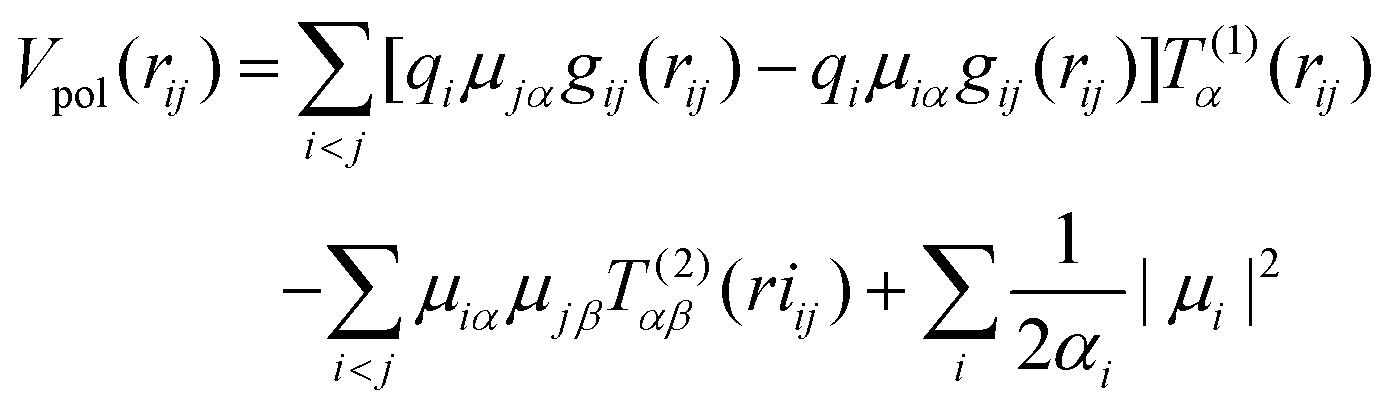 | (5) |
In the equation above, Tα(1) is the charge–dipole interaction tensor, Tαβ(2) is the dipole–dipole interaction tensor, αi is the polarizability of ion i, and μi is the set of dipoles, while gij(rij) is a damping function given by eqn (6):
 | (6) |
The parameters were taken from different sources, and are listed in Tables 1, 2 and 3. Aside from LiF–ThF4, the parameters were not derived ab initio especially for the binary systems in this work, but rather for other fluoride-based ionic systems (e.g. pure alkali fluorides).
| Ion pair | A ij | a ij | C ij 6 | C ij 8 | b ij 6 | b ij 6 | Source |
|---|---|---|---|---|---|---|---|
| F−–F− | 282.3 | 2.44 | 15 | 150 | 1.9 | 1.9 | 16 |
| F−–U4+ | 70.623 | 1.666 | 38.7 | 387 | 1.9 | 1.9 | 23 |
| F−–Th4+ | 70.148 | 1.634 | 38.7 | 387 | 1.9 | 1.9 | 16 |
| F−–Na+ | 52.83 | 1.97 | 13.25 | 88.15 | 1.9 | 1.9 | 25 |
| F−–K+ | 138.8 | 2.043 | 3.9 | 38.7 | 1.9 | 1.9 | 25 |
| F−–Cs+ | 151.12 | 1.874 | 10.95 | 109.5 | 1.9 | 1.9 | 21 |
| An4+–An4+ | 1 | 5 | 100 | 1000 | 1.9 | 1.9 | 23 |
| Th4+–Li+ | 1 | 5 | 3.16 | 31.6 | 1.9 | 1.9 | 23 |
| Th4+–Na+ | 1 | 5 | 0.001 | 0.001 | 1.9 | 1.9 | 17, 21 and 26 |
| U4+–Na+ | 1 | 5 | 10 | 100 | 1.9 | 1.9 | 21, 25 and 27 |
| An4+–A+ (An = U, Th; A = K, Cs) | 1 | 5 | 10 | 100 | 1.9 | 1.9 | 21 and 27 |
| Na+–Na+ | 1 | 5 | 11.7 | 5.18 | 1.9 | 1.9 | 25 |
| K+–K+ | 1 | 5 | 1 | 10 | 1.9 | 1.9 | 25 |
| Cs+–Cs+ | 5000 | 3 | 8 | 80 | 1.9 | 1.9 | 28 |
| Interaction | b D ij | c D ij | Source | |
|---|---|---|---|---|
| a Modified from cDUF = −0.84905 in ref. 23. b Not defined elsewhere, set arbitrarily. | ||||
| F−–U4+ | b DFU = bDUF = 1.7516 | c DFU = 1.8148 | c DUF = 0.84905a | 23 |
| F−–Th4+ | b DFU = bDUF = 1.938 | c DFU = 2.791 | c DUF = −0.60906 | 16 |
| F−–Na+ | b DFNa = bDNaF = 1.84 | c DFNa = 2.54 | c DNaF = −0.19 | 25 |
| F−–K+ | b DFK = bDKF = 1.75 | c DFK = 2.5 | c DKF = −0.31 | 25 |
| F−–Cs+ | b DFCs = bDCsF = 1.93 | c DFCs = 3.391 | c DCsF = 0.485 | 28 |
| An4+–A+b | b DAnA = 10.0 | c DAAn = 0.001 | — | — |
The systems were equilibrated for 500 ps in the NPT ensemble at 0 GPa and the corresponding temperature 50 K above the liquidus (Table 5), from which the equilibrium volume was taken. This was followed by a 100 ps equilibration and finally a 500 ps production run in the NVT ensemble at the same temperature. Time steps in all runs were set to 0.5 fs, while the relaxation time for both the Nosé–Hoover thermostat and barostat (for the NPT run) was set to 10 ps. The cubic simulation cell contained 600–800 ions in periodic boundary conditions. Cut-offs for the real space part of the Ewald sum and short-range potential were both set to less than half the length of the cell. After the production run, the extracted MD trajectories were used as input for the ab initio code FEFF8.4029 to compute a simulated EXAFS spectrum, to be compared with experimental data. The effect of anharmonic vibrations and Debye–Waller factor were accounted for by accumulating about 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ionic coordinates as input.
000 ionic coordinates as input.
4 Results and discussion
4.1 Local structure of uranium and thorium tetrafluoride
 | ||
| Fig. 1 (a) Experimental (fluorescence, room temperature: □, transmission, T = 1357 K: ■), fitted (green, blue) and simulated (red) k2χ(k) oscillations of UF4. (b) Fourier transform moduli |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 2 (a) Experimental (transmission, room temperature: □, transmission, T = 1433 K: ■), fitted (green, blue) and simulated (red) k2χ(k) oscillations of ThF4. (b) Fourier transform moduli |χ(R)| of the EXAFS spectra. Room temperature experimental data and fit taken from Smith et al.9 | ||
Uranium tetrafluoride is isostructural with thorium tetrafluoride, with space group C2/c. There are two uranium sites, both 8-coordinated, with U–F distances within ∼0.1 Å of each other (see Table 4). In EXAFS, the threshold above which it is possible to discriminate between neighboring shells depends on the k-range of the spectra, Δk, as ΔR = π/(2Δk). For the solid-state measurement in Fig. 1a, ΔR = 0.17 Å. This means that the resolution in this case is not sufficient to distinguish between more than one U–F first coordination shell and more than one U–U second-nearest neighbor shell, so only one of each was included in the fit. The results are summarized in Table 4 and compared with the structural model used. The bond lengths are within error of each other.
| Bond | CN | R/Å | σ 2/Å2 |
|---|---|---|---|
| Neutron data from ref. 30 | |||
| U1–F6 | 2 | 2.256(4) | |
| U1–F4 | 2 | 2.268(4) | |
| U1–F7 | 2 | 2.280(4) | |
| U1–F3 | 2 | 2.328(4) | |
| U2–F5 | 1 | 2.237(2) | |
| U2–F1 | 1 | 2.247(2) | |
| U2–F7 | 1 | 2.273(2) | |
| U2–F6 | 1 | 2.281(2) | |
| U2–F3 | 1 | 2.286(2) | |
| U2–F5 | 1 | 2.288(2) | |
| U2–F2 | 1 | 2.308(2) | |
| U2–F4 | 1 | 2.313(2) | |
| U1–U2 | 2 | 4.516(2) | |
| U2–U2 | 2 | 4.510(2) | |
| Averaged neutron data | |||
| U–F | 8 | 2.28(2) | |
| U–U | 2 | 4.51(4) | |
| EXAFS data | |||
| U–F | 8 | 2.26(3) | 0.008(6) |
| U–U | 2 | 4.50(2) | 0.0001(5) |
In the molten state, the loss of long-range order gives rise to strong damping of the EXAFS signal, as can be seen in Fig. 1. The disorder is also manifest in the Fourier transform (FT) modulus of the signal, where the intensity of the first peak in the liquid state is much diminished with respect to that of the solid. Furthermore, the second shell of U neighbors, displayed by the crystal phase as a peak centered at ∼4.5 Å, can no longer be resolved in the liquid. The same is true in the case of ThF4: the crystal has its second shell centered around ∼4.6 Å9 and in the liquid it is not visible anymore.
 | ||
| Fig. 3 Pairwise radial distribution functions of molten UF4 and ThF4 calculated with MD at T = 1357 K and T = 1433 K, respectively. | ||
The EXAFS oscillations in the molten state become considerably noisy at high wavenumbers, but there is a fair agreement between the first three oscillations in the experimental k2χ(k) EXAFS signal, the simulation, and the fit. The MD-generated signals are slightly out of phase with the EXAFS signal, which in many cases translates into an over or underestimation of the radial distance of the maximum of |χ(R)|. The fits using the standard EXAFS equation match the phase and the intensity of the experimental signal more closely. However, the fits themselves were directed by the quantitative information which could be directly extracted from the MD trajectories, namely the coordination numbers (Table 5). These tended to be underestimated by the fitting algorithm, a common problem arising when fitting EXAFS signals of liquids.12 Hence, if a fitting process is deemed appropriate for a given disordered system, it is good practice to combine it with another suitable structural model, e.g. MD, to gauge the results.
| System | X(AnF4)d | CNAn–F | CNAn–F | R An–F | R An–F | E[RAn–F]b | E[RAn–F]b | ΔE | σ 2 | R f | K/Te |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MD | Fit | MD/Å | MD/Å | MD/Å | Fit/Å | Fit/eV | Fit/Å2 | Fit | |||
| a Most probable distance. b Expected value. c Bond cut-off = maximum An–F distance. d Standard uncertainty on the composition of starting reagents was u(X(AnF4)) = 0.005. e Standard uncertainty on the temperature is estimated to be u(T) = 20 K. | |||||||||||
| LiF–ThF4 | 0.10 | 7.88 | 2.21 | 3.14 | 2.32 | 2.34(1) | 4(1) | 0.020(6) | 0.056 | 1133 | |
| 0.25 | 7.73 | 2.22 | 3.15 | 2.26 | 2.32(1) | 4.59(72) | 0.02(2) | 0.015 | 973 | ||
| 0.50 | 7.78 | 2.22 | 3.15 | 2.33 | 2.31(1) | 1.31(79) | 0.02(3) | 0.027 | 1193 | ||
| NaF–ThF4 | 0.33 | 7.52 | 7.4(4) | 2.21 | 3.17 | 2.31 | 2.30(1) | 6.75(43) | 0.020(1) | 0.016 | 1073 |
| 0.50 | 7.89 | 7.8(8) | 2.20 | 3.18 | 2.33 | 2.34(1) | 4.08(86) | 0.021(2) | 0.035 | 1108 | |
| 0.67 | 7.83 | 7.9(7) | 2.21 | 3.18 | 2.33 | 2.31(1) | 2.52(77) | 0.019(1) | 0.024 | 1252 | |
| NaF–UF4 | 0.50 | 7.75 | 7.2(7) | 2.15 | 3.07 | 2.27 | 2.26(1) | 1.61(87) | 0.020(2) | 0.036 | 1033 |
| 0.67 | 7.76 | 7.2(7) | 2.16 | 3.05 | 2.27 | 2.23(1) | 2.26(57) | 0.019(2) | 0.025 | 1153 | |
| KF–UF4 | 0.50 | 7.42 | 6.9(4) | 2.13 | 3.09 | 2.25 | 2.26(1) | 7.07(50) | 0.020(1) | 0.012 | 1098 |
| 0.67 | 7.72 | 7.8(7) | 2.16 | 3.07 | 2.27 | 2.23(1) | 2.56(94) | 0.02(2) | 0.049 | 1090 | |
| KF–ThF4 | 0.167 | 7.12 | 7.0(7) | 2.22 | 3.23 | 2.28 | 2.29(1) | 3.71(94) | 0.018(2) | 0.033 | 1060 |
| 0.33 | 6.99 | 6.5(8) | 2.19 | 3.15 | 2.27 | 2.30(1) | 2(1) | 0.017(3) | 0.048 | 1123 | |
| 0.50 | 7.53 | 7.6(8) | 2.18 | 3.23 | 2.31 | 2.31(1) | 3.17(92) | 0.024(2) | 0.040 | 1209 | |
| 0.67 | 7.70 | 8.0(4) | 2.20 | 3.20 | 2.33 | 2.33(5) | −1(1) | 0.020(3) | 0.089 | 1266 | |
| CsF–ThF4 | 0.25 | 6.24 | 6.6(7) | 2.15 | 3.16 | 2.21 | 2.29(1) | 3.65(92) | 0.020(2) | 0.043 | 1301 |
| 0.50 | 6.97 | 6.9(5) | 2.14 | 3.25 | 2.27 | 2.33(1) | 4.48(64) | 0.016(1) | 0.028 | 1201 | |
| CsF–UF4 | 0.25 | 6.2 | 7.0(6) | 2.12 | 3.11 | 2.17 | 2.25(1) | 7.48(78) | 0.018(2) | 0.027 | 1293 |
| 0.50 | 7.14 | 7.8(6) | 2.12 | 3.11 | 2.23 | 2.26(1) | 2.75(71) | 0.020(2) | 0.019 | 1058 | |
| 0.67 | 7.46 | 8.2(8) | 2.14 | 3.08 | 2.26 | 2.26(1) | −0.72(99) | 0.021(2) | 0.048 | 1191 | |
| ThF4 | 1 | 7.92 | 8.0(8) | 2.26 | 3.17 | 2.34 | 2.32(1) | 2.38(87) | 0.020(2) | 0.035 | 1433 |
| UF4 | 1 | 7.94 | 8.0(7) | 2.21 | 3.06 | 2.28 | 2.27(1) | 3.52(71) | 0.030(2) | 0.031 | 1357 |
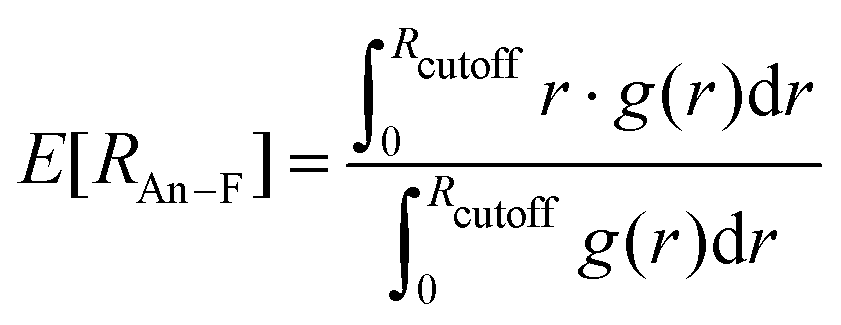 | (7) |
The spread between Rmax,Th–F = 2.26 Å and Rcutoff,Th–F = 3.17 Å is 0.91 Å, hence more than 6 times greater than in the solid state (0.141 Å), as derived from neutron diffraction data.9 In the case of UF4(l), Rcutoff,Th–F − Rmax,U–F = (3.06–2.21) Å = 0.85 Å, about 12 times greater than in UF4(cr) (0.072 Å, see Section 4.1.1 above). In that sense, there is an expansion of the coordination cage, as remarked by Bessada et al.10 Nevertheless, as Liu et al. suggested after finding Rmax to be smaller than the sum of the Shannon radii of Th4+ and F−, there is a concordant strengthening of the local structure. Ocádiz-Flores et al.31 reported the same phenomenon by comparing solid phases occurring in the LiF–UF4 system with the melts at the corresponding mole fractions. On average, the strengthening can be observed because even though longer bond lengths close to Rcutoff become available, shorter bond lengths close to Rmax are much more populated, with the result that the average U–F distance in the liquid mixtures is equal to or shorter than in the solids. For ThF4: E[R] = 2.26 < 2.324(19) Å in the solid‡. For UF4: E[R] = 2.28 = 2.28(2) Å in the solid§.
Bessada et al.10 calculated average bond lengths in the LiF–ThF4 melt as a function of coordination number, and the former are seen to consistently decrease as the latter increases. This can be explained mainly by an increased repulsion between the F− ligands with increasing coordination. Decreased shielding from the 2nd shell, evidenced by the absence of an An–An peak in the plots of |χ(R)| (Fig. 1a and 2b), is likely to contribute to the shortening of the An–F distance as well.32 In the liquid phase, the coordination numbers are dominated by CN = 7, 8, 9, with an average close to 8, yet CN = 7 weighs more in the distribution than CN = 9 (see Table 6), allowing for tightening of the cage. It is 8 in the solid state. Finally, in the gas phase, where fluorides are not shared between actinide centers, it is found that E[RTh–F,gas] = 2.08 Å,33 and E[RU–F,gas] = 2.017(5) Å.34
| System | X(AnF4) | Anionic fraction [AnFx]4−x | Expected value | ||||
|---|---|---|---|---|---|---|---|
| 6 | 7 | 8 | 9 | 10 | CN | ||
| LiF–ThF4 | 0.10 | 0.008 | 0.243 | 0.612 | 0.134 | 0.003 | 7.88 |
| 0.25 | 0.037 | 0.322 | 0.518 | 0.121 | 0.004 | 7.73 | |
| 0.50 | 0.040 | 0.315 | 0.476 | 0.157 | 0.012 | 7.78 | |
| NaF–ThF4 | 0.33 | 0.055 | 0.432 | 0.442 | 0.070 | 0.002 | 7.52 |
| 0.50 | 0.0271 | 0.271 | 0.505 | 0.184 | 0.0134 | 7.89 | |
| 0.67 | 0.0367 | 0.294 | 0.484 | 0.171 | 0.0137 | 7.83 | |
| NaF–UF4 | 0.5 | 0.036 | 0.323 | 0.503 | 0.133 | 0.006 | 7.75 |
| 0.67 | 0.038 | 0.322 | 0.491 | 0.141 | 0.007 | 7.76 | |
| KF–ThF4 | 0.167 | 0.836 | 0.163 | 0.001 | 0.0 | 0 | 7.12 |
| 0.33 | 0.233 | 0.551 | 0.206 | 0.010 | 5.85 × 10−5 | 6.99 | |
| 0.5 | 0.089 | 0.396 | 0.412 | 0.098 | 0.005 | 7.53 | |
| 0.67 | 0.060 | 0.343 | 0.449 | 0.139 | 0.010 | 7.70 | |
| KF–UF4 | 0.5 | 0.106 | 0.440 | 0.388 | 0.065 | 0.002 | 7.42 |
| 0.67 | 0.041 | 0.333 | 0.491 | 0.129 | 0.006 | 7.72 | |
| CsF–UF4 | 0.25 | 0.806 | 0.193 | 0.003 | 0.00 | 0.00 | 6.20 |
| 0.5 | 0.209 | 0.475 | 0.283 | 0.033 | 7.48 × 10−4 | 7.14 | |
| 0.67 | 0.103 | 0.422 | 0.392 | 0.079 | 0.003 | 7.46 | |
| CsF–ThF4 | 0.25 | 0.771 | 0.223 | 0.006 | 3.31 × 10−5 | 0 | 6.24 |
| 0.5 | 0.294 | 0.469 | 0.212 | 0.025 | 7.94 × 10−4 | 6.97 | |
| ThF4 | 1 | 0.034 | 0.262 | 0.473 | 0.210 | 0.021 | 7.92 |
| UF4 | 1 | 0.023 | 0.250 | 0.509 | 0.201 | 0.018 | 7.94 |
for both actinides, Fig. 3). The network structure found for molten UF4 with a distribution of 70% corner-sharing, 27% edge-sharing, and 3% face-sharing is very similar to that of ThF4, confirming the view of the melt provided by the RDFs. An MD snapshot of the networks, emphasizing An–An connectivity, is shown in Fig. 4.
 | ||
| Fig. 4 (MD) snapshot of (a) molten UF4 and (b) ThF4 at the EXAFS measurement temperatures, T = 1357 K and 1433 K, respectively, emphasizing An–An connectivity (U: green, Th: blue). Fluoride ions shown in gray. | ||
4.2 Local structure of the AF–UF4 and AF–ThF4 melts
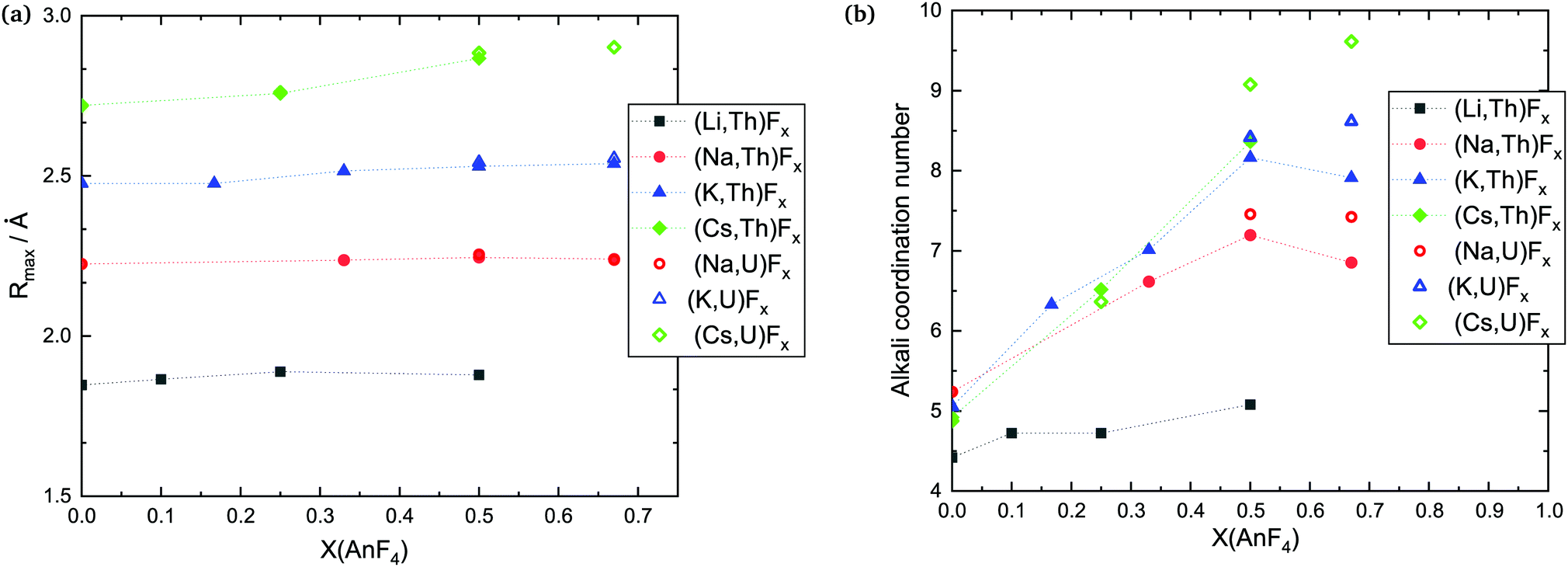 | ||
| Fig. 5 (a) Most probable A–F (A = Li, Na, K, Cs) distance and (b) average coordination number around the alkali cations in the (A,An)Fx solutions as a function of composition, calculated with MD in this work. Temperatures are variable; they are indicated in Table 6. For the pure AF melts, T = 1200 K for all except NaF with T = 1270 K (above the melting point). | ||
:ThF4 = (0.75:0.25) (Fig. 6), which can be singled out because of its importance as Molten Salt Fast Reactor (MSFR) fuel, is compared with data by Bessada et al.10 Although recorded at different temperatures, the spectra are comparable to each other. The experimental data for all compositions can be found in the ESI.†
 | ||
| Fig. 6 (a) Experimental (■, ref. 9), simulated (red), and fitted (blue) k2χ(k) oscillations of (LiF:ThF4) = (0.75:0.25) (fluorescence, T = 898 K). Dotted green line: experimental data by Bessada et al.10 at the same composition (absorption, T = 973 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 7 (a) Experimental (■) and calculated (red), and fitted (blue) k2χ(k) oscillations of (NaF:ThF4) = (0.33:0.67) (absorption, T = 1252 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 8 (a) Experimental (■) and calculated (red), and fitted (blue) k2χ(k) oscillations of (NaF:UF4) = (0.33:0.67) (absorption, T = 1153 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 9 (a) Experimental (■), simulated (red), and fitted (blue) k2χ(k) oscillations of (KF:ThF4) = (0.67:0.33) (fluorescence, T = 1123 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 10 (a) Experimental (■), simulated (red), and fitted (blue) k2χ(k) oscillations of (KF:UF4) = (0.50:0.50) (absorption, T = 1098 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 11 (a) Experimental (■), simulated (red), and fitted (blue) k2χ(k) oscillations of (CsF:UF4) = (0.75:0.25) (absorption, T = 1293 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
 | ||
| Fig. 12 (a) Experimental (■), simulated (red), and fitted (blue) k2χ(k) oscillations of (CsF:ThF4) = (0.75:0.25) (absorption, T = 1301 K). (b) Fourier transform modulus |χ(R)| of the EXAFS spectra. | ||
Table 6 lists the distribution and expected values of coordination numbers as calculated in this work. They are also plotted in Fig. 13. In agreement with previous studies,9,10,16,27,35,37 the distribution ranges between 6 and 10. From the works related to LiF–ThF4 and LiF–UF4 melts,8,9,16,23,27,31,35 it is clear that the dominant contributions are from CN = 7, 8, and 9 at all temperatures and compositions. Guo et al.,27 who performed MD calculations of dilute solutions of ThF4 in AF (A = Li, Na, K) systems, observed a bimodal distribution with virtually only [ThF7]3− and [ThF8]4− complexes present, and very small concentrations of [ThF9]5− (mean values at 1373 K indicated in cyan in Fig. 13). Bessada et al. report the following mean CN in the NaF–ThF4 system at 1223 K: 7.6 for X(ThF4) = 0.10, ∼7.8 for X(ThF4) = 0.45, and ∼8.0 for X(ThF4) = 0.6.10 These results correlate well with those in Table 5, where it can be seen that NaF–UF4 behaves similarly.
 | ||
| Fig. 13 Average coordination number in the (A,An)Fx solutions as a function of temperature, and composition, calculated with MD. Unless otherwise indicated, the data is from this work. Where the temperature is not indicated, it is variable, indicated in Table 5. | ||
In the KF–ThF4 system it was found previously,37 also with a combined EXAFS-MD approach, that the dominant complexes at X(ThF4) = 0.25, 1073 K, were [ThF6]2− (14.5%), [ThF7]3− (68.7%), and [ThF7]3− (16.8%), for an expected CN of 7.03.37 In a later work10 the authors reported a mean coordination of 7.5 in the KF–ThF4 system at X(ThF4) = 0.45, T = 1173 K, and slightly higher at T = 1073 K. These values are in excellent agreement with the data collected in this work, i.e. E[CN] = 7.12 at X(ThF4) = 0.167, T = 1060 K, E[CN] = 6.99 at X(ThF4) = 0.33, T = 1123 K, and E[CN] = 7.53 at X(ThF4) = 0.50, T = 1209 K (Table 6).
Finally, there are, to the best of our knowledge, no EXAFS, MD, or otherwise stuctural studies available in the literature on the CsF–AnF4 (A = U, Th) systems. Like for the other AF–AnF4 systems, the CN distribution is calculated to be in the 6–10 range, with a small contribution from CN = 9 and a negligible one from CN = 10. The atomic environment for the An4+ cations in the crystal phases occurring in the CsF–AnF4 binary systems is mostly a tricapped trigonal prism (CN = 9)38 except for an octa-coordinated site in Cs2UF6 (space group Cmcm,39) and an exceptionally rare 1-coordinated site in one of the polymorphs of CsThF5 (space group P4/nmm40). Hence, as discussed earlier for thorium and uranium tetrafluorides, thermal disorder reduces the coordination number in the first solvation shell, as CN = 6 becomes more prevalent than in melts based on other alkali fluorides.
As for the An–F bond length, which was varied freely during the fitting process without constraints based on the MD simulations, the agreement between the fits and the simulations is quite good: in all cases, the average value of the fit falls between the most probable distance and the bond cutoff distance extracted from the MD trajectories, i.e., it is close to the expected value of the simulations.
4.3 Main structural features of the liquid mixtures
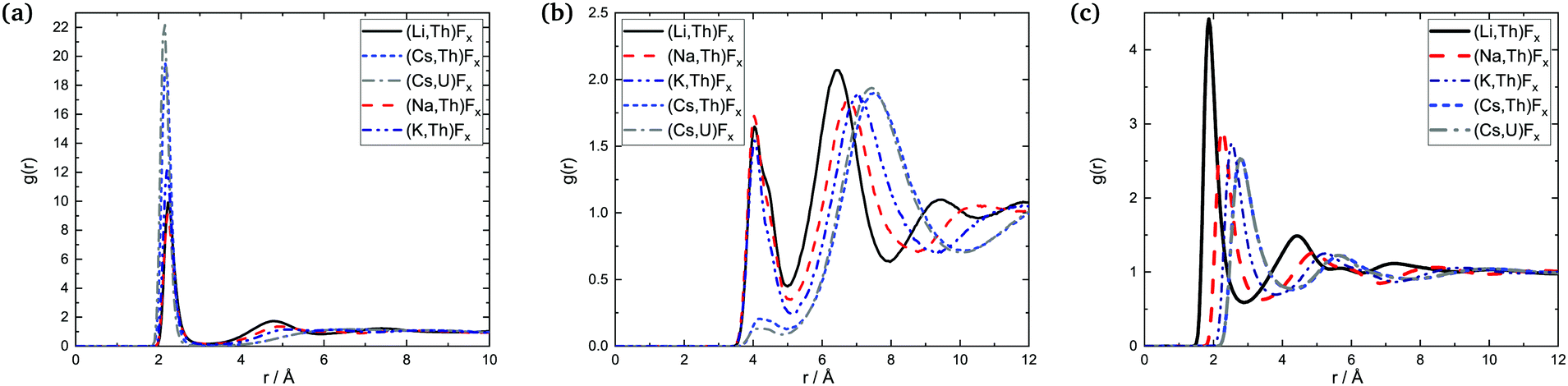 | ||
| Fig. 14 Radial distribution functions: (a) An–F, (b) An–An, and (c) A–F in the AF–AnF4 melts at composition X(AnF4) = 0.25 (Li and Cs-based melts) and X(AnF4) = 0.33 (Na and K-based melts). | ||
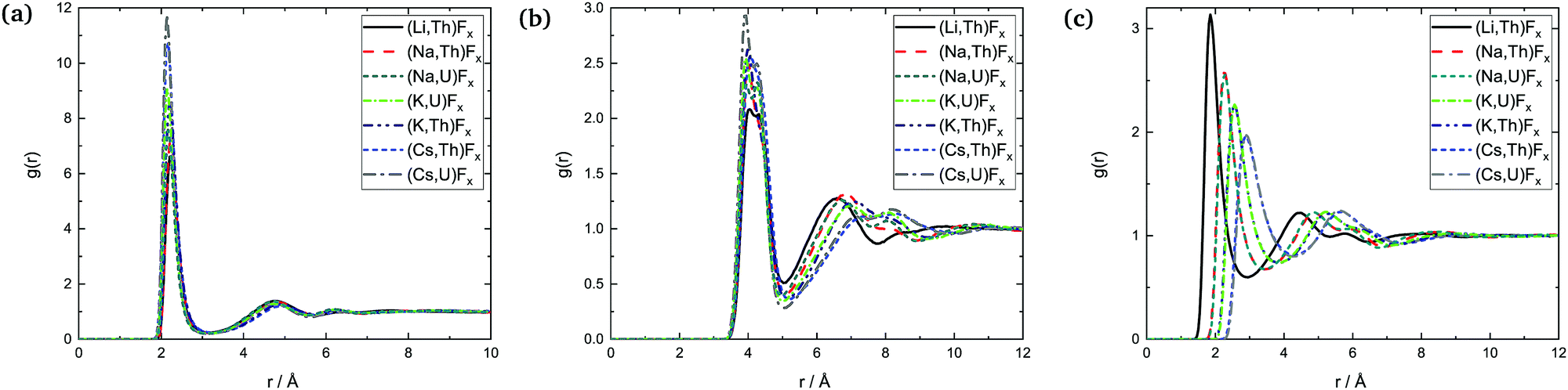 | ||
| Fig. 15 Radial distribution functions: (a) An–F, (b) An–An, and (c) A–F in the AF–AnF4 melts at composition X(AnF4) = 0.50. | ||
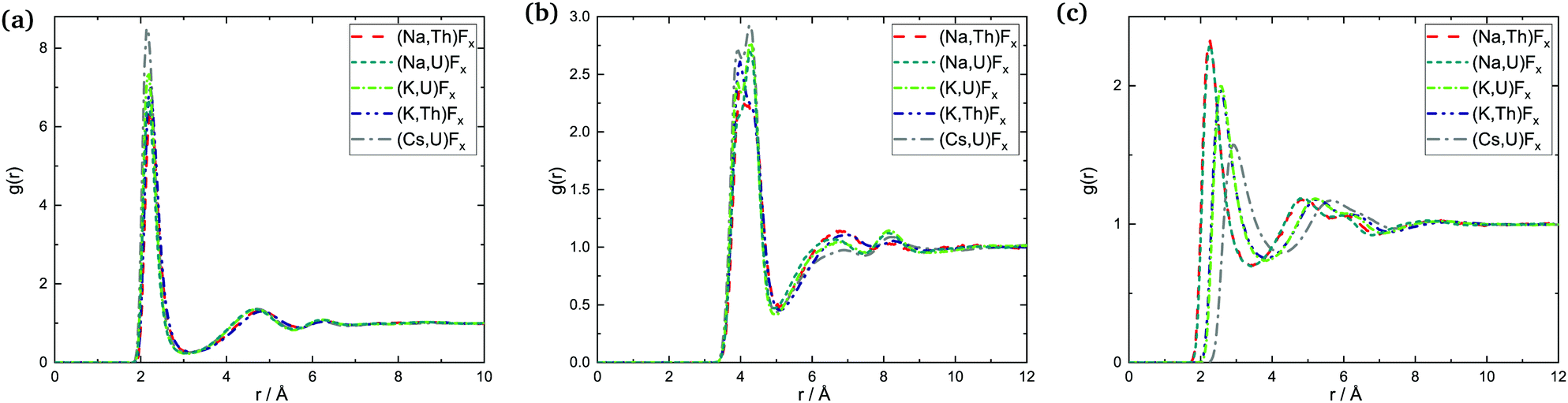 | ||
| Fig. 16 Radial distribution functions: (a) An–F, (b) An–An, and (c) A–F in the AF–AnF4 melts at composition X(AnF4) = 0.67. | ||
The data available, with varying compositions across systems, allow for three main trends to be observed: (i) trends across composition, (ii) trends as a function of the actinide metal, and (iii) trends as a function of the alkali metal.
which is of lower intensity than the following one centered between 6 and 7 Å: it is more probable at that composition for An centers to be farther away from each other than second-nearest neighbors (SNN) because it is more favorable for alkali cations to occupy SNN sites. In melts richer in AnF4 the An number density makes it more likely that actinide pairs will be SNN, so likely indeed that fluoride bridging is still possible, even in the presence of bulky Cs+ cations. This change is apparent in the An–F RDFs when going from being dissolved in CsF (X(AnF4) = 0.25) to AnF4 being the solvent (X(AnF4) = 0.67): in the former case only the first shell is distinguishable, while at the other two compositions there is a second shell centered at ∼4.8 Å where the majority of the fluorides meet the fluoride bridging criteria for r: r < 5.1 Å ∼ RAn–An,max and r < 6.2 Å ∼ 2R·An–F,max.
The An–F bond cutoffs are not very sensitive to composition, a result which agrees with the analysis of Th–F,41 U–F,31 and Zr–F7 bond lengths in mixtures with LiF. Rcutoff,An–F, however, becomes slightly more populated as the actinide cations start competing with each other for the fluoride counter ions, which results in an increase in the average bond length and the average coordination number. This increase is not monotonic, and actually both quantities were found to go through a minimum between 20 and 30 mol%, (where F:Th ∼7) in isothermal studies of AF–ThF4 (A = Li, Na, K) mixtures.10 This effect can be seen in Fig. 13 as well.
A second measure of the stabilization of the [AnFx]4−x complexes in alkali fluoride melts is given by the negative mixing enthalpies in these systems. The liquid–liquid mixing enthalpies in AF–ZrF4 (A = Li, Na, K, Rb) mixtures, are convex curves which become more negative in the order Li, Na, K, Rb.44 The AF–ThF4 (A = Li, Na, K) systems26,45,46 obey the same trend. Moreover, the depth of the curves is greater in ZrF4-based systems, due to the strength of the shorter Zr–F bond. The progressive tightening of the [AnFx]4−x cage down the alkali family corresponds well to the trend in liquid–liquid mixing enthalpies.
Another important conclusion from the study by Pauvert et al.7 was that large alkali cations favor lower coordination numbers around zirconium. In NaF and KF-based systems, [ZrF8]4− complexes were only formed when the concentration of ZrF4 exceeded 40–45 mol%. More recently, Bessada et al.10 pointed out that shorter AF–AnF4 (A = Li, Na, K) bond lengths and lower CN are favored as the radius of the alkali cation grows. In the case of CsF-based systems, the trend is accentuated, such that the average CN around the tetravalent actinides is heretofore unseen: as low as 6.2 at X(AnF4) = 0.25 (Table 5).
4.4 Implications for the transport properties: viscosity and electrical conductivity
Bessada et al.10 pointed out that the viscosity of LiF–AnF4 (A = Th, U) mixtures should be greater than that of LiF–ZrF4 at a the same molar composition, since in the latter system the emergent ionic network is weaker, having instead more stable coordination anions. At a given temperature, a dissociated melt should be less viscous than a molecular one, which in turn should be less viscous than a polymerized one. Nevertheless, it is interesting to consider the role of the alkali cation: the heavier and larger cations stabilize the coordination shell around the tetravalent cation, yet make the network more fragile.7,47 These two trends should have opposite effects, resulting in a non-monotonic increase of viscosity with AnF4 concentration. This is what is observed in Fig. 17, adapted from ref. 48. At a temperature of 1230 K, LiF–UF4, with relatively weak complexes but little ability to disrupt the ionic network, displays a viscosity increasing monotonically with UF4 concentration. The KF–UF4 system on the other hand, displays a local maximum in the vicinity of X(UF4) = 0.33. The MD trajectories of the analogous Th composition KF:ThF4 = (0.67:0.33) reveal a large fraction (∼0.5) of isolated [ThFx]4−x complexes. Then, there is a local minimum of the viscosity near X(UF4) = 0.5. The KF:UF4 = (0.5:0.5) simulation of the EXAFS sample, shows a large extent of network formation, (∼66.5%), but the network appears to be fragile enough that the composition with more long-lived isolated complexes (X(UF4 ∼ 0.33)) has a greater viscosity. From X(UF4) = 0.5 onwards, the network becomes more robust as K+ is diluted, and the viscosity increases, now monotonically.
 | ||
| Fig. 17 Molar viscosity (erg s mol−1) of AF–UF4 (A = Li, K) melts at an isotherm of T = 1230 K. Adapted from ref. 48. | ||
While the viscosities of CsF–AnF4 systems have not been measured, the electrical conductivities of AF–AnF4 (A = Li, Na, K, Rb, Cs; An = Th, U) are available in the literature,49–51 and a similar interpretation may be given. For isotherms across AnF4 mole fraction, the increasing alkali radius is reflected in decreasing electrical conductivities. The conductivities of AF–AnF4 (A = Li, Na; An = Th, U) do not go through local minima, while those of KF, Rb, and CsF-based melts have minima in the 30–50 mol% region. The reduced ionic mobilities in those areas suggest that the viscosity could go through a local maximum as seen in Fig. 17.
5 Conclusion
Structural studies of alkali fluoride/actinide fluoride salt mixtures in the molten state have been presented, using a combined EXAFS-MD approach which has proven to be a valuable tool to explore the rich chemistry of molten salt systems. Fitting the standard EXAFS equation was done in parallel, and was shown to be an adequate means to extract information about the first coordination shell of thorium and uranium, provided there is another structural model, e.g. obtained from MD, to constrain the fit to physically realistic values. Remarkably, the EXAFS spectra of pure molten ThF4 and UF4 were experimentally measured for the first time: the PIM potential parameters derived ab initio by Dewan et al.16 can approximate them well.PIM potentials derived for other AF-based systems (A = Na, K, Cs) have shown to have good applicability for the corresponding AF–AnF4 (An = U, Th) binaries. Fluorides are more loosely bound to alkali cations in mixtures with actinide fluorides than in the pure alkali fluoride melts, while the opposite is true for actinide cations. The observed trends of decreasing average bond length and coordination around the actinide with increasing alkali radius are followed when Cs+ is present in the melt; surprisingly, Cs+ can even promote an expected coordination number close to 6, something which had not been observed for Li+, Na+, or K+-based systems. The role alkali cations play in stabilizing coordination complexes while destabilizing networks in AF–AnF4 mixtures, and the opposing effects that might have on the viscosity, was discussed. In future works the role of charge and electronegativity of other families of solvents in molten salt environments will be explored.
6 CRediT author statement
J. A. Ocádiz-Flores: Conceptualization, Methodology, Investigation, Formal analysis, Visualization, Data Curation, Writing – Original Draft preparation A. E. Gheribi: Investigation J. Vlieland: Investigation D. de Haas: Investigation K. Dardenne: Investigation J. Rothe: Investigation R. J. M. Konings: Conceptualization, Supervision, Writing – Review & Editing A. L. Smith: Conceptualization, Methodology, Investigation, Supervision, Funding acquisition, Resources, Project Administration, Writing – Review & Editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Pavel Souček and Ondrej Beneš for providing us with ThF4 of high purity. We also thank Mathieu Salanne for the use of his Molecular Dynamics code, and fruitful discussions on these systems. We acknowledge the KIT light source for provision of instruments at the INE-Beamline of the Institute for Nuclear Waste Disposal and we would like to thank the Insitute for Beam Physics and Technology (IBPT) for the operation of the storage ring, the Karlsruhe Research Accelerator (KARA). A. L. Smith acknowledges financial support from the Netherlands Organisation for Scientific Research (NWO) (project 722.016.005). J. A. Ocádiz-Flores acknowledges CONACYT-SENER for financial support.References
- A. R. Kamali, RSC Adv., 2020, 10, 36020–36030 RSC.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS.
- W. Weng, L. Tang and W. Xiao, J. Energy Chem., 2019, 28, 128–143 CrossRef.
- J. J. Laidler, J. Battles, W. Miller, J. Ackerman and E. Carls, Prog. Nucl. Energy, 1997, 31, 131–140 CrossRef CAS.
- E. S. Bettis, R. W. Schroeder, G. A. Cristy, H. W. Savage, R. G. Affel and L. F. Hemphill, Nucl. Sci. Eng., 1957, 2, 804–825 CrossRef CAS.
- C. Renault, M. Hron, R. J. M. Konings and D.-E. Holcomb, GIF Symposium, Paris(France), 9–10 September 2009, pp. 191–200.
- O. Pauvert, M. Salanne, D. Zanghi, C. Simon, S. Reguer, D. Thiaudière, Y. Okamoto, H. Matsuura and C. Bessada, J. Phys. Chem. B, 2011, 115, 9160–9167 CrossRef CAS PubMed.
- C. Bessada, D. Zanghi, O. Pauvert, L. Maksoud, A. Gil-Martin, V. Sarou-Kanian, P. Melin, S. Brassamin, A. Nezu and H. Matsuura, J. Nucl. Mater., 2017, 494, 192–199 CrossRef CAS.
- A. L. Smith, M. N. Verleg, J. Vlieland, D. D. Haas, J. A. Ocadiz-Flores, P. Martin, J. Rothe, K. Dardenne, M. Salanne and A. E. Gheribi, et al. , J. Synchrotron Radiat., 2019, 26, 124–136 CrossRef CAS PubMed.
- C. Bessada, D. Zanghi, M. Salanne, A. Gil-Martin, M. Gibilaro, P. Chamelot, L. Massot, A. Nezu and H. Matsuura, J. Mol. Liq., 2020, 307, 112927 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- A. Filipponi, J. Phys.: Condens. Matter, 2001, 13, R23 CrossRef CAS.
- Y. Okamoto, Nucl. Instrum. Methods Phys. Res., Sect. A, 2004, 526, 572–583 CrossRef CAS.
- P. A. Madden and M. Wilson, Chem. Soc. Rev., 1996, 25, 339–350 RSC.
- P. Jemmer, M. Wilson, P. A. Madden and P. W. Fowler, J. Chem. Phys., 1999, 111, 2038–2049 CrossRef CAS.
- L. C. Dewan, C. Simon, P. A. Madden, L. W. Hobbs and M. Salanne, J. Nucl. Mater., 2013, 434, 322–327 CrossRef CAS.
- R. J. Heaton, R. Brookes, P. A. Madden, M. Salanne, C. Simon and P. Turq, J. Phys. Chem. B, 2006, 110, 11454–11460 CrossRef CAS PubMed.
- J. Rothe, S. Butorin, K. Dardenne, M. Denecke, B. Kienzler, M. Löble, V. Metz, A. Seibert, M. Steppert and T. Vitova, et al. , Rev. Sci. Instrum., 2012, 83, 043105 CrossRef CAS PubMed.
- P. Souček, O. Beneš, B. Claux, E. Capelli, M. Ougier, V. Tyrpekl, J. Vigier and R. Konings, J. Fluorine Chem., 2017, 200, 33–40 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Salanne, B. Rotenberg, S. Jahn, R. Vuilleumier, C. Simon and P. A. Madden, Theor. Chem. Acc., 2012, 131, 1143 Search PubMed.
- M. Salanne, C. Simon, P. Turq, R. J. Heaton and P. A. Madden, J. Phys. Chem. B, 2006, 110, 11461–11467 CrossRef CAS PubMed.
- L. Dewan, PhD thesis, Massachusetts Institute of Technology, 2013.
- K. Tang and J. Toennies, J. Chem. Phys., 1984, 80, 3726–3741 CrossRef CAS.
- M. Salanne, C. Simon, P. Turq and P. A. Madden, J. Fluorine Chem., 2009, 130, 38–44 CrossRef CAS.
- M. Schreuder, MSc thesis, Delft University of Technology, 2019.
- X. Guo, H. Qian, J. Dai, W. Liu, J. Hu, R. Shen and J. Wang, J. Mol. Liq., 2019, 277, 409–417 CrossRef CAS.
- O. Beneš, P. Zeller, M. Salanne and R. J. Konings, J. Chem. Phys., 2009, 130, 134716 CrossRef PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- S. Kern, J. Hayward, S. Roberts, J. Richardson Jr, F. Rotella, L. Soderholm, B. Cort, M. Tinkle, M. West and D. Hoisington, et al. , J. Chem. Phys., 1994, 101, 9333–9337 CrossRef CAS.
- J. A. Ocádiz-Flores, A. E. Gheribi, J. Vlieland, K. Dardenne, J. Rothe, R. Konings and A. L. Smith, J. Mol. Liq., 2021, 331, 115820 CrossRef.
- M. Walz and D. Van der Spoel, Phys. Chem. Chem. Phys., 2019, 21, 18516–18524 RSC.
- G. Girichev, A. Krasnov, N. Giricheva and O. Krasnova, J. Struct. Chem., 1999, 40, 207 CrossRef CAS.
- R. Konings, A. Booij, A. Kovács, G. Girichev, N. Giricheva and O. Krasnova, J. Mol. Struct., 1996, 378, 121–131 CrossRef CAS.
- J. Dai, D. Long, P. Huai and Q. Li, J. Mol. Liq., 2015, 211, 747–753 CrossRef CAS.
- H. Bilz and W. Kress, Phonon Dispersion Relations in Insulators, Springer, 1979, pp. 27–47 Search PubMed.
- C. Bessada, D. Zanghi, O. Pauvert, L. Maksoud, A. Gil-Martin, V. Sarou-Kanian, P. Melin, S. Brassamin, A. Nezu and H. Matsuura, J. Nucl. Mater., 2017, 494, 192–199 CrossRef CAS.
- P. Villars and K. Cenzual, Crystal Structure Database for Inorganic Compounds, Materials Park (OH), ASM International, 2012, 2009 Search PubMed.
- V. Volkov, I. Suglobova and D. E. Chirkst, Koord. Khim., 1980, 6, 417–422 CAS.
- C. C. Underwood, M. Mann, C. D. McMillen, J. D. Musgraves and J. W. Kolis, Solid State Sci., 2012, 14, 574–579 CrossRef CAS.
- J.-B. Liu, X. Chen, Y.-H. Qiu, C.-F. Xu, W. E. Schwarz and J. Li, J. Phys. Chem. B, 2014, 118, 13954–13962 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Pennenvan, R. Ryan and A. Rosenzweig, Rare Earths, Springer, 1973, pp. 1–52 Search PubMed.
- G. Hatem, F. Tabaries and M. Gaune-Escard, Thermochim. Acta, 1989, 149, 15–26 CrossRef CAS.
- E. Capelli, O. Beneš, M. Beilmann and R. J. M. Konings, J. Chem. Thermodyn., 2013, 58, 110–116 CrossRef CAS.
- J. Ocádiz-Flores, E. Carré, J.-C. Griveau, E. Colineau, E. Capelli, P. Souček, O. Beneš, R. Konings and A. Smith, J. Chem. Thermodyn., 2020, 145, 106069 CrossRef.
- M. Wilson and P. Madden, Phys. Rev. Lett., 1994, 72, 3033 CrossRef CAS PubMed.
- V. Desyatnik, A. Nechaev and Y. F. Chervinskii, At. Energy, 1979, 46, 408–409 CrossRef.
- R. Meaker, B. Porter and D. Kesterke, Electrical Conductivity and Density of Fluoride Systems, U.S. Department of Interior, Bureau of Mines, 1971 Search PubMed.
- V. Desyatnik, A. Koverda and N. Kurbatov, At. Energy, 1983, 55, 544–546 CrossRef.
- V. Desyatnik, A. Koverda and N. Kurbatov, Atomic Energy, 1983, 55, 487–488 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00566a |
| ‡ From neutron diffraction data, ref. 9. |
| § Averaged from the crystallographic data in ref. 30. |
| This journal is © the Owner Societies 2021 |