
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the spectral features of dangling bonds in CH4/H2O amorphous ice mixtures
Belén
Maté
 *a,
Miguel Á.
Satorre
b and
Rafael
Escribano†
c
*a,
Miguel Á.
Satorre
b and
Rafael
Escribano†
c
aInstituto de Estructura de la Materia, IEM-CSIC, and Unidad Asociada Physical Chemistry UCM-CSIC, Serrano 123, 28006 Madrid, Spain. E-mail: belen.mate@csic.es
bDepartamento de Física Aplicada, EPS Alcoi, Centro de Tecnologías Físicas, Universitat Politècnica de València, Placeta Ferrándiz-Carbonell s/n, 03801 Alcoi, Spain
cDepartamento de Química Física, Facultad de CC. Químicas, Universidad Complutense, and Unidad Asociada Physical Chemistry UCM-CSIC, 28040 Madrid, Spain
First published on 26th March 2021
Abstract
Dangling bond (DB) bands in IR spectra, above 3600 cm−1, are a source of information on structural properties of amorphous water ice, and especially on ice mixtures of water and other frozen gases. We deal in this paper with the spectroscopic behavior of DB bands of CH4/H2O mixtures. We use ab initio methodology to predict theoretical results which are compared with experimental results. Our model mixtures are created by inserting a variable number of molecules of either species into a cell of appropriate size to reach an initial density of 1 g cm−3, which can be modified by including an empty space at the top, to simulate pores. The cell is taken as a unit cell for a solid state calculation The structure of the mixture is optimized and the IR spectrum is calculated for the converged geometry. We find two different kinds of dangling bonds, in which the O–H stretching responsible for this mode is directed either to an empty space of a pore or towards a nearby CH4 molecule, with which some interaction takes place. The spectral characteristics of these two DB types are clearly different, and follow satisfactorily the pattern observed in experimental spectra. Estimated band strengths for these DB bands are given for the first time.
1. Introduction
Amorphous solid water (ASW) is a disordered molecular solid that can have different porosity depending on the formation conditions.1,2 Although it is not naturally found on Earth, it is present in many astrophysical environments, as in dense clouds in the interstellar medium or on the surface of some Solar System objects. In the laboratory it can be formed by water vapor deposition on cold surfaces. Like other condensed phases of water, its structure is mainly formed by 4-coordinated hydrogen bonded water molecules.3 In the IR spectrum, the most outstanding feature is a broad and intense band, at ∼3300 cm−1, related to O–H stretching vibrations of all water molecules in the sample, either from individual molecules, or as part of the network of H-bonded aggregates. There are of course other bands related to bending and combination modes, and also to low-energy librations in the far-IR zone of the spectrum. On top of those, some interesting weak features in the high-frequency slope of the strong 3300 cm−1 IR band have been identified as due to water molecules with non-saturated hydrogen bonding capacity.4 These water molecules are on the surface of pores of the ASW ice structure. Their O–H bonds oriented towards the pore are called dangling bonds (DBs), and their absorptions are consequently termed dangling bond bands. In the pure ASW spectrum they present two characteristic features, at 3720 and 3696 cm−1. Buch and Devlin,4 from their molecular dynamics simulations of water clusters with up to 316 molecules, related these bands to surface water molecules where the O atom was 2-coordinated and 3-coordinated respectively, that is to say, having two or three H-bonds, respectively, where water molecules in the bulk are completely saturated, with four H-bonds. Buch and Devlin,4 who did not provide any theoretical spectrum to support their outstanding conclusions, dwelt also on the relative intensity of these vibrational modes, and we shall come back to this topic later on.When water ice is mixed with other molecular species, the absorption bands in the ice mixture spectrum may display wavenumber shifts, and bandwidth and intensity variations with respect to those in the spectrum of the pure species.5,6 Moreover, modes that are IR forbidden for a pure species could become active in the mixture.7 In particular, the DB feature of water ice is strongly modified in ice mixtures, when the foreign species interacts with the unbound OH bonds available on the surfaces of the ASW structure (pores or top surface).8,9 Interestingly, the position and shape of the DBs can inform about which molecule (N2, CO, CO2, CH4,…) is trapped in the pores of ASW.9
In this work we will concentrate on CH4:H2O ice systems. Methane is a molecule of special astrophysical relevance, since CH4 is considered a bio-tracer when found in atmospheres of planets in our solar system10 or in exoplanets.11 Before sublimation from CH4:H2O ice mixtures, CH4 molecules diffuse through the pores of ASW interacting with the dangling bonds.12 During this trip they can react with other trapped species or with the ice surface, making it possible to form species such as methanol. Understanding the mechanisms of methane–water interaction is a key to comprehend volatile retention in porous structures and to explore possible chemical reaction paths.
Although, due to its weakness, the DB features have not been detected in interstellar ices with the sensitivity of the instruments used to date, this does not prevent its presence. If detected, these DB features will provide information about the porosity of ASW formed on dust grains, and on the existence of other species trapped on its pores.
The present work focuses on the DB features of CH4/H2O mixtures, where, besides the above-mentioned pure ASW DB bands, a new peak appears,13,14 shifted to lower wavenumber and with stronger intensity (henceforth termed DBM). This paper presents an ab initio study on spectral characteristics of DB and DBM modes. The calculations will be compared with CH4/H2O IR spectra recorded in our laboratory. We present for the first time a physico-chemical explanation of the origin of this new peak DBM, giving an estimate of its infrared absorption strength, together with those of free DB and the fully coordinated OH modes.
2. Experimental
Experimental method
The experimental setup has been described in previous studies.12 It consists of a high vacuum chamber, with a base pressure in the 10−8 mbar range, that is provided with a closed cycle He cryostat and coupled to an FTIR spectrometer through KBr windows. A Si plate attached to the cold head of the cryostat was the surface where the ices were grown. Water vapor and CH4 entered the chamber via independent lines, provided with a precision leak valve and a mass flow controller, respectively. Ice mixtures were grown by simultaneous deposition of both gases on the cold surface (codeposition), or in a layered way, introducing one gas after the other into the chamber (sequential deposition). The ices were grown at 30 K with 0.5 K accuracy. This temperature is low enough to avoid significant CH4 sublimation, but also high enough to maximize CH4 diffusion through ASW pores. IR spectra were recorded in a normal transmission configuration, with 2 cm−1 resolution and adding 500 scans. The CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O ratio was derived from the ice mixture IR absorbance spectra. The absorption coefficients for an OH-stretching band at 3200 cm−1 and the ν3 band at 1300 cm−1 were taken from Mastrapa et al.16 (2.00 × 10−16 cm molecule−1), and Molpeceres et al.15 (6.5 × 10−18 cm molecule−1), respectively.
:H2O ratio was derived from the ice mixture IR absorbance spectra. The absorption coefficients for an OH-stretching band at 3200 cm−1 and the ν3 band at 1300 cm−1 were taken from Mastrapa et al.16 (2.00 × 10−16 cm molecule−1), and Molpeceres et al.15 (6.5 × 10−18 cm molecule−1), respectively.
Experimental observations
Fig. 1 shows the IR spectrum of a CH4/H2O ice mixture at 30 K. The mid IR phonons of this molecular solid can be assigned to vibrational modes of the component species. In this way, the broad absorptions at 3300 cm−1, 2100 cm−1 and 1600 cm−1 correspond to stretching, combination, and bending motions, respectively, of the water molecules in the ice. The absorption at 800 cm−1 is assigned to a libration movement of H2O molecules. The narrow peaks at 3000 cm−1 and 1300 cm−1 correspond to the ν3 and ν4 modes of CH4. The dangling bond features, assigned to free OH bonds, appear around 3700 cm−1, in the spectral region highlighted in the inset in Fig. 1.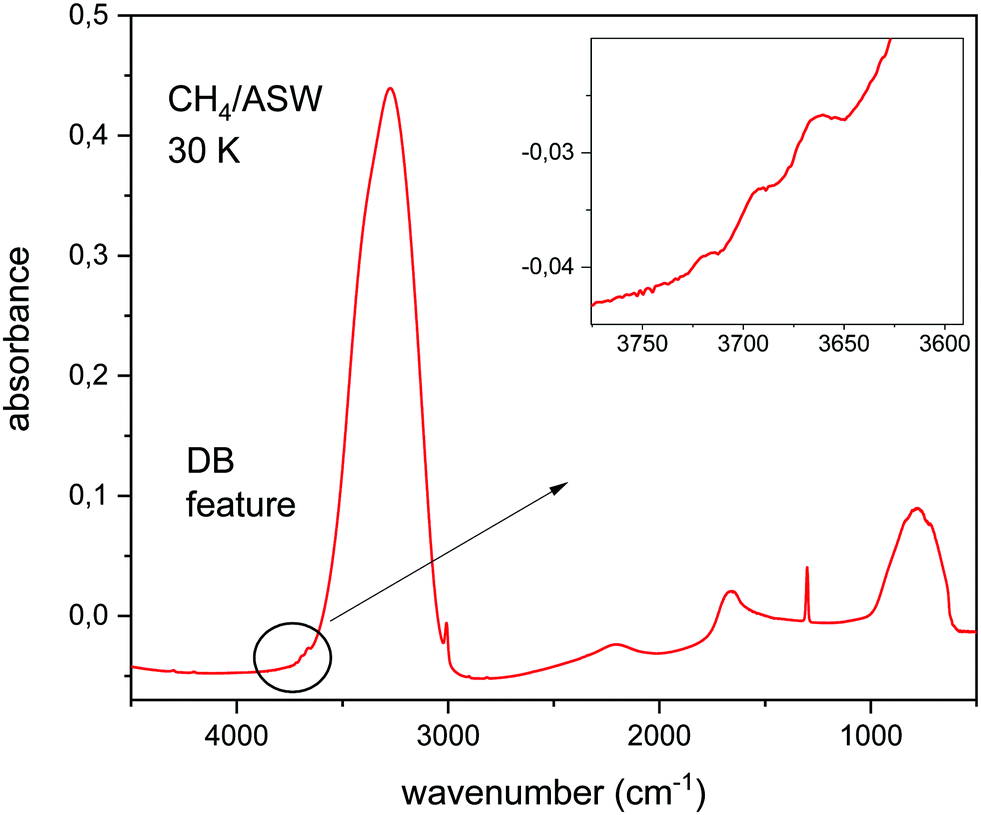 | ||
| Fig. 1 IR spectra of a CH4/H2O ice mixture grown by sequential deposition at 30 K, CH4/H2O = 1/11. | ||
This figure is presented to illustrate the relative intensity in the IR absorbance spectrum, of the OH stretching modes of water for fully coordinated bonds, and for dangling bonds. This intensity ratio depends on the ice generation conditions, mainly on deposition temperature. The lower the temperature the larger the porosity of the ASW ice, and the larger the intensity of the DB features observed. At this point, it is important to notice that the relative intensity of fully coordinated versus DB bands depends not only on the number ratio of DB versus fully coordinated bonds present in the ice, but also on the intrinsic IR band strength of each particular mode. In this work we have calculated the IR spectrum of CH4/H2O mixtures and we will concentrate on the analysis of the DB region, between 3800 and 3600 cm−1.
The variation of the DB in CH4/H2O mixtures with respect to that of pure ASW is presented in Fig. 2. The blue lines correspond to pure ASW DB. The observed two peaks, at 3720 cm−1 and 3690 cm−1, will be called DB1 and DB2. When the stretching of free OH bonds of water interacts with CH4 molecules a new DB feature appears, shifted to lower frequencies and with larger intensity than the ASW DB modes. We have labelled this new peak that appears at 3666 cm−1, as DBM. The intensity of this DBM feature in CH4/H2O mixtures depends on the relative amount of CH4, but it is observed to be always higher than that of the pure ice (see Herrero et al. 2010).
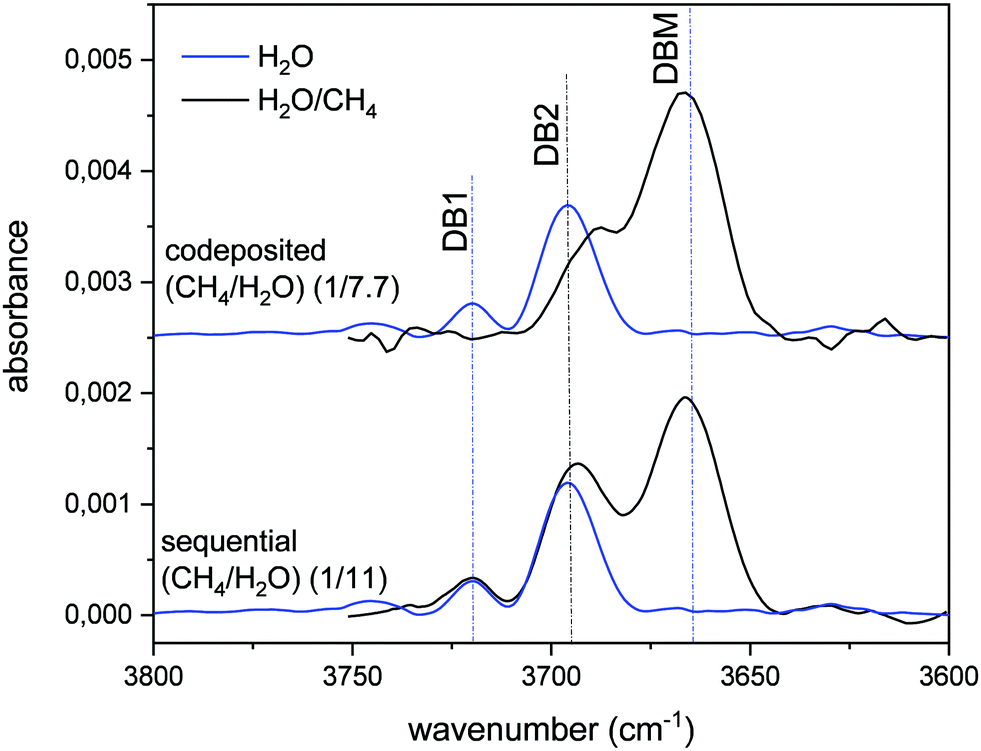 | ||
| Fig. 2 Absorbance infrared spectrum in the DB spectral zone. A baseline has been subtracted from all the spectra shown. Blue lines: pure ASW grown at 30 K. Black top line: ice mixture grown by simultaneous vapor deposition of both gases (codeposited), (1/7.7) (CH4/H2O) ratio. Black bottom line: layered ice mixture grown by sequential deposition at 30 K, first CH4 and then ASW, (1/11) (CH4/H2O) ratio. All the spectra correspond to an equivalent ASW layer thickness of 200 nm, estimated from the IR intensity of the OH stretching feature and the IR band strength given by Mastrapa et al.16 | ||
3. Theoretical methodology
All theoretical calculations are carried out at the Density Functional Theory (DFT) level using the Castep17 and Amorphous Cell modules from Materials Studio.18 This software has proved to be adequate for this type of calculation.7,19–21 Details of its application have been given in more detail by Gómez et al.21 and we refer to that article for further information.Our samples contain CH4/H2O mixtures in a ratio of 1:9. In order to simulate the porous surface present in experimental samples, the cells have been forced to have an empty space on top, as a way to generate a surface. The detailed procedure to build them is as follows. Within a cubic cell of side 8.3955 Å all H2O and CH4 molecules are introduced, giving a mixture of intrinsic density 1 g cm−3. To this cell an empty space is added along one of the three indistinguishable axes, the C axis, thus reaching 8.3955 × 8.3955 × 25 Å3. The molecular structure is then amorphized, that is, with the amorphous cell module all molecules are moved at random with a given kinetic energy, or temperature, for a number of steps. In our case, we have used two kinetic temperatures, 40 K and 200 K, with similar results. Two different 200 K and one 40 K original structures were prepared, and the four amorphous structures of lowest energy after the amorphization process are chosen for the subsequent optimization process. In this way, we use a data set of 12 structures, all with 18 water molecules and 2 methane molecules.
Each selected amorphous sample is allowed to refine its molecular structure searching for a minimum in the Potential Energy Surface (PES). Once the minimum is attained, vibrational modes are calculated. All modes in the DB spectral region, i.e. >3600 cm−1, are carefully scrutinized and classified according to their characteristics, into the following categories:
DB: O–H stretching modes of molecules near the upper or lower surfaces of the bulk, when the displacement of the H atom is directed towards the empty space, and does not approach any other molecule;
DBM: similar but where the H atom of the O–H vibration is directed to a nearby CH4 molecule, reaching a short distance from one or more H atoms of methane. We understand by short distance a value of <1.7 Å;
Bulk: O–H modes of H2O molecules located inside the bulk of the sample, away from the surfaces.
This classification is based on the graphical display afforded with Castep. Two examples are shown in Fig. 3(a). On the left panel, a typical DB mode, at 3660 cm−1 and intensity 56 km mol−1, is depicted, with the O–H motion directing the H atom towards the empty space in the cell that simulates the pore. On the right panel, a DBM mode at 3616 cm−1 and 282 km mol−1 intensity is shown, with the H atom of H2O moving towards a CH4 molecule. Note that, for the same molecular system, the DBM mode appears at lower wavenumber and higher intensity than the DB mode, as is frequently the case. We can follow with Castep the animation picture of DBM modes, and it can be seen that, during the O–H vibration, as the H atom of H2O moves closer to, and away from, the nearest CH4 molecule, the C–H bonds of CH4 also show some in-phase response discussed below. We can focus on the latter DBM mode, illustrated in more detail in Fig. 3(b). The arrow represents the stretching motion of the O–H bond of water. The initial and final interatomic distances for the atoms involved in this mode are collected in Table 1, normalized to a value of 1.098 Å, the C–H bond length in an undisturbed CH4 molecule. A normalization factor is required because the eigenvectors that conform the description of a normal mode are, by definition, affected by an unknown arbitrary factor. We have labelled C–H1 that methane bond which is most altered by the motion of the H atom of H2O. It undergoes a shrink of 0.018 Å when the water H atom moves closest. The C–H2 bond also shrinks in minor proportion, and the other C–H bonds are little affected. This kind of motion bears large resemblance to a typical asymmetric C–H stretch within the ν3 mode, with some damping that reduces the extent of the atomic motions. The last two rows in Table 1 indicate how close the water H atom gets to the most affected methane H atoms, H1 and H2. The interaction that causes these disturbances will be discussed below, in the Results section, but we advance that it conveys the explanation for the wavenumber redshift and the increase in intensity and width of DBM modes with respect to “pure”, water-only, DB modes.
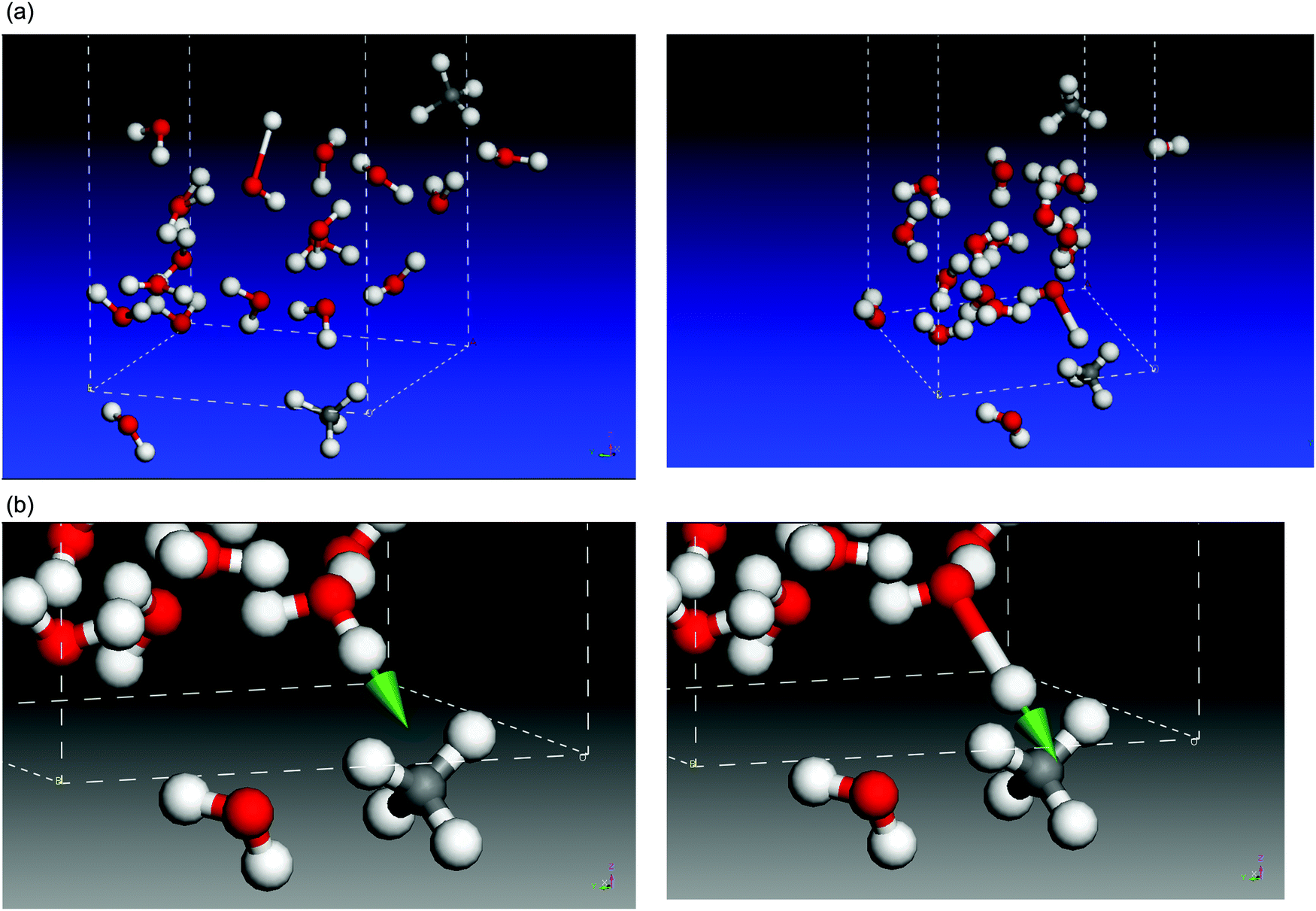 | ||
| Fig. 3 (a) Two examples of dangling bonds in our cell model, where the large empty space along the C axis is cut off the figure. Left: DB mode of a molecule near the upper occupied surface, at the center of the image, with O–H stretching motion directed towards empty space, clear of any other molecules. Right: DBM mode of a water molecule, near the cell bottom, on the right, with O–H stretching motion towards a nearby CH4 molecule. (b) This DBM mode is focussed on the enlarged views of initial (left) and final (right) atomic positions during the corresponding O–H stretching motion. The strongest interaction takes place between the water–H and the methane–H pointing north east. | ||
| At rest | End of O–H stretch/normalized to 1.098 Å | Net effect | |
|---|---|---|---|
| C–H1 | 1.098 | 1.080 | Shrink 0.018 |
| C–H2 | 1.100 | 1.092 | Shrink 0.008 |
| C–H3 | 1.097 | 1.095 | Shrink 0.002 |
| C–H4 | 1.100 | 1.102 | Enlarge 0.002 |
| H(H2O)–H1 | 2.108 | 1.174 | Shrink 0.934 |
| H(H2O)–H2 | 2.155 | 1.328 | Shrink 0.827 |
4. Results
Table 2 collects the results of the calculations. A total of 38 DB modes and 14 DBM modes were identified in the 12 structures investigated. On average, about 3 DB and 1 DBM were found in each simulated structure. In Fig. 4, the modes listed in Table 2, characterized by its wavenumber and intensity, are represented with points, where the squares correspond to DB modes, and the circles to DBM modes. In order to facilitate a comparison with experimental observations, each mode has been represented with a Gaussian profile centered on the calculated wavenumber, with an area equal to the intensity of the mode, and with a full width at half maximum (FWHM) chosen to match the experimental values of Fig. 2. FWHMs of 10 and 25 cm−1 were adopted for the peaks falling in the zones labelled DB (DB1 and DB2, see below) and DBM, respectively. The number of structures sampled is not enough to provide a statistical description of all the possible geometries, and therefore do not allow an appropriate estimation of the bandwidth of the DB or DBM features.| Wavenumber (cm−1) | km mol−1 | Wavenumber (cm−1) | km mol−1 | Wavenumber (cm−1) | km mol−1 | Wavenumber (cm−1) | km mol−1 | |
|---|---|---|---|---|---|---|---|---|
| DB exp | 3720 | |||||||
| 3690 | ||||||||
| DB theo | 3686 | 40 | 3671 | 55 | 3648 | 78 | 3693 | 55 |
| 3694 | 60 | 3691 | 79 | 3702 | 60 | 3682 | 75 | |
| 3686 | 66 | 3681 | 61 | 3670 | 56 | 3672 | 116 | |
| 3680 | 54 | 3677 | 66 | 3685 | 61 | 3658 | ||
| 3664 | 112 | 3672 | 99 | 3683 | 76 | 3679 | 78 | |
| 3693 | 78 | 3658 | 31 | 3667 | 64 | 3670 | 68 | |
| 3666 | 91 | 3681 | 76 | 3686 | 66 | 3643 | 87 | |
| 3665 | 80 | 3669 | 103 | 3660 | 85 | 3641 | 61 | |
| 3684 | 62 | 3665 | 55 | 3656 | 76 | 118 | ||
| 3685 | 74 | 3637 | 98 | 3686 | 45 | |||
| DBM exp | 3666 | |||||||
| DBM theo | 3632 | 143 | 3662 | 201 | 3638 | 231 | 3637 | 165 |
| 3644 | 232 | 3656 | 224 | 3641 | 127 | 3646 | 216 | |
| 3631 | 126 | 3645 | 112 | 3632 | 178 | |||
| 3667 | 81 | 3616 | 282 | 3634 | 189 |
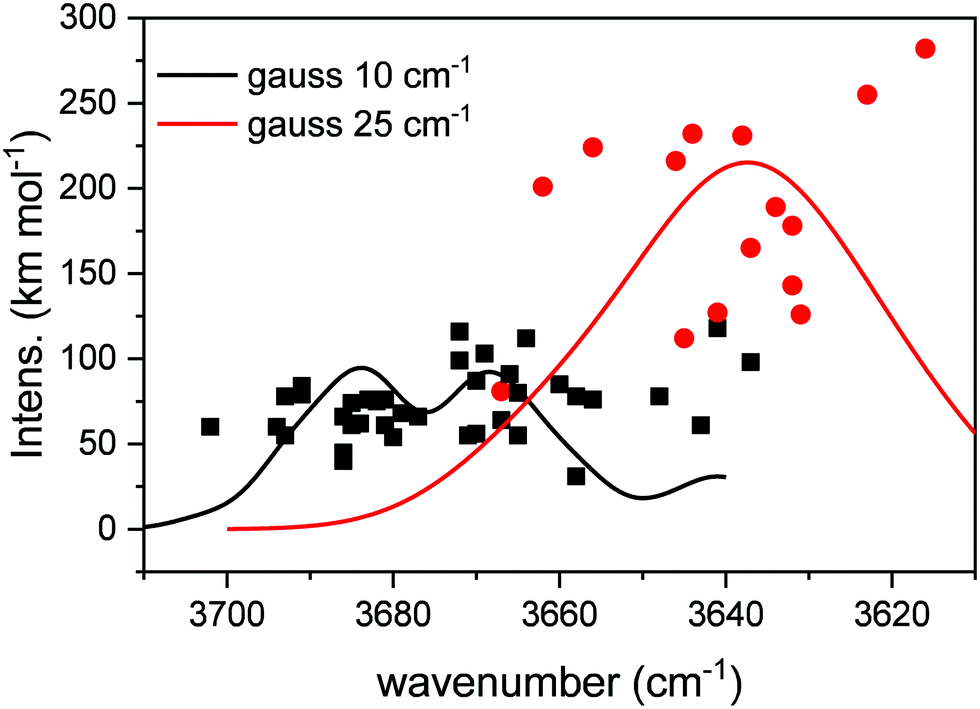 | ||
| Fig. 4 Summary of DB modes (isolated O–H stretchings), black squares; and DBM modes (CH4-involved O–H stretchings), red circles. The black and red traces were obtained using Gaussian profiles to represent each mode; FWHM of 10 cm−1 for DB modes, and 25 cm−1 for broader DBM modes, to match experimental widths. | ||
From Fig. 4, we can extract some conclusions. On average, our calculated DB modes appear in the 3700–3640 cm−1 zone, and have intensities in the 50–75 km mol−1 range, although some values fall outside those boundaries. In turn, the DBM modes appear mostly in the 3670–3610 cm−1 region, with much stronger intensities, that can almost reach 300 km mol−1. In the profile obtained from the Gaussian broadening of the modes, three peaks stand out that can be related in good agreement to the experimental bands DB1, DB2 and DBM in Fig. 2. The similarity with our experimental observations, as far as the general behavior of the whole area is concerned, is therefore pleasing. The wavenumber range in our calculations is systematically lower than the observed one by about 20 cm−1 or less (see Table 2). This is not infrequent when comparing experimental and calculated wavenumbers, especially when the focus of the calculations is not so much on the wavenumber reproducibility but on the general behavior of the modes in the whole region, as in the present study. Other literature studies, using different theoretical approaches, simulated the redshift and intensity enhancement of the ASW DB feature due to the interaction of adsorbed molecules.22–24 Scaling factors of 0.8522 or 0.9424 were applied to their calculated DB wavenumbers in order to agree with the experimental ones. Manca and Allouche22 predicted DB redshifts due to N2, CO or Ar absorptions that range between 13 and 72 cm−1. Hujo et al.23 predicted a redshift of 18 cm−1 for N2 molecules adsorbed on water clusters and larger shifts, up to 85 cm−1, were computed by Michoulier et al.24 for adsorption of aromatic molecules of ASW. In our calculations, the averaged redshift observed for CH4 adsorption is 35 cm−1 whereas the experimental redshift measured between DB2 and DBM is 24 cm−1 (see Fig. 2).
Since the calculations provide infrared intensities for the different modes, it is possible to estimate the infrared band strength of the two types of DB features identified in the calculations and compare them with the intensity of the bulk OH modes of water ice. The results are presented in Table 3, where the band strengths of the DB and DBM modes have been calculated averaging over the 39 and 15 points represented in Fig. 4, respectively.
To complete the picture, we include in this Table 3 the band strength of the bulk OH modes ν(OH) calculated at wavenumbers above 2500 cm−1, which correspond to symmetric and antisymmetric OH stretching of the 18 water molecules present in the unit cell (see Fig. 3). From the total number of OH stretching modes, 36, those already identified as DB or DBM were not included. Similarly, the eight CH modes corresponding to the asymmetric and symmetric ν3 and ν1 vibrations of the two CH4 molecules in the unit cell that also appear in this wavenumber region, have been excluded from the intensity addition. Once all the specified intensities were added, the number was divided by 18, in order to give the band strength of the mode per unit molecule. This procedure has been made for the 12 structures investigated and the average value is presented in Table 3. The close resemblance between calculated and experimental values for the band strength of the bulk O–H mode, gives us confidence in our calculated intensities for the DB and DBM modes, which, as far as we know, has not been estimated either theoretically or experimentally before.
Therefore, the general rule is that of wavenumber red-shift, and band intensity and bandwidth enhancement in DBM modes as compared to DB modes. In the following, we discuss these effects in terms of molecular vibrational modes and molecular interactions rather than the wholesome effect in the cells that conform the solid, but the extrapolation to the latter would be straightforward. We have mentioned above that the O–H vibration in a DBM mode prompts the C–H bonds of the methane involved to vibrate in phase with the water mode, with one or two C–H bonds bearing the largest alteration. Thus, an effective dipole arises in methane, similar to that found in the asymmetric stretching modes. If we think of a bond dipole moment along the O–H bond, this dipole would meet an in-phase response from the altered charge distribution of methane. The motion in phase of the two bond dipoles, that of H2O which wears most of the charge, and the smaller one induced on CH4, is likely to increase the variation of the O–H dipole along its displacement, or, in other words, its derivative with respect to the stretching coordinate, which is the main contribution to the band strength of the mode. On DB modes, this kind of interaction does not take place, and therefore their intensity is not especially enhanced.
Moreover, the possible interaction between the O–H dipole and a distorted charge distribution in methane, which also vibrates in-phase, can be responsible for a large delocalization of the water H atom, which would explain an increase in bandwidth of these DBM vibrations. Lastly, the water H atom involved in this bond will be less tightly bound to its O atom, and the corresponding loss of binding strength induces a reduction in the wavenumber of the vibration, always compared to O–H bonds in “pure” DB modes.
Analogous effects are found in H-bond modes in condensed phases with respect to similar vibrations in the gas phase, in terms of wavenumber red-shift and intensity enhancement. Although the water H atom is not expected to be actually shared with CH4 in our samples, whereas a H atom is shared between two H2O molecules in pure water condensed phases, the interaction between H atoms of H2O and CH4 molecules in DBM resembles H-bonding liaisons, whereas the DB modes would be physically closer to those of gas phase systems.
As mentioned in the Introduction, Buch & Devlin 1991, interpret the increasing intensity and decreasing wavenumber values of O–H vibrations in terms of growing coordination number of the O atom of water. The highest intensity corresponds to 4-coordinated O atoms, like those in bulk water, and weaker intensities are associated with 3- and 2-coordinated water molecules, respectively. Our results seem to support that interpretation, if we accept that the DBM modes correspond to vibrations in which the O atom establishes some kind of link with the CH4 molecule involved, leading to an interaction that might resemble a 3-coordinated molecule. Then, the DB modes will contain a mixture of 2- and 3-coordinated water molecules, with weaker intensity, which might evolve to DB1 or DB2 modes under certain experimental conditions.
From the band strengths collected in Table 3 we see that the bulk O–H mode is one order of magnitude stronger than the DB features. The measured band strength of the free O–H mode in the gas phase is 4.2 × 10−19 cm mol−1,25 whereas that of the bulk ice is 2.9 × 10−16 cm mol−1.16 The parallelism with our calculated values for the DB and bulk O–H modes seems therefore evident.
5. Conclusions
We study the spectral characteristics of dangling bond bands in CH4/H2O ice mixtures with an ab initio model that allows following the displacement of the H atom during the O–H vibration for individual molecules. Focusing on molecules near the surface of a pore in a sample, we identify two main types of vibrations of the water unbound O–H bonds, differing in the orientation of the corresponding stretching motion. The well-known dangling bond (DB) band is generated when the H atom is directed towards the empty space of the pore, like in pure amorphous water. On the other hand, when the H atom approaches a nearby CH4 molecule, some interaction takes place between both molecules and the corresponding band (termed DBM band in this work) has different spectral behavior.In a nutshell, DB bands appear at higher wavenumber and with smaller intensity than DBM bands. In turn, O–H stretching vibrations in bulk water molecules, where no dangling bonds are present, are stronger and cover a lower wavenumber region. This is clearly displayed from our experimental spectra. And this pattern is satisfactorily reproduced in our calculations in general terms.
Our results allow providing a theoretical estimation for the band strengths of DB and DBM bands, which had never been reported before, as far as we know. Extension of our calculations to the full O–H band of the bulk shows a pleasing agreement with previously published results, which give us confidence in our estimations for the band strengths of the DB bands.
We advance a possible mechanism to explain the observed spectral effects, based on the interaction between the bond dipole moment of the interacting O–H with the close-by methane molecule. The response of the most affected C–H bond in methane resembles a typical ν3 vibration of this molecule.
Our calculations are in good agreement with the interpretation provided by Buch and Devlin,4 based on the different coordination numbers that the O atom may adopt for surface molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funds from the Spanish MINECO/FEDER FIS2016-77726-C3-1-P and C3-3-P projects are acknowledged. We are indebted to V. J. Herrero for technical assistance with the CASTEP calculations, performed at SGAI-CSIC.References
- Z. Dohnálek, G. A. Kimmel, P. Ayotte, R. S. Smith and B. D. Kay, J. Chem. Phys., 2003, 118, 364 CrossRef.
- U. Raut, B. D. Teolis, M. J. Loeffler, R. A. Vidal, M. Famá and R. A. Baragiola, J. Chem. Phys., 2007, 126, 244511, DOI:10.1063/1.2746858.
- A. H. Narten, C. G. Venkatesh and S. A. Rice, J. Chem. Phys., 1976, 64, 1106, DOI:10.1063/1.432298.
- V. Buch and J. P. Devlin, J. Chem. Phys., 1991, 94, 4091, DOI:10.1063/1.460638.
- D. M. Hudgins, S. A. Sandford, L. J. Allamandola and A. G. G. M. Tielens, Astrophys. J., 1993, 86, 713 CAS.
- O. Gálvez, I. K. Ortega, B. Maté, M. A. Moreno, B. Martín-Llorente, V. J. Herrero, R. Escribano and P. J. Gutiérrez, Astron. Astrophys., 2007, 472, 691–698, DOI:10.1051/0004-6361:20077421.
- R. Escribano, V. Timón, O. Gálvez, B. Maté, M. A. Moreno and V. J. Herrero, Phys. Chem. Chem. Phys., 2014, 16, 16694 RSC.
- M. E. Palumbo, Astron. Astrophys., 2006, 453, 903–909, DOI:10.1051/0004-6361:2004238.
- J. He, S. M. Emtiaz and G. Vidali, Astrophys. J., 2018, 863, 156 CrossRef.
- E. W. Knutsen, G. L. Villanueva, G. Liuzzi, M. M. J. Crismani, M. J. Mumma, M. D. Smith, A. C. Vandaele, S. Aoki, I. R. Thomas, F. Daerden, S. Viscardy, J. T. Erwin, L. Trompet, L. Neary, B. Ristic, M. Angel Lopez-Valverde, J. Juan Lopez-Moreno, M. R. Patel, O. Karatekin and G. Bellucci, Icarus, 2021, 357, 114266 CrossRef CAS.
- T. S. Barman, Q. M. Konopacky, B. Macintosh and C. Marois, Astron. Astrophys., 2015, 804, 61 Search PubMed.
- B. Maté, S. Cazaux, M. A. Satorre, G. Molpeceres, J. Ortigoso, C. Millán and C. Santonja, Astron. Astrophys., 2020, 643, A163 CrossRef.
- V. J. Herrero, O. Gálvez, B. Maté and R. Escribano, Phys. Chem. Chem. Phys., 2010, 12, 3164–3170, 10.1039/b922598f.
- B. Maté, S. Cazaux, M. Ángel Satorre, G. Molpeceres, J. Ortigoso, C. Millán and C. Santonja, Astron. Astrophys., 2020, 643, A163, DOI:10.1051/0004-6361/202038705.
- G. Molpeceres, M. A. Satorre, J. Ortigoso, A. Zanchet, R. Luna, C. Millán, R. Escribano, I. Tanarro, V. J. Herrero and B. Maté, Mon. Not. R. Astron. Soc., 2017, 466, 1894–1902, DOI:10.1093/mnras/stw3166.
- R. Mastrapa, S. A. Sandford, T. L. Roush, D. P. Cruikshank and C. M. Dalle Ore, Astrophys. J., 2009, 701, 1347–1356 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnito, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- Materials Studio: Materials Studio, 2014, http://accelrys.com/products/materials.studio.
- G. Molpeceres, M. Á. Satorre, J. Ortigoso, C. Millán, R. Escribano and B. Maté, Astrophys. J., 2016, 825, 156, DOI:10.3847/0004-637X/825/2/156.
- P. C. Gómez and R. Escribano, Phys. Chem. Chem. Phys., 2017, 19, 26582, 10.1039/c7cp04695b.
- P. C. Gómez, M. Á. Satorre and R. Escribano, Chem. Phys. Lett., 2020, 745, 137222, DOI:10.1016/j.cplett.2020.137222.
- C. Manca and A. Allouche, J. Chem. Phys., 2001, 114, 4226, DOI:10.1063/1.1331106.
- W. Hujo, M. Gaus, M. Schultze, T. Kubar, J. Grunenberg, M. Elstner and S. Bauerecker, J. Phys. Chem. A, 2011, 115, 6218, DOI:10.1021/jp111481q.
- E. Michoulier, C. Toubin, A. Simon, J. Mascetti, C. Aupetit and J. A. Noble, J. Phys. Chem. C, 2020, 124, 2994, DOI:10.1021/acs.jpcc.9b09499.
- R. A. Toth, J. Quant. Spectrosc. Radiat. Transfer, 1973, 13, 1127 CrossRef CAS.
Footnote |
| † Author is now deceased. |
| This journal is © the Owner Societies 2021 |