
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormic acid dehydrogenation over PdNi alloys supported on N-doped carbon: synergistic effect of Pd–Ni alloying on hydrogen release†
Rizcky
Tamarany‡
a,
Dong Yun
Shin‡
b,
Sukho
Kang
b,
Hyangsoo
Jeong
 a,
Joohoon
Kim
cd,
Jun
Kim
a,
Chang Won
Yoon
*ac and
Dong-Hee
Lim
*b
a,
Joohoon
Kim
cd,
Jun
Kim
a,
Chang Won
Yoon
*ac and
Dong-Hee
Lim
*b
aCenter for Hydrogen and Fuel Cell Research, Korea Institute of Science and Technology (KIST), 5 Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: cwyoon@kist.re.kr
bDepartment of Environmental Engineering, Chungbuk National University, Chungdae-ro 1, Seowon-gu, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: limkr@cbnu.ac.kr
cKHU-KIST Department of Converging Science and Technology, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea
dDepartment of Chemistry, Research Institute for Basic Sciences, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul 02447, Republic of Korea
First published on 30th April 2021
Abstract
Bimetallic Pd1Nix alloys supported on nitrogen-doped carbon (Pd1Nix/N–C, x = 0.37, 1.3 and 3.6) exhibit higher activities than Pd/N–C towards dehydrogenation of formic acid (HCO2H, FA). Density functional theory (DFT) calculations provided electronic and atomic structures, energetics and reaction pathways on Pd(111) and Pd1Nix(111) surfaces of different Pd/Ni compositions. A density of states (DOS) analysis disclosed the electronic interactions between Pd and Ni revealing novel active sites for FA dehydrogenation. Theoretical analysis of FA dehydrogenation on Pd1Nix(111) (x = 0.33, 1 and 3) shows that the Pd1Ni1(111) surface provides optimum H2-release efficiency via a favorable ‘HCOO pathway’, in which a hydrogen atom and one of the two oxygen atoms of FA interact directly with surface Ni atoms producing adsorbed CO2 and H2. The enhanced efficiency is also attributed to the blocking of an unfavorable ‘COOH pathway’ through which a C–O bond is broken and side products of CO and H2O are generated.
1. Introduction
The development of sustainable energy technology is currently one of the most urgent and important challenges in mitigating global energy and environmental issues related to climate change.1–4 A universal transition to renewable energy is required to provide sustainable resources in future societies. To efficiently utilize renewable energy produced in an intermittent and unpredictable manner, a carbon-free and high-capacity energy carrier is needed. In the regard, hydrogen (H2) has long been recognized as a viable and consistent energy carrier owing to its high gravimetric energy density (ca. 33.3 kW h kg−1-H2).5 In addition, use of hydrogen in a fuel cell that produces electricity and water as the only byproducts is effective in many portable, stationary and transportation applications.6–10 However, because gaseous hydrogen has a very low volumetric energy density, current pressurized storage technologies (e.g., 700 bar H2, 40 g H2 per L)11 may not meet the geographical and temporal gaps between the hydrogen supply and demand site. For this reason, hydrogen storage in liquid chemicals has received particular attention for high-capacity storage and long-distance transport because liquid hydrogen carriers allow to store large amounts of hydrogen and to affordably deliver it to desired sites using the currently existing infrastructure.5,9,10,12,13A promising candidate for liquid-based hydrogen storage materials is formic acid (HCO2H, FA). Formic acid is a relatively nontoxic liquid with a large volumetric hydrogen storage density (53 g L−1), which allows it to be economically stored and transported. The hydrogen atoms of FA can be liberated with an appropriate catalyst under ambient conditions (HCO2H → H2 + CO2). Moreover, FA can also be produced by biomass processing6,14–19 and catalytically or electrocatalytically generated by hydrogenation of carbon dioxide (H2 + CO2 → HCO2H) using excess renewable energy.16,18,20 These properties make FA appealing as a hydrogen carrier applicable to decentralized fuel cell power generation, hydrogen refueling stations and large-scale renewable energy storage systems.21
A highly efficient catalyst is needed to release hydrogen from FA in a controllable manner and to facilitate its application in various energy-requiring scenarios. Successful heterogeneous catalysts were developed to selectively catalyze FA dehydrogenation include numerous Pd-based nanomaterials: Pd nanoparticles (NPs),15–17,19,22–24 PdM (M = Ag, Au, Ni, Co) core–shells25–30 and Pd-based alloys.25,27,29,31–35 The superior performance of these catalysts is thought to originate from electronic structural modifications that create active sites by alloying with a different metal27,32–34 and/or by using supports with nitrogen dopants or nitrogen-containing (amine) functionalities.16,17,19,28 Pd-Based alloy catalysts without other noble metals also have been reported to reduce the cost of catalyst implementation. For example, a PdNi@Pd catalyst exhibits good FA dehydrogenation activity at room temperature.29 Pd–Co-based nanoparticles are multifunctional catalysts for FA dehydrogenation and methanol oxidation.30 A boron-doped Pd catalyst displays greater activity than bimetallic Pd–Au catalysts for hydrogen generation from aqueous formic acid/formate solutions at room temperature.15 However, a remaining challenge is to develop relatively inexpensive catalysts that can rapidly produce hydrogen by dehydrogenation. Increased temperature can improve the rate of H2-release from FA, but the accompanying dehydration reaction (HCO2H → H2O + CO) could also take place to produce carbon monoxide which deactivates the Pt catalysts in polymer electrolyte membrane fuel cells (PEMFCs). Therefore, understanding the pathways of FA dehydrogenation over low-cost, bimetallic Pd-based materials is of significant importance in gaining insight to the development of highly active and selective heterogeneous catalysts.
In this paper, we report experimental and theoretical studies of FA dehydrogenation over Pd1Nix alloy catalysts (x = 0.37, 1.3 and 3.6) supported on nitrogen-doped carbon (Pd1Nix/N–C) as model Pd based alloy catalysts possessing an inexpensive transition metal. The nanostructured PdNi active sites prepared by a simple wet chemical method are characterized by a number of different analytical techniques. The PdNi/N–C catalysts exhibit greater FA dehydrogenation activity than a Pd/N–C catalyst. The electronic structure of the PdNi alloys and the FA decomposition mechanisms are elucidated using density functional theory (DFT) calculations. The results demonstrate the cooperative role of the Pd–Ni active sites in rotating a key HCOO intermediate as well as in activating the C–H bonds of FA during dehydrogenation. In addition, the Pd1Ni1.3 active sites prove to prevent a H2O and CO formation step (dehydration) over the H2 and CO2 formation step (dehydrogenation).
2. Experimental and computational methodology
2.1. Synthesis of nitrogen-doped carbon support (N–C) and Pd/N–C, PdNi/N–C and Ni/N–C catalysts
Palladium(II) nitrate dihydrate [Pd(NO3)2·2H2O], nickel(II) nitrate hexahydrate [Ni(NO3)2·6H2O] and dicyandiamide [NH2C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NHCN] were purchased from Sigma-Aldrich. Dicyandiamide was used to prepare a nitrogen doping precursor. Carbon black (Ketjen) was purchased from Akzo Nobel. Unless noted, other chemicals were used without further purification.
NH)NHCN] were purchased from Sigma-Aldrich. Dicyandiamide was used to prepare a nitrogen doping precursor. Carbon black (Ketjen) was purchased from Akzo Nobel. Unless noted, other chemicals were used without further purification.
For the materials synthesis, the nitrogen-doped carbon support was initially prepared by adding dicyandiamide (1.0 g, 12 mmol) to an aqueous solution (50 mL) containing carbon black (1.0 g) followed by heating at 100 °C for 4 h with vigorous stirring to vaporize the water. Black solids were obtained upon complete drying. The resulting solids were ground and pyrolyzed at 550 °C for 4 h under a N2 flow yielding the final N–C products as black powders. The N–C support was characterized using XRD.
Pd1Nix alloys supported on N–C (Pd1Nix/N–C) were prepared by dispersing N–C into aqueous solutions containing Pd(NO3)2·2H2O and Ni(NO3)2·6H2O at different Pd/Ni mole ratios (1/0.33, 1/1 and 1/3). The distilled water in the heterogeneous mixture was evaporated slowly by stirring. The resulting black powders were collected and reduced under a H2/N2 flow (20% H2) at 450 °C for 4 h, affording Pd1Nix/N–C. The Pd1Nix nanoparticles on the N–C supports were determined to have Pd/Ni mole ratios of Pd1Ni0.37, Pd1Ni1.3, Pd1Ni3.6, respectively, by ICP-OES (ESI,† Section S1 and Table S1). Pd/N–C and Ni/N–C materials were synthesized for comparison using solely Pd(NO3)2·2H2O and Ni(NO3)2·6H2O, respectively, by the Pd1Nix/N–C procedure. Pd1Ni1.3/C also was prepared by the above procedure using a commercial carbon support (Ketjen). The Pd/Ni ratio in Pd1Ni1.3/C was determined using TEM-EDS.
2.2. Characterization
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was used to analyze the chemical composition of the PdNi nanoparticles (NPs). Measurements were performed on an ICP-OES Varian 720ES Agilent instrument using a cross-flow nebulizer with the following parameters: plasma, 18.0 L Ar per min; auxiliary, 0.3 L Ar per min; nebulizer, 0.73 L Ar per min; power, 1500 W; peristaltic pump rate, 1.40 mL min−1.High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to determine the morphology of the catalysts. Elemental distributions in PdNi NPs were obtained by energy dispersive X-ray spectroscopy (EDS). Samples were prepared by drop-casting an ethyl alcohol suspension of carbon-supported NPs onto a carbon-coated Cu grid followed by solvent evaporation at room temperature. Measurements were performed on an FEI Talos G2 80–200 Chemi-STEM electron microscope at 200 kV.
X-Ray photoelectron spectroscopy (XPS) analysis was conducted ex situ using a PHI 5000 Versa Probe (Ulvac-PHI). The system used a focused monochromatic Al Kα (1486.6 eV) source for excitation and a spherical section analyzer. The 100 W, 100 μm diameter X-ray beam was rastered over a 1.4 mm × 0.2 mm rectangular spot on the sample. The beam was incident to the sample normally, and the detector was placed at 45° from the normal. The binding energy (BE) scale was calibrated using the C 1s peak (284.6 eV) as an internal standard. XRD measurements were performed using Cu Kα radiation (λ = 0.154 nm) at the 11-ID-C beamline with a Rigaku Miniflex II X-ray diffractometer. For ex situ experiments, PdNi/N–C samples were loaded into thin glass capillaries open at one end. The scan rate was set at 4°/min with a scan range of 10° ≤ 2θ ≤ 90°. Crystallite sizes of Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) and Ni/N–C were calculated using the Scherrer method, and summarized in Table S2 (ESI†). In addition, XPS Pd3d data for Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) were analyzed and relevant peak positions were listed in Table S3 (ESI†).
2.3. Catalytic activity measurements
Catalytic reactions were performed at a desired temperature of 30 °C in a two-neck glass tube. One neck of the reactor was connected to an Ar gas supply. The other was connected to a gas burette to determine the quantity of gas released in real time. The gas burette is filled with oil to prevent potential CO2 dissolution in water. The reaction tube was cleaned with Ar gas prior to reaction. Catalytic FA dehydrogenation was initiated by introducing a 10 mL mixture of FA (HCO2H, 1.0 M) and sodium formate (HCO2Na, 1.0 M) to the reaction tube containing a desired catalyst followed by stirring under ambient conditions. The Pd quantities in a series of Pd1Nix (x = 0, 0.37, 1.3 and 3.6) supported on nitrogen-doped carbon were listed as follows: Pd/N–C, nPd = 12.7 μmol; Pd1Ni0.37/N–C, nPd = 12.5 μmol and nNi = 4.61 μmol; Pd1Ni1.3/N–C, nPd = 12.2 μmol and nNi = 15.9 μmol; Pd1Ni3.6/N–C, nPd = 11.6 μmol and nNi = 41.7 μmol. The volume of gas produced was determined using an automatic gas burette system directly connected to the reactor for real-time volumetric measurements. In a separate experiment, the influence of temperature on FA dehydrogenation over Pd1Ni1.3/N–C and Pd/N–C was monitored by varying reaction temperatures (25 °C, 45 °C and 65 °C) using the identical quantities of HCO2H and NaHCO2. The Pd1Ni1.3/N–C material was prepared at a fixed Pd content (nPd) of 8.9 μmol, followed by testing the temperature-dependent FA dehydrogenation. Unless otherwise noted, initial TOFs were calculated using the total amount of gas produced at 5 min by the following equation (eqn (1) and (2)). TOF values for FA dehydrogenation over Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) obtained at different temperatures were listed in Table S4 (ESI†). Further, the catalytic activities of the as-prepared catalysts toward formic acid decomposition were compared to those of other Pd based bi- and trimetallic catalysts reported previously (Table S5, ESI†).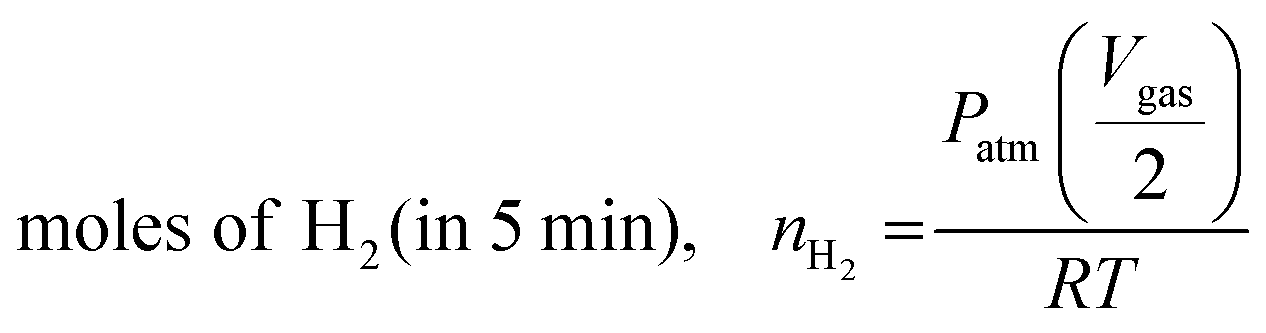 | (1) |
 | (2) |
2.4. Computational methodology
Plane-wave periodic DFT calculations were performed using the Vienna ab initio simulation package (VASP).36–38 The projector-augmented wave (PAW)39,40 was used to describe electron–ion interactions. The exchange–correlation function was described by PW91-GGA with spin polarization.41,42 The Kohn–Sham one-electron states were expanded into a plane-wave basis set up with a 400 eV cut-off energy. Brillouin zone sampling was employed using a Monkhorst–Pack grid.43 The k-point samplings were set at 16 × 16 × 16 and 4 × 4 × 1 for bulk and surface calculations, respectively. Methfessel–Paxton smearing with a value of 0.1 eV was applied.44The energy of gas-phase molecules was calculated using a single molecule in a 12 Å cube with gamma-point sampling. The metal (111) surfaces were modeled by a periodic slab containing four atomic layers with full relaxation of the uppermost two layers. A p(4 × 4) super cell with an 18 Å vacuum space in the z direction was used for the surface (111) slab models to prevent interaction with periodic images. The result of surface coverage is shown in Table S6 (ESI†).
The nudged elastic band (NEB) method was used to find a minimum energy path between the initial and final states where both states are local minima on the potential energy surface.45,46 In this approach, the reaction path was discretized with the distinct configurations or images between the energy minima connected by elastic springs to prevent the images from sliding to the minima during optimization. Convergence criteria for electronic and atomic relaxations were set at 10−4 and 10−2 eV, respectively. The k-points were selected as 2 × 2 × 1 to optimize computing time and performance. To verify the use of the smaller number of k-points for the activation energy calculations, the NEB calculations were conducted by using k-points of 4 × 4 × 1 for a key step of the reactions on both Pd(111) and Pd1Ni1(111). There was little difference in the activation energies (difference smaller than 0.02 eV), indicating that k-points grids of 2 × 2 × 1 would not affect the accuracy of activation energy. The calculated activation energies for a key step are shown in Table S7 (ESI†).
Bader charge calculations47,48 were conducted for the most stable models of Pd(111), Pd1Ni0.33(111), Pd1Ni1(111) and Pd1Ni3(111) surfaces containing an adsorbed H atom. The electron charge transfer between adsorbate and surface was determined by subtracting the Bader charge of the free adsorbate plus clean surface from that of surfaces containing adsorbates to obtain the excess Bader charge. Positive and negative excess Bader charge values represent the gain and loss of charge, respectively, upon adsorption to the catalytic surfaces.
3. Results and discussion
3.1. Synthesis and characterization of Pd1Nix materials
The bulk compositions of the Pd1Nix/N–C catalysts determined by ICP-OES are represented in terms of mole ratios as Pd1Ni0.37, Pd1Ni1.3 and Pd1Ni3.6 (Table S1, ESI†). The structures of the bimetallic Pd1Nix/N–C alloy materials (x = 0.37, 1.3 and 3.6) were first verified by HAADF-STEM and XRD analyses. Representative HAADF-STEM images indicate that Pd1Ni1.3/N–C has a smaller average PdNi NP size (3.1 nm) with higher particle dispersion compared to Pd1Ni1.3/C (7.9 nm) suggesting that the N-doped carbon support helps form relatively small PdNi NPs in a well-dispersed manner (Fig. 1). Pd1Ni0.37/N–C and Pd1Ni3.6/N–C likewise exhibit smaller average metallic sizes of 3.0 and 2.5 nm, respectively (Fig. S1, ESI†). This result is consistent with previous reports that Pd supported on mesoporous graphitic carbon nitride (mpg-C3N4)16 or N-doped carbon provides better Pd dispersion than Pd/C. The line profiles of Pd1Ni1.3 supported on C and N–C indicate formation of PdNi alloys over the entire surfaces (Fig. 1c and d). Elemental mapping of Pd1Ni1.3/N–C by HAADF-STEM confirms overlap of the Pd and Ni positions consistent with the formation of PdNi alloys (Fig. S2, ESI†).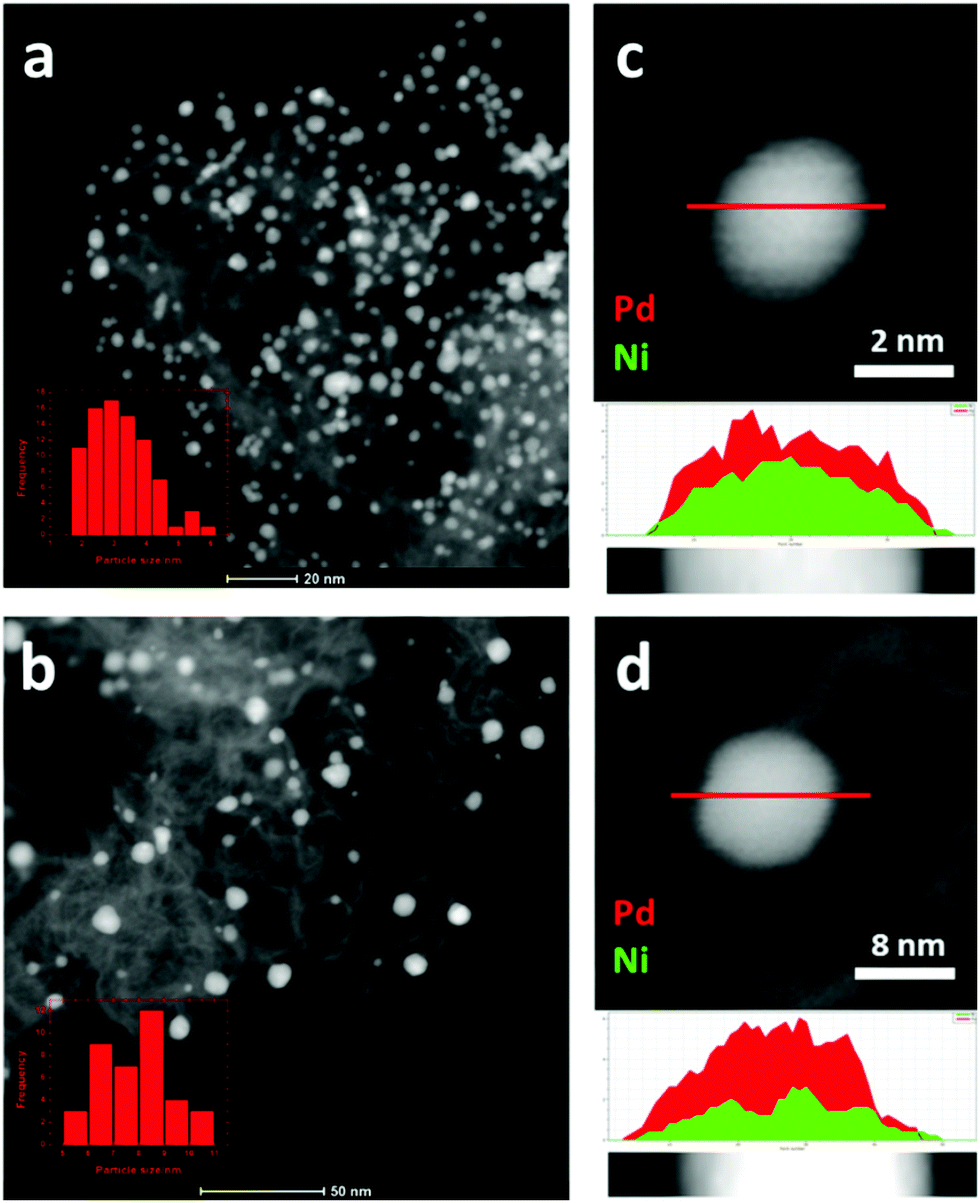 | ||
| Fig. 1 HAADF-STEM images of (a) Pd1Ni1.3/N–C and (b) Pd1Ni1.3/C and line profiles for (c) Pd1Ni1.3/N–C and (d) Pd1Ni1.3/C. | ||
The structure of the Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) alloys was further confirmed by XRD analysis. Fig. 2a shows XRD patterns of Pd1Nix/N–C as a function of the Pd/Ni ratio. The diffraction peak centered at 39.1° corresponding to the crystalline Pd nanoparticles in Pd/N–C shifts to larger diffraction angles as the Ni content increases. This indicates a gradual decrease in lattice constants due to incorporation of the smaller Ni atoms into the Pd lattice upon forming the Pd1Nix alloys: Pd, 39.1°; Pd1Ni0.37, 39.4°; Pd1Ni1.3, 39.9°; and Pd1Ni3.6, 40.0°. It has been reported that Pd and Ni form an isomorphous alloy with a face-centered cubic (fcc) structure over the entire composition range.49,50 Crystallite sizes calculated using the Scherrer method and based on the XRD results (Table S2, ESI†) exhibit similar particle sizes close to those observed by TEM. The electronic (Pd 3d) states of the alloyed catalysts were further characterized by XPS. Fig. 2b shows that the Pd(3d5/2) and Pd(3d3/2) binding energies in Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) increase slightly relative to those of Pd/N–C with increasing Ni content. In the XPS spectra, the peaks were further deconvoluted to give two separable peaks attributed to Pd0 (blue peaks) and Pd2+ (green peaks) species, respectively, presenting increased binding energies as Ni content increased. The binding energies (eV) as a function of composition are summarized in ESI† (Table S3). These results are consistent with increases in the Pd 3d XPS binding energies of Pd1Nix alloys (x = 0.33, 1 and 3) employed for CO oxidation51 and further indicate the presence of electronic interactions between Pd and Ni in the PdNi alloys.
 | ||
| Fig. 2 (a) XRD analyses of Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) and (b) XPS Pd 3d spectra of Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6). The blue and green curves represent deconvoluted peaks attributed to Pd0 and Pd2+, respectively. | ||
3.2. Catalytic activity towards FA dehydrogenation over Pd1Nix (x = 0, 0.37, 1.3 and 3.6)
Five materials (Pd/N–C, Pd1Ni0.37/N–C, Pd1Ni1.3/N–C, Pd1Ni3.6/N–C and Ni/N–C) were employed to initially examine the influence of Pd/Ni ratio on H2-release rate from aqueous 1.0 M HCO2H plus 1.0 M sodium formate as a Brønsted base at 30 °C. Fig. 3a shows that Ni/N–C exhibits negligible FA dehydrogenation activity, whereas Pd/N–C displays much higher activity with ca. 100 mL of gases (H2 + CO2) being released within 100 min of reaction time. Upon utilization of Pd1Nix/N–C (x = 0.37, 1.3 and 3.6), their catalytic activities were further increased compared to that of Pd/N–C. In addition, the catalysts with three different Pd/Ni ratios showed volcano-type behavior where the highest activity was obtained over Pd1Ni1.3/N–C (Fig. 3b). These results emphasize the important role of Ni in perturbing the Pd electronic structure, thereby facilitating H2-release from aqueous FA solution. In line with the results, XPS analysis (Fig. 2b) also suggests constructive interaction between Pd and Ni atoms possibly leading to the formation of new active sites responsible for the enhanced FA dehydrogenation activity. To determine composition of the released gases during formic acid decomposition, separate experiments were carried out using newly prepared Pd1Nix catalysts (x = 0.37, 1.3 and 3.6). Analysis with gas chromatography (GC) revealed the formation of H2 and CO2, with no CO gas being formed (Fig. S3, ESI†). The calculated turnover frequencies (TOFs) for Pd1Ni0.37/N–C, Pd1Ni1.3/N–C and Pd1Ni3.6/N–C of 369, 447 and 347 h−1, respectively, at 30 °C are greater than that of Pd/N–C (TOF = 232 h−1 at 30 °C) under identical reaction conditions (Table S4, ESI†). Likewise, the TOF of Pd1Ni1.3/C (TOF = 363 h−1) determined at 25 °C is much higher than those of Pd/N–C (TOF = 127 h−1), Pd1Ni1.3/C (TOF = 85.5 h−1) and Pd/C (TOF = 37.5 h−1) obtained at 25 °C.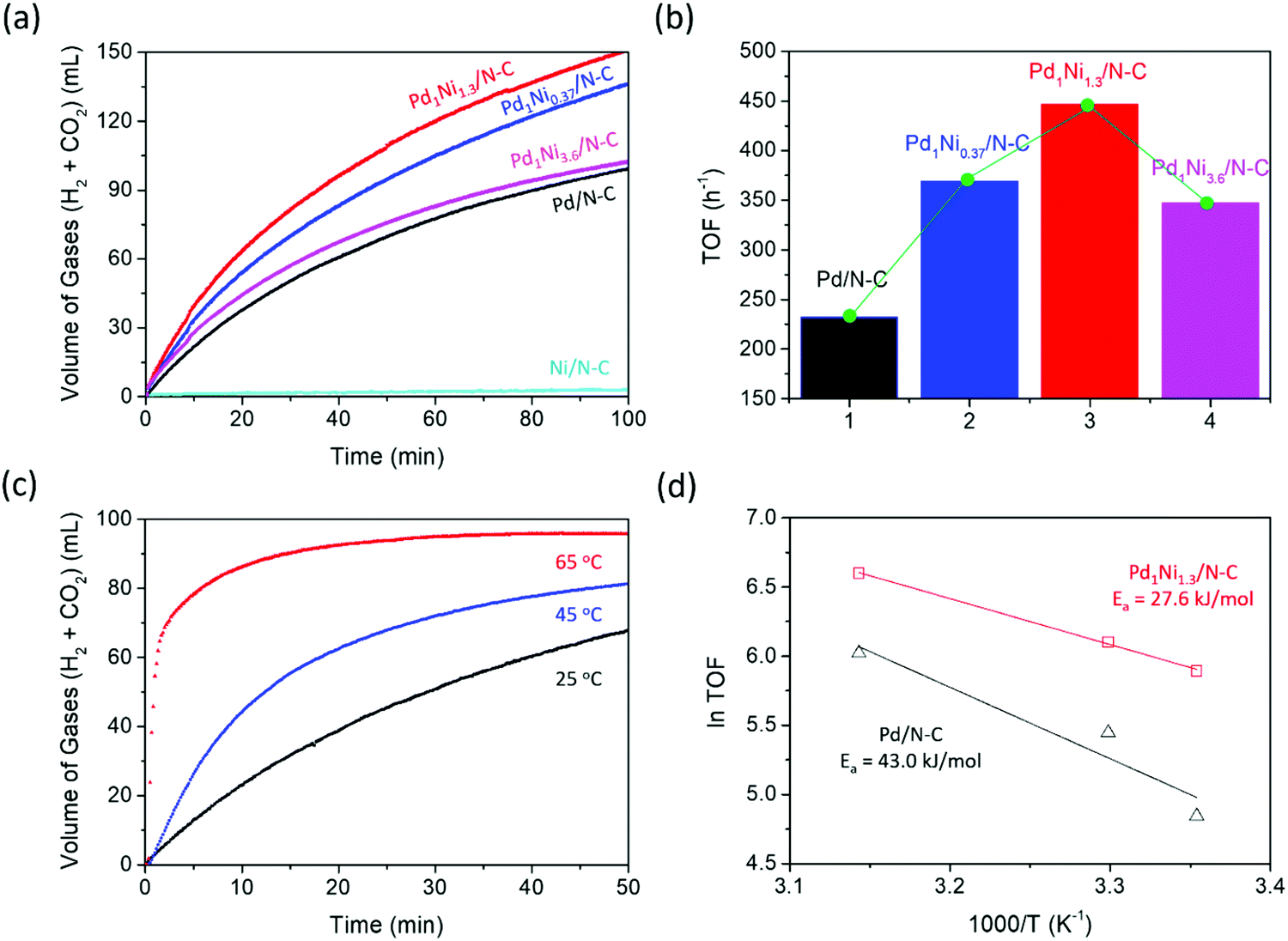 | ||
| Fig. 3 (a) Gas (H2 + CO2) evolution profiles from 10 mL of aqueous FA/formate (1.0 M HCO2H, 1.0 M HCO2Na) solution with Pd-based catalysts (Pd, 11.6–12.7 μmol) at 30 °C. (b) TOFs obtained using Pd1Nix/N–C (x = 0, 0.37, 1.3 and 3.6) (c) temperature dependent gas (H2 + CO2) evolution obtained using Pd1Ni1.3/N–C (Pd, 8.9 μmol). (d) Arrhenius plots based on initial rates at 25, 30 and 45 °C. Hollow squares and triangles represent Pd1Ni1.3/N–C and Pd/N–C, respectively. | ||
In a separate experiment, the influence of temperature on dehydrogenation activity was studied using Pd1Ni1.3/N–C as a function of temperature (Fig. 3c). TOFs of 363, 736 and 2195 h−1 were obtained using the initial 5 min data at 25, 45 and 65 °C, respectively. It is noted that the TOF of 2195 h−1 at 65 °C was underestimated because the rate of H2-release based on the initial 5 min data was estimated using both linear and curved sections; when considering the initial 2 min data for dehydrogenation, the TOF at 65 °C increases to 4804 h−1. Given that the added Brønsted base, sodium formate can potentially react with water to release hydrogen (HCO2Na + H2O → HCO3Na + H2) particularly at temperatures of >65 °C in the presence of a Pd based catalyst,21 the observed rapid H2-release at 65 °C not only originates from FA decomposition (HCO2H → CO2 + H2), but it could also come from formate dehydrogenation (HCO2− + H2O → HCO3− + H2). To avoid the potential formate dehydrogenation effect, Arrhenius plots were obtained based on the TOFs determined at the low temperatures of 25, 30 and 45 °C. The activation energies calculated from the temperature dependent rate of FA dehydrogenation (Fig. 3d) were 27.6 and 43.0 kJ mol−1 for Pd1Ni1.3/N–C and Pd/N–C, respectively. The results again support the positive effect of PdNi alloying on H2-release from FA. The apparent prefactor, A values were then calculated using the Arrhenius equation, presenting 2.5 × 107 s−1 and 4.9 × 109 s−1 for Pd1Ni1.3/N–C and Pd/N–C, respectively. Since pre-exponential factor is related to the amount of times molecules hit in the orientation necessary to cause a reaction, this collision factor is higher in Pd/N–C catalyst than that of Pd1Ni1.3/N–C catalyst although the activation energy value itself is lower for Pd1Ni1.3/N–C catalyst (27.6 kJ mol−1) than that of Pd/N–C catalyst (43.0 kJ mol−1).
Pd-Based bimetallic catalysts for FA decomposition have been developed to reduce Pd contents and the effect of Pd–M alloys (M = transition metals) on FA dehydrogenation has been studied. For instance, comparing dehydrogenation catalysts utilizing carbon-based supports, Pd59Au41/C32 and Pd25Au75/C52 were reported to liberate molecular hydrogen from FA with the TOFs of 230 h−1 (at 50 °C) and 212 h−1 (at 75 °C), respectively. Similarly, monodispersed Ag42Pd5853 alloy nanoparticles were also found to be active and durable catalysts for FA decomposition at 50 °C, with the TOF of 382 h−1. In addition to numerous PdAu and PdAg catalysts, Pd-based bimetallic catalysts alloyed with non-precious metals were also developed. A PdNi@Pd/GNs–CB29 (GNs–CB = graphene nanosheets–carbon black) catalyst was synthesized and tested for FA dehydrogenation at room temperature, presenting a high activity with the TOF of 577 h−1 calculated based on the number of active sites determined. Compared to the PdNi/GNs–CB catalyst, the as-developed Pd1Nix/N–C (x = 0.37, 1.3 and 3.6) material exhibits comparable TOFs at the similar temperature of 30 °C. Compared to catalysts possessing precious metals, our catalysts could likely be economically advantageous. Additionally, the nitrogen-doped carbon support employed in the Pd1Nix/N–C catalysts can readily be mass produced. In addition to the beneficial effect of PdNi alloys on FA dehydrogenation, nitrogen-doped carbon employed can also provide a positive effect in increasing catalytic activity by donating charge density to the PdNi sites. Catalytic activities of Pd-based bi- and tri-metallic catalysts for formic acid dehydrogenation were compared (Table S5, ESI†). To elucidate how Ni alloying affects the reactivity on FA decomposition, mechanistic pathways were further explored by theory with relevant energetic consideration.
3.3. Possible mechanism of FA dehydrogenation
O) and 1.353 (C–O) Å (Table S8, ESI†) in agreement with experimental values of 0.972 (O–H), 1.097 (C–H), 1.202 (CO) and 1.343 (C–O) Å, respectively.58
Fig. 4 shows that the d-band centers of Pd–Pd shift to more negative values with increasing Ni content implying that the metal–adsorbate interaction decreases as: Pd(111) > Pd1Ni0.33(111) > Pd1Ni1(111) > Pd1Ni3(111). However, the d-band centers of Pd–Ni shift to more positive values or closer to the Fermi level potentially leading to enhanced interaction between metal atoms and adsorbates. Considering that catalytic activity is high when the strength of substrate adsorption is moderate, the d-band centers that decrease (in the Pd–Pd case) and increase (in the Pd–Ni case) with increasing Ni mole fraction suggest that there is a balance point at which an appropriate Ni content maximizes activity. The changes in spin density of individual Pd and Ni atoms in the Pd–Ni alloys are inversely proportional to one another upon changing the Ni content. Previous studies reported that asymmetric spin density showed high catalytic activity for oxygen reduction reaction (ORR) and HCOO− dehydrogenation by improving the adsorption of intermediates.59,60 Thus, greater spin density, which is the difference between the electron densities of the spin-up and spin-down electrons, may correlate with higher catalytic activity. This result supports the existence of a balance point where there is optimum catalytic activity of the Pd1Ni1 alloy. The Pd1Ni1(111) surface also provides more Pd–Ni interactions compared to the Pd1Ni0.33(111) and Pd1Ni3(111) surfaces (Fig. 4b–d, insets). The effect of spin density on adsorption is discussed in ESI,† Section S2.
 | ||
| Fig. 4 The d-band structures and center values of neighboring Pd and Ni atoms represented by the local density of states (LDOS) for: (a) Pd(111), (b) Pd1Ni0.33(111), (c) Pd1Ni1(111) and (d) Pd1Ni3(111) surfaces. The grey and yellow spheres represent Pd and Ni atoms, respectively. The spin density (the sum of absolute value of difference between the electron densities of the spin-up and spin-down electrons) of individual Pd and Ni atoms in each system is shown in the second row of panels (e–h). | ||
Fig. 4d also shows that the degree of d-orbital hybridization of Pd and Ni on the Pd1Ni3(111) surface is somewhat lower than on the Pd1Ni0.33(111) and Pd1Ni1(111) surfaces. The degree of hybridization is verified by the 0.67, 0.78 and 0.95 eV gaps between the d-band centers of Pd and Ni on Pd1Ni0.33(111), Pd1Ni1(111) and Pd1Ni3(111), respectively. This suggests that electronic stimulation of FA dehydrogenation is greater on Pd1Ni1(111) and Pd1Ni0.33(111) than it is on Pd1Ni3(111), which is structurally more perturbed by greater amounts of Ni. This phenomenon may explain the volcano-type activity obtained experimentally with Pd1Ni0.37, Pd1Ni1.3 and Pd1Ni3.6 in Fig. 3b, and is supported by a recent report that the elevated catalytic activity observed experimentally for CO oxidation over Pd1Ni1 arises from increased interactions among adjacent Pd and Ni atoms based on HE-XRD/PDFs and a Monte Carlo simulation.51 The balance point and calculation methods for the gaps is shown in ESI,† Section S2.
Adsorbates are bound primarily at the Ni atoms on the Pd1Ni1(111) surface. This result is attributed in part to the difference in metal electronegativity. Carbon-containing adsorbates will interact more strongly with Ni than with Pd, because the electronegativity of Ni is smaller.65 This implies that electron charge transfer from Ni to C in key intermediates is easier than from Pd to C. The Pauling electronegativities of Ni, Pd, H, C and O are 1.91, 2.20, 2.20, 2.55 and 3.44, respectively. The calculated binding affinity of intermediates on the Pd(111) surfaces increases (i.e., the adsorption energy becomes more negative) in the order HCOOH < H ≪ HCOOb < CO < COOH < HCOOa < OH (Table S9, ESI†). The binding affinity is greater on the Pd1Ni1(111) surface than it is on Pd(111) for all key intermediates except CO* and H* (Table S9, ESI†). The adsorption strength of intermediates on the Pd1Ni1(111) surface increases as: HCOOH < H ≪ CO < HCOOb < COOH < HCOOa < OH. The adsorption configurations are shown in Fig. S7 (ESI†). HCOOH is preferentially adsorbed in the trans rather than cis conformation, which has been confirmed by theoretical and experimental studies.61–64,66 HCOOa has a high binding affinity on both surfaces due to the strong interaction between the metal surface and the two oxygen atoms of HCOO (Fig. S7, ESI†). The adsorption configuration of HCOOb leads to the formation of metal–O and metal–H bonds and to weaker binding energies than with HCOOa (Fig. S7, ESI†).
The initial adsorption configuration of a reactant may strongly influence the entire pathway of a reaction in the NEB calculation. Therefore, we calculated the geometrical parameters of relevant adsorbates as a function of their configurations. The calculated parameters of the O–H, C–H, CO and C–O bonds in gas-phase HCOOH, HCOOH*, HCOOa*, HCOOb* and COOH* on Pd(111) and Pd1Ni1(111) surfaces are listed in Table S8 (ESI†). The asterisk (*) denotes adsorbed species. Compared to gas-phase HCOOH, the adsorbed HCOOH* configurations on Pd(111) and Pd1Ni1(111) surfaces show variations in their O–H and C–H bond lengths. The O–H bonds of HCOOH* are stretched by 3.19 and 3.84%, whereas the C–H bonds are compressed by 0.21 and 0.32% on the Pd(111) and Pd1Ni1(111) surfaces, respectively. The elongation of O–H bonds of both surfaces suggests that O–H bond scission is the initial step in the production of H* and HCOO* from HCOOH* (HCOOH* → HCOO* + H*). The C–H bond lengths in HCOOa* are nearly identical at 1.110 Å on Pd(111) and 1.107 Å on Pd1Ni1(111), but the corresponding bonds in HCOOb* are elongated by 6.45 and 1.34% on Pd(111) and Pd1Ni1(111), respectively. Given the geometric information and binding affinities of HCOOa* and HCOOb* for metal surfaces, it is plausible that the HCOO* produced in the initial dehydrogenation of HCOOH is first bound to the surface as HCOOa*. However, actual C–H bond activation may occur via HCOOb* to produce CO2 and H* by the following process: HCOOa* + H* → HCOOb* + H* → CO2 + 2H*. This suggests the likely interconversion of HCOOa* to HCOOb* (vide infra). The two H* species produced in this process recombine to release a hydrogen molecule: H* + H* → H2. To validate the foregoing hypothesis, plausible mechanisms of FA dehydrogenation were further explored by calculating the activation energies of relevant transition states in the reaction pathways.
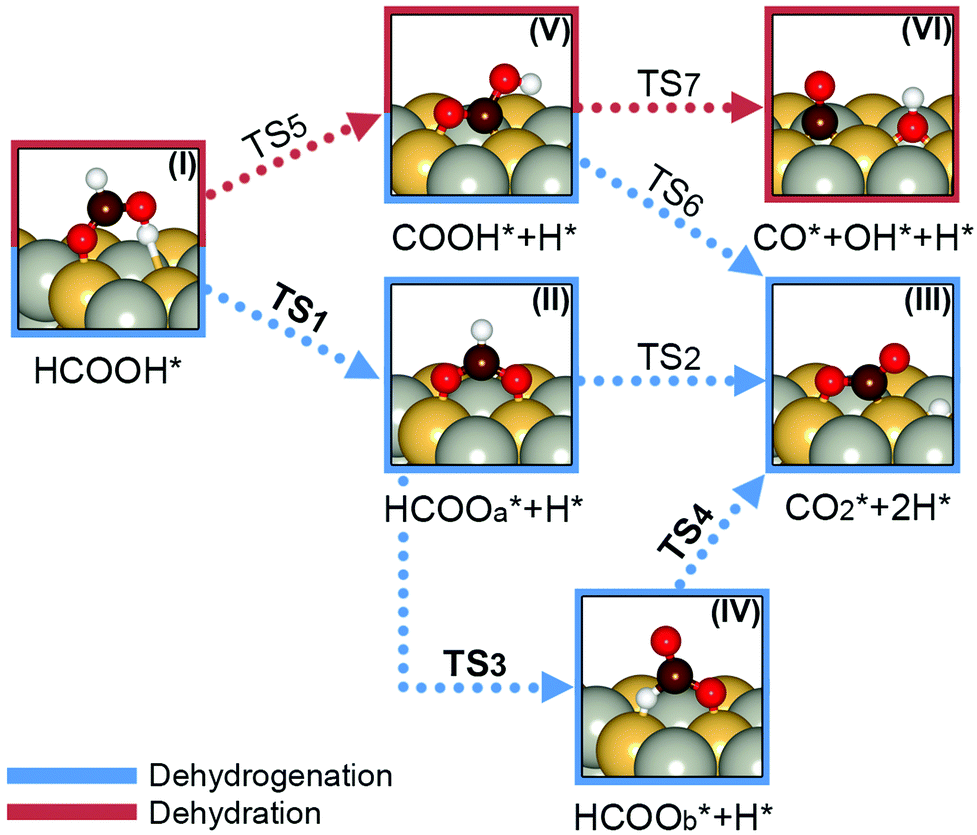 | ||
| Fig. 5 Schematic representation of probable reaction pathways of FA dehydrogenation at the Pd1Ni1(111) surface. Dashed blue and red lines indicate dehydrogenation and dehydration reactions, respectively. Grey, light yellow, dark brown, red and white spheres represent Pd, Ni, C, O and H atoms, respectively. | ||
3.3.4.1. FA dehydrogenation initiated by O–H bond scission (HCOO pathway). HCOOH dehydrogenation initiated by O–H bond cleavage produces the HCOOa intermediate (II). The geometries of HCOOH* ground state structures at Pd(111) and Pd1Ni1(111) surfaces (Table S8, ESI†) suggest that the O–H bonds of adsorbed HCOOH* are partially activated at both surfaces. This is supported by bond elongations of 3.19 and 3.84% on Pd(111) and Pd1Ni1(111), respectively. Fig. 6a shows the steps and activation energies of FA dehydrogenation by the HCOO pathway based on the initial HCOOa* configuration.
 | ||
| Fig. 6 Relative DFT energy diagrams for FA dehydrogenation proceeding by the (a) HCOO and (b) COOH pathways over Pd(111) (red) and Pd1Ni1(111) (blue) surfaces. The solid and dashed lines indicate the I → II → IV → III and I → II → III steps in the HCOO pathway and the I → V → VI and I → V → III steps in the COOH pathway, respectively, according to the notation in Fig. 5. | ||
The first step in the HCOO pathway is activation of the O–H bond of HCOO–H* to form HCOOa* + H* (II) via TS1 with activation energies of 0.78 and 0.61 eV for Pd(111) and Pd1Ni1(111), respectively (Table 1). The most stable configuration of the adsorbed HCOOa* intermediate has two Pd–O bonds on Pd(111) and two Ni–O bonds on Pd1Ni1(111). Other adsorption configurations comprising two Pd–O bonds and one Pd–O plus one Ni–O bond were optimized on Pd1Ni1(111), but these configurations were less stable than the intermediate with two Ni–O bonds (Fig. S7c, ESI†). The HCOOa* intermediate is converted into CO2* + 2H* (III) via C–H bond scission, which can proceed in two ways. The first path is direct C–H bond activation via TS2 yielding CO2* + 2H* (III), which requires greater activation energies of 2.66 and 3.34 eV on Pd(111) and Pd1Ni1(111) surfaces, respectively (dashed lines in Fig. 6a). The other path involves rotational conversion of HCOOa* + H* (II) into HCOOb* + H* (IV) via TS3 followed by C–H bond activation via TS4 to produce CO2* + 2H* (solid lines in Fig. 6a). The conformational rotation of HCOOa* requires activation energies of 1.19 eV on Pd(111) and 1.02 eV on Pd1Ni1(111). The HCOOb* + H* (IV) intermediate produced by rotation of HCOOa* + H* (II) has one Pd–O bond and one Pd–H bond on Pd(111) and one Ni–O bond and one Ni–H bond on Pd1Ni1(111) (Fig. 5 and Fig. S7c, ESI†). The consecutive C–H bond cleavage requires activation energies of 0.19 and 0.21 eV on Pd(111) and Pd1Ni1(111) surfaces, respectively. These results clearly indicate that the energetically lower HCOO pathway involves interconversion from HCOOH* (I) → HCOOa* + H* (II) → HCOOb* + H* (IV) to CO2* + 2H* (III). The greater activation energy for the direct C–H bond scission of HCOOa* is related to the unreactive character of its C–H bond at both surfaces, as evident from the negligible change in C–H bond length of adsorbed HCOOa* (Table S8, ESI†). These results further indicate that the rate determining step in the HCOO pathway of FA dehydrogenation on Pd(111) and Pd1Ni1(111) lies in the TS3 step involving rotation of HCOOa* to HCOOb*. This interpretation is in line with recent studies of FA dehydrogenation, which favor the rate limiting step of HCOO* + H* → CO2* + 2H* rather than HCOOH* → HCOO* + H*.67–69
| HCOO pathway | TS1 (eV) (I → II) | TS2 (eV) (II → III) | TS3 (eV) (II → IV) | TS4 (eV) (IV → III) | |
|---|---|---|---|---|---|
| I → II → III | Pd(111) | 0.78 | 2.66 | — | — |
| Pd1Ni1(111) | 0.61 | 3.34 | — | — | |
| I → II → IV → III | Pd(111) | 0.78 | — | 1.19 | 0.19 |
| Pd1Ni1(111) | 0.61 | — | 1.02 | 0.21 | |
| COOH pathway | TS5 (eV) (I → V) | TS6 (eV) (V → III) | TS7 (eV) (V → VI) | |
|---|---|---|---|---|
| I → V → III | Pd(111) | 1.08(5) | 1.15 | — |
| Pd1Ni1(111) | 1.07(6) | 2.89 | — | |
| I → V → VI | Pd(111) | 1.08(5) | — | 0.92 |
| Pd1Ni1(111) | 1.07(6) | — | 1.31 | |
3.3.4.2. FA dehydrogenation initiated by C–H bond scission (COOH and CO–OH pathways). HCOOH dehydrogenation via the COOH pathway is initiated by C–H bond scission and produces the COOH* intermediate. The C–H bond cleavage of HCOOH* via TS5 on Pd(111) and Pd1Ni1(111) surfaces requires greater activation energies of 1.085 and 1.076 eV, respectively (Table 1 and solid lines in Fig. 6b), than those of TS1. This view is supported by the compressed C–H bond of HCOOH* on Pd1Ni1(111) as well as the identical C–H bond length on Pd(111) (Table S8, ESI†). The HCOO pathway is favored over the COOH pathway on both surfaces. The COOH* species (V) in Fig. 5 can further be dehydrogenated by O–H bond cleavage via TS6 generating CO2* + 2H* (III) (dashed lines in Fig. 6b) or dehydrated by C–O bond cleavage via TS7 yielding CO* + OH* (VI) (solid lines in Fig. 6b). The barrier to O–H bond breaking (TS6) on both surfaces is greater than that of TS5. The activation energies of TS6 on Pd(111) and Pd1Ni1(111) are 1.15 and 2.89 eV, respectively. The kinetic barriers to C–O bond cleavage [TS7: 0.92 eV on Pd(111), 1.31 eV on Pd1Ni1(111)] are lower than those of TS6 at these surfaces. This indicates that the dehydration reaction (C–O bond cleavage) is favored over the dehydrogenation reaction (O–H bond cleavage) in the COOH pathway. Despite these negative findings, it is important that the activation energy of TS7 on Pd1Ni1(111) is greater than it is on Pd(111). This means that the Pd1Ni1(111) surface will have greater ability than the Pd(111) surface to control CO production.
The calculated energy diagrams in Fig. 6 reveal that the HCOO pathway is the most favorable HCOOH dehydrogenation pathway through the conversion of HCOOa* to HCOOb*. It is evident that the Pd1Ni1(111) catalyst displays lower kinetic barriers to the desired reaction than the Pd(111) catalyst. Thus, the computational results agree well with the experimental results that demonstrate the improved activity of PdNi/N–C compared to Pd/C for H2-release from aqueous FA solution (Fig. 3).
3.3.4.3. FA dehydrogenation on Pd1Ni0.33(111) and Pd1Ni3(111) surfaces. We further confirmed that HCOOH dehydrogenation is more favorable on Pd1Nix(111) surfaces than on Pd(111) by analyzing the activation energies of key steps in the optimal HCOO pathway and the initial step of the COOH pathway on Pd1Ni0.33(111) and Pd1Ni3(111) surfaces (Fig. 7 and Table S14, ESI†). The activation energies of the HCOO pathway [HCOOH* (I) → HCOOa* + H* (II) → HCOOb* + H* (IV) → CO2* + H* (III)] and the initial step of the COOH pathway [HCOOH* (I) → COOH* + H* (V)] on Pd1Ni0.33(111) and Pd1Ni3(111) demonstrate the favorability of the HCOO pathway [TS1: Pd1Ni0.33(111) = 0.77 eV, Pd1Ni3(111) = 0.60 eV] over the COOH pathway [TS5: 0.78 and 0.74 eV, respectively] (Fig. 7 and Table S14, ESI†). In the rate determining step (TS3), Pd1Ni0.33(111) (0.86 eV) and Pd1Ni3(111) (1.16 eV) exhibit lower activation energies than the Pd(111) surface (1.19 eV) (Fig. 7).
 | ||
| Fig. 7 Relative DFT energy diagram for FA dehydrogenation proceeding by the optimal HCOO pathway (I → II → IV → III steps, right) and the COOH pathway (initial step only, left) over Pd1Ni0.33(111) (orange), Pd1Ni1(111) (blue) and Pd1Ni3(111) (green) surfaces. | ||
Interestingly, the Pd1Ni0.33(111) surface has the lowest energy barrier for the rate determining step among the Pd1Nix(111) surfaces. This disagrees with the trend in efficiency of HCOOH dehydrogenation determined by our experimental findings (Pd1Ni1.3/N–C > Pd1Ni0.37/N–C > Pd1Ni3.6/N–C > Pd/N–C; Fig. 3b). However, there is a very small difference between the activation energies of the initial steps in the HCOO and COOH pathways (0.77 and 0.78 eV, respectively) at the Pd1Ni0.33(111) surface. This may lead to CO production via a side reaction in the COOH pathway due to poor selectivity between the HCOO and COOH mechanisms resulting in a decrease in the measured efficiency of HCOOH dehydrogenation. Similar cases have been reported in previous studies.63,68,70 These facts inspired us to analyze the detailed properties of Pd1Nix(111) surfaces to assess the efficiency of HCOOH dehydrogenation on each material.
3.4. Key factors of Pd1Nix(111) surfaces for enhancing FA dehydrogenation
Alloying Pd with Ni produces electronic and geometric effects arising from the relatively smaller electronegativity and lattice constants of Ni, respectively, which may affect HCOOH dehydrogenation in positive ways. Fig. 8 and Table S15 (ESI†) show that the key adsorbates in the HCOOH dehydrogenation reaction pathways are significantly influenced by the electronic effect of Pd1Nix(111) surfaces, especially when the O atoms of adsorbates interact directly with Ni atoms on the catalyst surfaces. The O atom with the greatest electronegativity prefers to interact with the relatively less electronegative Ni than Pd because electron transfer from O to the metal is easier.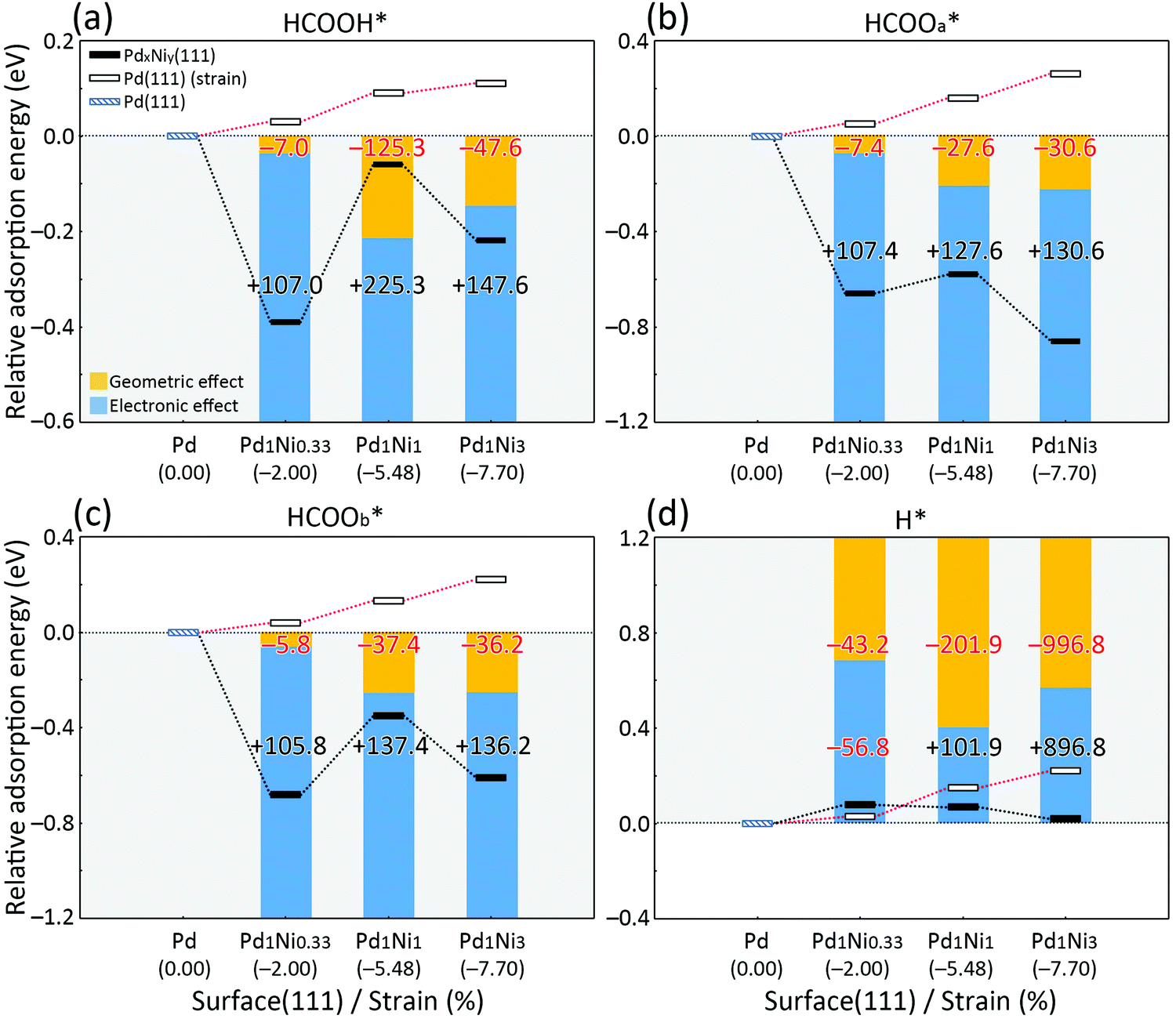 | ||
| Fig. 8 Relative adsorption energy diagrams of (a) HCOOH*, (b) HCOOa*, (c) HCOOb* and (d) H* as a function of surface composition and strain. The small blue, black and white rectangles represent the relative adsorption energy of adsorbates on Pd(111), Pd1Nix(111) and compressed Pd(111) surfaces, respectively. The yellow and blue bars indicate the relative contributions of geometric and electronic effects on the adsorption strength at Pd1Nix alloys; positive (black) and negative (red) values (%) indicate increasing and decreasing contributions to the strength of adsorption. The numbers in parentheses on the x-axis represent the percent compressive strain. | ||
We also found changes in the electronic properties of catalysts depending on the amount of Ni in Pd1Nix alloys by calculating work functions and spin densities of the four catalyst surfaces. The work function values of Pd(111), Pd1Ni0.33(111), Pd1Ni1(111) and Pd1Ni3(111) surfaces are 5.57, 5.30, 5.19 and 5.20 eV, respectively. Generally, the reduction in work function with increasing Ni content indicates easier electron transfer from the catalyst to adsorbate, which leads to a stronger adsorption interaction. Higher Ni content in Pd1Nix also produces greater spin densities on the surface layers of the catalysts (Fig. S7, ESI†), which can trigger strong adsorption.59,60 An interesting finding is that the differences in adsorption strength of intermediates on Pd(111) surface and Pd1Nix(111) surfaces were clearly attributed to the electronic properties, whereas those were not among Pd1Nix(111) surfaces. These results mean that the electronic properties of Pd1Nix alloys play an important role in the formation of intermediates on catalyst surfaces as evidenced by the more favorable adsorption of HCOOH and other intermediates on Pd1Nix(111) than on Pd(111) surfaces, and also imply the existence of possibility that other factors like geometric properties may affect the adsorption of adsorbates.
Adsorbates such as CO and H are adsorbed more weakly on Pd1Nix(111) than on Pd(111) (Table S9, ESI†). Because CO and H do not have an O atom that strongly interacts with the catalyst surfaces due to its high electronegativity, these adsorbates may be more influenced by factors beyond electronic effects unlike HCOOH, HCOOa and HCOOb (Fig. 8). Fig. 8d demonstrates that for H adsorption on PdNi surfaces the geometric effect is greater than the electronic effect relative to the behavior of other adsorbates (Fig. 8a–c). In the case of HCOOH dehydrogenation, which depends on the weak adsorption of hydrogen, desorption of H* from the surface may be an important step in producing H2(g) as the final product.60 The most stable adsorption configurations of H* on Pd(111), Pd1Ni0.33(111), Pd1Ni1(111) and Pd1Ni3(111) are shown in Fig. S10 (ESI†). The calculated adsorption energies are −0.60, −0.52, −0.53 and −0.58, respectively. The results do not correlate with the amount of Ni. An interesting and important finding is that—although electron transfer from the catalyst surface to H* increases with increasing Ni content as 0.11, 0.17, 0.21 and 0.26 e—H* exhibits weaker adsorption energies on the Pd1Ni0.33(111) and Pd1Ni1(111) surfaces that simulate the Pd1Ni0.37 and Pd1Ni1.3 catalysts, which show excellent FA dehydrogenation efficiency in our experiments. This evidence supports the assumption that factors other than electronic effects may influence FA dehydrogenation. Fig. 8d shows that electronic effects on H* adsorption are weak relative to adsorption of other substrates.
An important factor in FA dehydrogenation is the compressive strain caused by geometric differences in the lattice constants of Pd and Ni. To assess the contribution of this factor, we applied compressive strain to Pd(111) surfaces in increments of 2.00, 5.48 and 7.70% from the interatomic distances on a Pd(111) surface (0% compressive strain) and subsequently calculated the H* adsorption energies and conducted an excess Bader charge analysis. The strength of H* adsorption decreases to −0.57, −0.45 and −0.38 eV (Table S16, ESI† and Fig. 8d), and the electronic charge transferred from the surface to H* decreases to 0.118, 0.095 and 0.089 e as the compressive strain increases. Fig. 8a–c show that the adsorption strength of O-containing adsorbates (HCOOH, HCOOa and HCOOb) also decreases as the interatomic distance decreases, which agrees with previous studies.71,72 Detailed adsorption energies of these forms are collected in Table S16 (ESI†). The results indicate that addition of an appropriate amount of Ni to Pd can lower the activation energy for the FA dehydrogenation and enhance the reaction rate based on (i) the stable formation of key O-containing intermediates (an electronic effect) that interact directly with the surface (Fig. 8a–c) and (ii) weakened H* adsorption (a geometric effect), which facilitates more favorable H* desorption to produce H2(g) (Fig. 8d). Pd1Ni1(111) exhibits the most appropriate electronic and geometric properties for FA dehydrogenation among the Pd(111), Pd1Ni0.33(111), Pd1Ni1(111) and Pd1Ni3(111) surfaces. This conclusion is supported by the result that Pd1Ni1(111) presents the lowest activation barrier to H* desorption (2.80 eV) based on single point calculations of the four catalyst surfaces (Fig. S11, ESI†). It is also true that the energetic span (sum of endothermic reactions) from DFT calculations on the Pd1Ni1(111) surface (1.84 eV) in the overall HCOO pathway including the CO2* + 2H* formation step (TS4) leading to H2(g) production is slightly lower than that of Pd1Ni0.33(111) (1.85 eV) (Fig. 7).
4. Conclusions
Experimental and theoretical studies demonstrate that Pd–Ni alloys possess greater catalytic activities towards FA dehydrogenation than Pd metal. Electronic structural studies using XPS and d-band center calculations show that catalytic dehydrogenation activities improve as a result of synergistic interactions between Pd and Ni atoms, which suggests that the active site of FA dehydrogenation is associated with both Pd and Ni atoms rather than either metal alone. The heterogeneous Pd–Ni structure also plays a synergistic role in improving catalytic activity based on constructive delocalization of Pd and Ni d-bands. The improved catalytic activity of Pd1Nix/N–C (x = 0.37, 1.3 and 3.6) in FA dehydrogenation at ambient temperature is supported by calculations employing the DFT-NEB method. The HCOO pathway is favored over the COOH/CO–OH pathway at both Pd and Pd1Ni1 surfaces, which indicates that the as-prepared catalyst prefers the dehydrogenation over the dehydration pathway. Finally, the PdNi catalyst with the appropriate amount of Ni (i.e., the Pd1Ni1 catalyst in the DFT calculations) showed the highest catalytic activity not only by lowering the activation energy along the pathway of FA dehydrogenation reaction but also by suppressing the side reaction of CO production. The fundamental understanding of FA dehydrogenation over PdNi-alloy catalysts presented herein should contribute to the design of novel, highly capable, and inexpensive heterogeneous catalysts to achieve the rapid H2-release from FA for numerous hydrogen energy applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by a National Research Foundation (NRF) grant funded by the Korean government (Ministry of Science, ICT & Future Planning) [grant number NRF-2019M3E6A1064611 & 2019M3E6A1064940] and KIST Institutional Programs [No. 2E30993].References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- E. B. Fox, Z.-W. Liu and Z.-T. Liu, Energy Fuels, 2013, 27, 6335–6338 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- A. Boddien, F. Gärtner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- D. J. Durbin and C. Malardier-Jugroot, Int. J. Hydrogen Energy, 2013, 38, 14595–14617 CrossRef CAS.
- P. P. Edwards, V. L. Kuznetsov, W. I. F. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- M. Reuß, T. Grube, M. Robinius, P. Preuster, P. Wasserscheid and D. Stolten, Appl. Energy, 2017, 200, 290–302 CrossRef.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- K. Jiang, K. Xu, S. Zou and W.-B. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- J. H. Lee, J. Ryu, J. Y. Kim, S.-W. Nam, J. H. Han, T.-H. Lim, S. Gautam, K. H. Chae and C. W. Yoon, J. Mater. Chem. A, 2014, 2, 9490–9495 RSC.
- F.-Z. Song, Q.-L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141–5144 CrossRef CAS.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- K. Koh, J. E. Seo, J. H. Lee, A. Goswami, C. W. Yoon and T. Asefa, J. Mater. Chem. A, 2014, 2, 20444–20449 RSC.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- T. Asefa, K. Koh and C. W. Yoon, Adv. Energy Mater., 2019, 9, 1901158 CrossRef.
- Y. Kim and D. H. Kim, Appl. Catal., B, 2019, 244, 684–693 CrossRef CAS.
- H.-j. Jeon and Y.-M. Chung, Appl. Catal., B, 2017, 210, 212–222 CrossRef CAS.
- S. Akbayrak, Y. Tonbul and S. Özkar, Appl. Catal., B, 2017, 206, 384–392 CrossRef CAS.
- M. Hattori, H. Einaga, T. Daio and M. Tsuji, J. Mater. Chem. A, 2015, 3, 4453–4461 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122–5128 CrossRef CAS.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang, Y. Ping and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12721–12725 RSC.
- Y.-L. Qin, J. Wang, F.-Z. Meng, L.-M. Wang and X.-B. Zhang, Chem. Commun., 2013, 49, 10028–10030 RSC.
- Y.-L. Qin, Y.-C. Liu, F. Liang and L.-M. Wang, ChemSusChem, 2015, 8, 260–263 CrossRef CAS PubMed.
- C. Hu, X. Mu, J. Fan, H. Ma, X. Zhao, G. Chen, Z. Zhou and N. Zheng, ChemNanoMat, 2016, 2, 28–32 CrossRef CAS.
- Ö. Metin, X. Sun and S. Sun, Nanoscale, 2013, 5, 910–912 RSC.
- W.-Y. Yu, G. M. Mullen, D. W. Flaherty and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 11070–11078 CrossRef CAS PubMed.
- Z.-L. Wang, J.-M. Yan, Y. Ping, H.-L. Wang, W.-T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- H. Wang, Y. Chi, D. Gao, Z. Wang, C. Wang, L. Wang, M. Wang, D. Cheng, J. Zhang, C. Wu and Z. Zhao, Appl. Catal., B, 2019, 255, 117776 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 4978 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef PubMed.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673–9682 CrossRef CAS.
- Q. Guo, D. Liu, X. Zhang, L. Li, H. Hou, O. Niwa and T. You, Anal. Chem., 2014, 86, 5898–5905 CrossRef CAS PubMed.
- S. Shan, V. Petkov, L. Yang, J. Luo, P. Joseph, D. Mayzel, B. Prasai, L. Wang, M. Engelhard and C.-J. Zhong, J. Am. Chem. Soc., 2014, 136, 7140–7151 CrossRef CAS.
- J. L. Santos, C. León, G. Monnier, S. Ivanova, M. Á. Centeno and J. A. Odriozola, Int. J. Hydrogen Energy, 2020, 45, 23056–23068 CrossRef CAS.
- S. Zhang, Ö. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed.
- I. A. Pašti, N. M. Gavrilov and S. V. Mentus, Electrochim. Acta, 2014, 130, 453–463 CrossRef.
- S. Bhattacharjee, S. J. Yoo, U. V. Waghmare and S. C. Lee, Chem. Phys. Lett., 2016, 648, 166–169 CrossRef CAS.
- J. H. Lee, S. Kattel, Z. Jiang, Z. Xie, S. Yao, B. M. Tackett, W. Xu, N. S. Marinkovic and J. G. Chen, Nat. Commun., 2019, 10, 3724 CrossRef PubMed.
- H. Asonen, C. Barnes, A. Salokatve and A. Vuoristo, Appl. Surf. Sci., 1985, 22-23, 556–564 CrossRef CAS.
- A. Staykov, T. Kamachi, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2008, 112, 19501–19505 CrossRef CAS.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- D. Y. Shin, M.-S. Kim, J. A. Kwon, Y.-J. Shin, C. W. Yoon and D.-H. Lim, J. Phys. Chem. C, 2019, 123, 1539–1549 CrossRef CAS.
- C. Hu, S.-W. Ting, K.-Y. Chan and W. Huang, Int. J. Hydrogen Energy, 2012, 37, 15956–15965 CrossRef CAS.
- Y. Wang, Y. Qi, D. Zhang and C. Liu, J. Phys. Chem. C, 2014, 118, 2067–2076 CrossRef CAS.
- J. A. Herron, J. Scaranto, P. Ferrin, S. Li and M. Mavrikakis, ACS Catal., 2014, 4, 4434–4445 CrossRef CAS.
- J. S. Yoo, F. Abild-Pedersen, J. K. Nørskov and F. Studt, ACS Catal., 2014, 4, 1226–1233 CrossRef CAS.
- J. A. Kwon, M.-S. Kim, D. Y. Shin, J. Y. Kim and D.-H. Lim, J. Ind. Eng. Chem., 2017, 49, 69–75 CrossRef CAS.
- W. H. Hocking, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1976, 31, 1113–1121 Search PubMed.
- J. Cho, S. Lee, J. Han, S. P. Yoon, S. W. Nam, S. H. Choi, K.-Y. Lee and H. C. Ham, J. Phys. Chem. C, 2014, 118, 22553–22560 CrossRef CAS.
- J. Cho, S. Lee, S. P. Yoon, J. Han, S. W. Nam, K.-Y. Lee and H. C. Ham, ACS Catal., 2017, 7, 2553–2562 CrossRef CAS.
- J. H. Lee, J. Cho, M. Jeon, M. Ridwan, H. S. Park, S. H. Choi, S. W. Nam, J. Han, T.-H. Lim, H. C. Ham and C. W. Yoon, J. Mater. Chem. A, 2016, 4, 14141–14147 RSC.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- B. Hammer and J. K. Nørskov, Advances in Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00236h |
| ‡ First authors equally contributed to this work. |
| This journal is © the Owner Societies 2021 |