
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixing divalent ionic liquids: effects of charge and side-chains†
Eduards
Bakis
 a,
Adriaan
van den Bruinhorst
b,
Laure
Pison‡
b,
Ivan
Palazzo
a,
Thomas
Chang
a,
Marianne
Kjellberg
a,
Cameron C.
Weber
c,
Margarida
Costa Gomes
*b and
Tom
Welton
*a
a,
Adriaan
van den Bruinhorst
b,
Laure
Pison‡
b,
Ivan
Palazzo
a,
Thomas
Chang
a,
Marianne
Kjellberg
a,
Cameron C.
Weber
c,
Margarida
Costa Gomes
*b and
Tom
Welton
*a
aDepartment of Chemistry, Imperial College London, White City Campus, 80 Wood Lane, London W12 0BZ, UK. E-mail: t.welton@imperial.ac.uk
bLaboratoire de Chimie de l’ENS Lyon, CNRS and Université de Lyon, 46 allée d’Italie, 69364 Lyon, France. E-mail: margarida.costa-gomes@ens-lyon.fr
cSchool of Chemical Sciences, University of Auckland, 23 Symonds St, Auckland, New Zealand
First published on 17th February 2021
Abstract
We have prepared novel divalent ionic liquids (ILs) based on the bis(trifluoromethylsulfonyl)imide anion where two charged imidazolium groups in the cations are either directly bound to each other or linked by a single atom. We assessed the influence of the side-chain functionality and divalency on their physical properties and on the thermodynamics of mixing. The results indicate that shortening the spacer of a divalent IL reduces its thermal stability and increases its viscosity. Mixtures of divalent and monovalent ILs show small but significant deviations from ideality upon mixing. These deviations appear to depend primarily on the (mis)match of the nature and length of the cation side-chain. The non-ideality imposed by mixing ILs with different side-chains appears to be enhanced by the increase in formal charge of the cations in the mixture.
Introduction
Estimates place the number of possible ionic liquids (ILs) available at considerably over one million.1 Amongst such a vast number of alternatives there should exist an ideal candidate for every application. Rather than endeavouring to prepare a new IL for each application, we2–5 and others6–8 have explored the possibility of mixing ILs in order to control the physicochemical properties of these fluids and to determine whether unique systems can be accessed by simply combining already established ILs. Most salts mix as molecular compounds, but— depending on the nature of the salts—the properties of the resulting mixtures can be radically different from those of the pure constituents. For example, mixing salts can significantly lower the melting point or result in complex speciation. In this work, the ILs used to form the mixtures are well characterised and are expected to mix as molecular compounds. Understanding their mixing behaviour enables the properties of the resultant mixtures to be predicted.Interactions between the individual ions in the ILs are dominated by long-range Coulombic forces.9,10 However, the liquid phase shows more complex behaviour due to differences in ion size, shape, conformation, flexibility, or specific interactions, such as hydrogen bonding and π-stacking. Consequently, the liquid cohesive energies might depend as much upon van der Waals interactions as electrostatics.11 The molar volumes of IL mixtures typically follow nearly ideal behaviour, since the liquid phase of ILs generally has low compressibility and reduced free volume. The molar volumes of IL mixtures have been, so far, relatively easy to predict from the pure IL molar volumes. The excess volumes being often less than one percent of the magnitude of the property itself.12 However, the same is not true for the energetics of mixing.13
The first IL mixtures studied showed slightly endothermic or exothermic mixing enthalpies (less than 1 kJ mol−1), but more recent studies revealed stronger deviations from ideal mixing with reported excess enthalpies as low as −3 kJ mol−1.8 The prediction of the excess enthalpy is not straightforward, even when the IL mixtures are regular.14 The excess enthalpy macroscopically reflects the microscopic effects of adding a third or fourth ion to the liquid phase. Furthermore, precisely measured partial molar enthalpies can be extrapolated to infinite dilution conditions, hence providing unique experimental insight into the interactions between the two salts.
Mixing ILs also allows the control of their transport properties, for example by reducing the IL viscosity upon dilution with a second, less viscous, salt. Such mixtures have been used to improve the performance of several IL-based processes, such as CO2 capture15,16 in terms of the kinetics of gas absorption and absorbent regeneration.17 The use of IL mixtures has also led to breakthroughs in the capacitive performance of electrical double layer (EDL) capacitors18,19 outperforming systems consisting of simple ILs or molecular liquids. So far, relatively high melting points limit IL application at low temperatures while relatively low diffusion coefficients, implied by the high viscosity, reduce their conductivity. High temperature devices using surfactant ILs20 or IL mixtures have been shown to resolve both these issues,19 whilst taking advantage of the properties of these liquid salts which are inherently conductive, often possess broad electrochemical windows and have negligible vapour pressures. These aspects provide ILs with safety benefits over volatile, flammable organic electrolytes combined with markedly improved electrochemical windows as compared to aqueous solutions.
Given the importance of Coulomb forces in ILs, it might be speculated that increasing the charge density on one or more of the ions in a mixture of ILs could lead to greater effects than when all of the ions are singly charged.
Most of the known divalent ILs (DILs, where either the cation, anion or both are divalent) in the condensed phase suffer from high melting points due to their high lattice energies,21,22 or are too viscous for practical use. The majority of room temperature ILs that comprise a formal divalent ion contain two formal monovalent ions that are joined by an aliphatic or aromatic linker.22,23 Accordingly, the two charged sites of the cation are distant from one another and do not localise the charge density.
Shortening the size of the aliphatic linker has proven to have important advantages, as far as the properties and performance of the divalent salt are concerned. In liquid chromatography, sorbents functionalised with DIL cations exhibited enhanced performance over their monovalent counterparts.24 Decreasing the aliphatic linker led to an enhanced band separation,24 which might be related to the increased localisation of the charge density in the cation. Also, EDL capacitance is affected by the DIL cation linker size. Molecular dynamics of spatially isolated DILs, such as [(C1im)2C6][BF4]2, suggest these provide similar capacitance to the monovalent [C6C1im][BF4].25 However, shortening the linker length to C3 in [(C1im)2C3][BF4]2 brings the capacitance above that of the [(C1im)2C6]2+ analogue.25 Moreover, Matsumoto et al. recently demonstrated that using oligoether linkers to join together four imidazolium cations (with [NTf2]− as counterion) to form a tetravalent ion leads to “exceptionally high” EDL capacitance, exceeding that of [C4C1im][NTf2] by more than 6 times.26 The authors hypothesised that a relatively smaller entropy loss upon self-organization of the tetravalent cation around the electrode, as well as multivalent electrostatic interactions account for this remarkable boost in capacitive performance. Oligoether-functionalised ILs, depending on the anion,27 are usually less viscous than their alkylated counterparts.28,29 It is therefore of great interest whether the desirable electrochemical effects arise from the increased cation charge, conformational flexibility of cations introduced by ether linkages, or a combination thereof.
Another important physical property with respect to the use of ILs as electrolytes, materials, and solvents is their thermal stability. It has been argued that DILs are more thermally stable than monovalent ILs, with improvements in thermal stability ranging from 40 K to approximately 150 K.30–32 These findings have focused on DILs that contain two singly charged head groups linked by an aliphatic spacer of at least 3 carbons in length, i.e. headgroups that are largely independent of each other. Several groups have successfully utilized such DILs as media for high-temperature organic transformations.22 While the mechanism of the improved DIL thermal stability is not clear, it is important to note that such liquids are substantially more viscous than their monovalent counterparts, which may influence the rate of decomposition. When used in gas chromatography (GC) stationary phases, whether as individual ILs or mixtures thereof,33 IL thermal stability is of utmost importance and determines the upper column operation temperature boundary.34
The [NTf2]− anion is the archetypal anion for air- and water-stable ILs with low viscosities and excellent electrochemical properties, hence it has been chosen for this study. The lowest-melting [NTf2]− DIL with a single 2+ charge centre remains [(C4im)4Cu][NTf2]2, which melts at 46 °C, although lower melting DILs based on this anion where two singly charged heads are joined via a linker were reported before.35 Armstrong and co-workers have introduced a family of ILs with formally divalent cations (Fig. 1A), among which there are [NTf2]− based salts that are fluid at room temperature.32 The corresponding cations consist of two charged heads that are joined with (CH2)m (m ≥ 3)32 or oligoether linkers. Molecular dynamics studies by Yeganegi et al. show that anion coordination around the charged heads closely resembles that in classical monovalent imidazolium ILs.36 Xiao et al. reported37 ILs containing monovalent and divalent (Fig. 1B) cations formed from 2,2′-biimidazole with [NTf2]− and their application in Suzuki cross-coupling reactions. There have been no subsequent studies of this unusual IL-forming divalent cation.
 | ||
| Fig. 1 General structure of imidazolium-based dications. | ||
Herein, we aim to develop an understanding of the physicochemical properties of DILs, both with and without oligoether functionalisation, and their mixtures with both di- and monovalent ILs. To this end, several novel DILs containing divalent cations have been prepared (Table 1) where we have used the structural motifs discussed above but shortened the distance between the charged heads to a minimum in order to provide an increase in charge density, and/or modified the cation symmetry. These combined changes allowed these ILs to remain liquid at ambient temperature. Moreover, we have assessed the effect of both aliphatic and oligoether side chains on the DIL properties.
| No. | Structure | Chg | Abbreviation | m.p. |
|---|---|---|---|---|
| a Novel compounds. | ||||
| 1 |
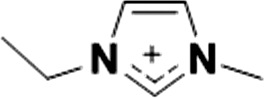
|
1+ | [C2C1im]1+ | <20 °C |
| 2 |
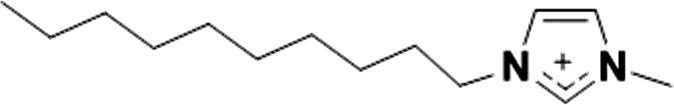
|
1+ | [C10C1im]1+ | <20 °C |
| 3 |
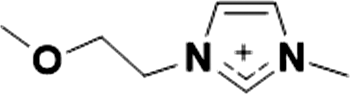
|
1+ | [(C3O)C1im]1+ | <20 °C |
| 4 |

|
1+ | [(C5O2)C1im]1+ | <20 °C |
| 5 |

|
1+ | [(C7O3)C1im]1+ | <20 °C |
| 6 |

|
1+ | [C10C1C1biim]1+ | <20 °Ca |
| 7 |

|
2+ | [C10C1C1C1biim]2+ | 60.0–63.0 °Ca |
| 8 |
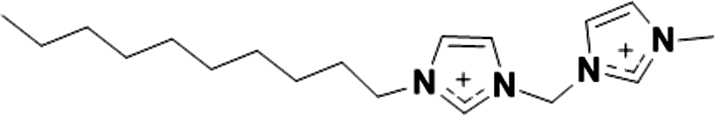
|
2+ | [C10imC1imC1]2+ | <20 °Ca |
| 9 |

|
2+ | [(C7O3)imC1imC1]2+ | <20 °Ca |
| 10 |
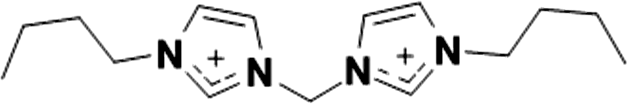
|
2+ | [(C4im)2C1]2+ | 58.6–59.3 °C |
| 11 |

|
2+ | [((C3O)im)2C1]2+ | <20 °Ca |
Mixing these ILs allows the tuning of their properties. For example, it is possible to improve their viscosity without significantly affecting their density or thermal stability. In order to understand how the properties of the mixtures depend upon those of the pure ILs and of their composition in the mixture, we conducted a thorough and original study of the thermodynamic properties of mixing. After characterisation of the properties of the pure ILs, both the excess molar volumes and the excess enthalpies were measured. These investigations represent the first calorimetric studies of mixtures of divalent salts with monovalent ILs and present a valuable insight into the properties of ILs with high charge densities. The ion dynamics and glass transition temperatures of mixtures containing divalent and a monovalent IL have been studied previously23 but only at a few compositions and including families of ILs that contained divalent salts having large spacers between the charged head-groups.
In the current manuscript we explore a conceptually novel family of DILs that comprise ions with high charge density in the divalent headgroup. Furthermore, we studied the effect of the functionalisation of the DILs and monovalent ILs upon mixing over wide composition ranges.
Experimental
The synthetic procedures for all of the ILs prepared are provided in the ESI.† Mixtures of ILs were prepared gravimetrically with the uncertainty on the mole fraction composition being estimated as ±0.0001. Hereinafter, the following nomenclature is used for the mixtures: [cation1]+(1−x)[cation2]+x[anion]− and [cation1]2+(1−x)[cation2]+x[anion]−(2−x), where x stands for the mole fraction composition. For the sake of simplicity and whenever it does not affect the understanding of the data, the mole fraction and/or the common anion will be omitted.Density measurements
Densities were measured at atmospheric pressure and in the temperature range of 293.15–353.15 K using a U-shaped vibrating-tube densimeter (model DMA 5000M; Anton Paar). The calibration of the equipment was verified with air and tridistilled water before and after each measurement. The estimated uncertainties of this equipment are 0.00001 g cm−3 and 0.01 K for density and temperature, respectively.Molar volume
Molar volumes of the pure ILs were determined experimentally from the density data and predicted using a group contribution method:38 | (1) |
The excess molar volumes of the IL mixtures were interpolated using a zero-order expansion of the Redlich–Kister polynomial:
| VE = (1 − xIL2)xIL2A0 | (2) |
Viscosity measurements
Viscosity measurements were performed at atmospheric pressure and in the temperature range 293.15–353.15 K using a falling-ball-based microviscosimeter Lovis 2000 M/ME from Anton Paar. The temperature was controlled to within 0.005 K and measured with an accuracy better than 0.02 K. A capillary tube of 1.8 mm diameter previously calibrated, as a function of temperature and angle of measurement, with reference oils was used for the measurements. The overall uncertainty on the viscosity was estimated to be 2%.Thermogravimetric analysis
Temperature-ramped Thermogravimetric Analysis (TGA) experiments were performed on a PerkinElmer ‘Pyris 1 TGA’ thermogravimetric analyser, using platinum sample pans of 6 mm diameter. Samples of 5–15 mg were heated at 10 °C min-1 from 70 to 600 °C under a nitrogen flow of 20 ml min−1. Each IL was dried isothermally at 70 °C for one hour prior to measurement.Isothermal titration calorimetry
An isothermal titration nanocalorimeter (TA Instruments) equipped with a thermal activity monitor—TAM III from TA Instruments—was used for measuring the excess molar enthalpies (HE, or mixing enthalpies, ΔmixH) at atmospheric pressure and at 313.15 K. The temperature of the thermostat was controlled to within 10−5 K.Two 1 mL Hastelloy cells were used. Both the measuring cell and the reference cell were initially filled with identical amounts of the same liquid. During the titration experiments, small volumes (between 3 and 10 μL) of a pure IL or a mixture of ILs at a known composition were injected (from a 250 μL or 100 μL gastight Hamilton syringe) into the measuring cell containing an accurately known quantity of approximately 0.8 mL of liquid. To ensure effective mixing, the liquid in the cell was stirred at 60 or 100 rpm. A motor-driven pump (thermometric 3810 syringe pump) was used for the automatic injections (around 5 per experiment), with each injection taking either 10 or 60 s. Between injections the system was allowed to equilibrate for 25–60 min to re-establish the baseline.
After each injection, a peak corresponding to the heat generated or absorbed by the mixing process is recorded. The area of the peak, Qi, is proportional to the heat involved and is used to calculate the excess molar enthalpy. The integration of the peaks recorded during the titration experiments was done using the TAM assistant software. The consistency of the results was checked by measuring the mixing enthalpies at several mole fraction concentrations along the whole composition range.
The partial molar excess enthalpy of IL2 added to IL1, can be calculated as described previously,39 through the measured heat effects (QIL2) when small quantities of IL2 (ΔnIL2) are added to larger quantities of IL1:
 | (3) |
The enthalpy of mixing can be represented as a function of mole fraction composition by a Redlich–Kister equation:
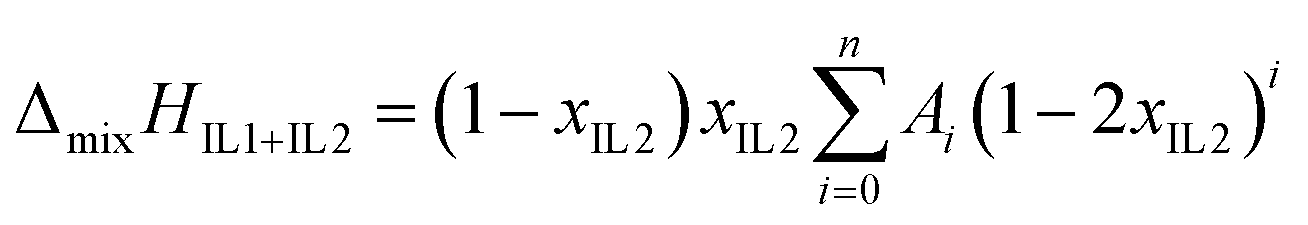 | (4) |
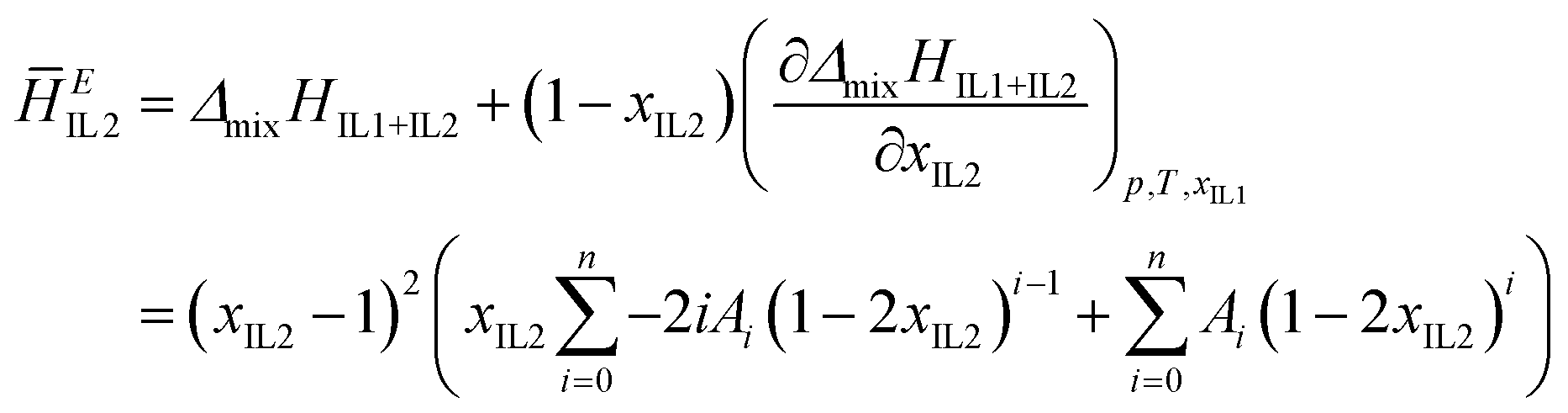 | (5) |
 | (6) |
 | (7) |
Results and discussion
Ionic liquid selection and synthesis
In order to assess the effect of the side-chain and the linker between imidazolium cationic groups, we prepared the compounds listed in Table 1 using the procedure outlined in Scheme 1. A graphical overview of these compounds and the properties that have been characterised is given in Fig. 2. Three key structural motifs were employed. The first is the presence of symmetric side-chains on each imidazolium headgroup for the divalent cation (No 10 and 11 Table 1, 5th row Fig. 2). | ||
| Scheme 1 Synthesis of geminal dicationic ILs. (1) NaOH, PhMe, 24 h reflux with Dean–Stark, drying at 100 °C; (2) R-Cl, MeCN, r.t./17 h reflux, vac. distillation; (3) CH2Br2, MeCN, 96 h reflux, recryst.; (4) LiNTf2 (aq), H2O/EtOAc extraction; (5) NaOH, Bu4NCl, DCM, 24 h reflux, sublimation; (6) MeI (0.55 eq.), THF, 2 h, 80 °C, re-precipitation) R′–X, MeCN, 1–8 days, 80–100 °C. | ||
 | ||
| Fig. 2 Overview of synthesised ionic liquids for which the physicochemical properties were evaluated. The ILs have an alkyl or oligoether sidechain R, where Cn and C2n+1On correspond to a –(CH2)n−1CH3 and –(CH2CH2O)nCH3 chain, respectively. Symbols correspond to those used in Fig. 3 and 5. For ionic liquid 10, only the decomposition temperature was studied. Numbers correspond to entry in Table 1. | ||
Syntheses of these ILs were straightforward and scalable, proceeding through the preparation of the dibromide salts (via the reaction of alkylimidazoles with dibromomethane) followed by an aqueous anion exchange (Scheme 1). In addition, a multi-gram alkylation protocol for the preparation of various alkylimidazoles, such as 1-(2-methoxyethyl)imidazole, from readily available precursors was developed and is discussed in greater detail in the ESI.† Both ether and aliphatic sidechains were chosen to have approximately the same sidechain length in terms of number of consecutive atoms to enable a direct comparison of the role of the ether functionality on the properties of the DIL.
The second motif was the formation of an asymmetric DIL featuring a methylimidazolium cation directly attached to an imidazolium cation with a larger sidechain (no 8 and 9 Table 1, 4th row Fig. 2). These were prepared using the mono-quaternisation protocol of bis-imidazolylmethane established by Weiss et al.40 We also prepared an [NTf2]− salt of a divalent cation without carbon linkers between the imidazolium rings (no 7 Table 1, 3rd row Fig. 2), leading to a DIL with the greatest charge density in the cationic ‘head-group’ amongst the ILs studied in this work. The monovalent analogue with only one of the imidazole rings quaternised was also prepared (No 6 Table 1, 2rd row Fig. 2), providing access to similar structures that differ only with the formal charge of the cation. Preparation of these 2,2′-biimidazolium cations was performed from 1,1′-dimethyl-2,2′-biimidazole, which was straightforward to mono-quaternise using bromodecane (Scheme 2). The second alkylation required the use of dimethylsulfate, perhaps because of the reduced nucleophilicity and steric hindrance around the remaining neutral imidazole moiety. For the synthesis of this series of cations we also developed a multi-gram protocol for 1,1′-dimethyl-2,2′-biimidazole (see ESI†), based on catalytic oxidative homocoupling of azoles and using readily available precursors.
 | ||
| Scheme 2 Synthesis of 2,2′-biimidazolium-based ILs. (1) CuCl, NaO2Py, air, xylenes, 8–15 h reflux, sublimation; (2) n-C10H21Br, 6 h, 120 °C, recryst.; (3) LiNTf2 (aq), H2O/DCM extraction; (4) Me2SO4, 21 h, 130 °C, (5) LiNTf2 (aq), H2O/EtOAc extraction. | ||
Outside of these three DIL motifs, ILs containing monovalent cations either with or without ether groups were also prepared (no 1–5 Table 1, 1st row Fig. 2). These were quaternised using the conventional two-step route using an alkyl halide substitution followed by an aqueous ion exchange step (see ESI†).
Physical properties of the pure ionic liquids
ILs typically do not show clear structure–property relationships for their melting points, owing to the many factors that govern the thermodynamic equilibrium between their liquid and solid phase.41,42 As a result, simply extending a side-chain by one extra carbon atom can have a significant effect on the melting temperature in a way that is difficult to predict. Nonetheless, the melting temperatures of the studied divalent ILs revealed some interesting trends.[(C1im)2C1][NTf2]2 ILs, those with a divalent cation containing a methylene linker and methyl substituents on each imidazolium ring (i.e. R1 = R2 = C1), are solids at room temperature with a melting point of 90–94 °C (Table S1, ESI†). Similarly to monovalent ILs, the melting point is reduced when either the linker or side chains are substituted for more flexible butyl chains although they remain above 50 °C (Table S1, ESI,† and no 10 Table 1). This could indicate that the melting temperature of these DILs is affected similarly whether it is the linker or the side-chain that is modified. The melting points of the DILs are reduced further if ether, rather than aliphatic, side chains are present, e.g. [(C3Oim)2C1][NTf2]2versus [(C4im)2C1][NTf2]2 (no 11 vs. 10 in Table 1). The ether chains increase the conformational flexibility of the side-chain to a greater extent than the presence of butyl chains. [(C3Oim)2C1][NTf2]2 was liquid at room temperature and remained fluid after rigorous drying and prolonged storage. To the best of our knowledge this is the lowest molecular weight aprotic room temperature DIL.
The other DILs and monovalent ILs explored were all liquid at 20 °C with the exception of [C10C1C1C1biim][NTf2]2 (no 7, Table 1) which has a melting point above 60 °C. Its singly charged counterpart, [C10C1C1biim][NTf2], is a liquid at room temperature. However, it can be seen that many factors influence the melting temperature of these ionic liquids, of which charge is only one. For example, [C10C1C1C1biim][NTf2]2 has a considerably higher melting point than [C10imC1imC1][NTf2]2, probably due to the greater rigidity and charge delocalisation of the biimidazolium moiety, whereas [(C4im)2C1][NTf2]2 has a similar melting point to [C10C1C1C1biim][NTf2]2, suggesting perhaps that the effect of the symmetry of the cation on the melting point is of a similar order as that of the presence of the methylene linker.
Fig. 3 shows that, as expected, the molar volumes of all pure ILs increase with temperature over the investigated temperature range. The data was derived from density measurements and interpolated using a second order polynomial, with regression parameters listed in Table S4 (ESI†). For the majority of studied ILs a first order (linear) fit yielded minor, but non-random residuals (Fig. S4, ESI†). The derivative of the interpolations is very similar for all ILs. Hence, the introduction of a divalent cation does not seem to have a pronounced effect on the thermal expansion within the studied temperature range.
 | ||
| Fig. 3 Molar volume (Vm) of the synthesised [NTf2]-based ILs as a function of temperature (T). Cation abbreviations and structures are listed in Table 1, symbols and corresponding structures are clarified in Fig. 2. Lines represent a second order polynomial interpolation of the data (Table S3, ESI†); fitting parameters are listed in Table S4 (ESI†). | ||
Previous studies have shown that the molar volume of an IL is additive with that of its ions.38,43 This allows for the description and prediction of the molar volume using group contribution methods. Moreover, it was found that the additivity held for sub-groups of ions for which the side-chain was extended or modified.38,43 Following this approach, the molar volumes of the ILs highlighted in Fig. 2 were predicted using eqn (1). The corresponding group coefficients and the number of groups per IL can be found in Tables S5 and S6 (ESI†), respectively. Fig. 4a and b show the resulting molar volumes and the relative difference between experiment and prediction. The presence of ether groups on the alkyl chain systematically increases the density (and decreases Vm) of the ILs, regardless of whether divalent or monovalent. This effect has been reported previously32,43 and has been attributed to the improved packing of ether chains due to their greater flexibility.
 | ||
| Fig. 4 (a) Molar volume (Vm) as function of molar mass (Mw) at 353.15 K calculated from the experimental density data (exp) and described/predicted using the group contribution method (GCM). Cation charge (divalent/monovalent) and the nature of the imidazolium side-chain (aliphatic/ether) are distinguished. (b) Relative difference between the experimental and calculated molar volume using the GCM (ΔexpGCMVm) at different temperatures. | ||
The additivity of the molar volume is valid for the monovalent ILs, with maximum relative deviations in the order of 1%, typical for these kinds of models.38 For the DILs, however, the molar volume is over-estimated when two methylimidazolium ([C0imC1]+) groups are used to describe the cationic charge centres. This is also the case when predicting molar volumes for a reported32 series with increasing linker size m (Fig. S5A and B, ESI†) with R1 = R1 = C1 and m = 3–12, following the nomenclature of Fig. 1A. The densities of this series were measured using a volumetric flask, and therefore reflect a significantly higher uncertainty than the data presented in this work. Despite these uncertainties, a clear negative deviation from the GCM-predicted molar volumes can be observed for these DILs as well. Hence, the increased charge density of the cations under study is not the sole cause for the break-down of the additivity.
This is emphasized when a new group, [(C0imC1)2]2+: a non-functionalised divalent cation consisting of two imidazolium rings linked by a single carbon atom, is introduced to correct for the over-estimation. The parameters for this group were obtained using the molar volumes of [C10imC1imC1][NTf2]2 (Table S3, ESI†). The resulting predictions were surprisingly accurate for the structurally different [C10C1C1C1biim][NTf2]2 and [((C3Oim)2C1][NTf2]2 (Fig. S5C and D, ESI†). However, [(C7O3im)C1imC1][NTf2]2 which is of similar size and symmetry as [C10imC1imC1][NTf2]2 showed significant systematic negative deviations. The new group performed well for DILs reported elsewhere that featured a larger linker chain, despite the increased distance between the imidazolium rings (Fig. S5, ESI†).
The differences between the predictions based on the original parametrisation and the actual molar volumes are in the order of a single CH2 group for most DILs (Fig. S5E and F, ESI†). This indicates that the differences are likely to originate from the side-chains rather than the imidazolium groups. One main difference between the monovalent alkylimidazolium cations used for the original parametrisation and the divalent cations studied here is the nature of the alkyl chain linker between the imidazolium rings. When the imidazolium cations are linked, the alkyl chain linkers are not capped by a CH3 group, unlike the terminal alkyl chains. Whether this is the main cause for the observed discrepancies needs to be confirmed by studying more ILs, but GCMs for non-ILs showed that the CH3 contribution is nearly 60% larger than that of CH2.44 Tail end-groups are therefore best considered as separate moieties. However, this would require the current parameters and groups reported in the open literature for the IL-GCM to be revisited, which is outside the scope of this work.
The viscosities of the simple ILs were measured over various temperature ranges, limited by the melting temperature of the IL. They were interpolated using the Vogel–Fülcher–Tamman–Hesse equation:45–47
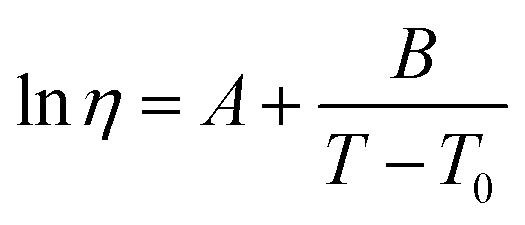 | (8) |
 | ||
| Fig. 5 Viscosity (η) of the synthesised [NTf2]-based ILs as a function of temperature (T). Cation abbreviations and structures are listed in Table 1, symbols and corresponding structures are clarified in Fig. 2. Lines represent an interpolation of the data using eqn (8); data and fitting parameters are listed in Tables S10 and S11 (ESI†), respectively. | ||
Ether functionalisation of the sidechain resulted in a decrease in viscosity for the monovalent ILs, as shown by comparing [(C7O3)C1im][NTf2] with [C10C1im][NTf2]. This effect has been reported previously.48 However, for the corresponding DILs, [C10imC1imC1][NTf2]2 and [(C7O3)imC1imC1][NTf2]2, the opposite effect is observed and the viscosity of the aliphatic-based DIL is lower than that of the ether functionalised DIL. Although many factors can influence the dynamics of ILs, the higher proportion of anions to cations in the DIL (2 to 1) seems to overcome the fluidifying effect of ether chains, common in monovalent ILs.48 The more concentrated charges increase Coulombic interactions that often strongly affect the viscosity. As a result, interactions between ether chains and imidazolium rings49 could be forced, thus additionally increasing the viscosity. However, it is difficult to ascertain the generality of these findings as only one ether/aliphatic DIL combination was examined.
The thermal stability of the prepared ILs was examined by thermogravimetric analysis (TGA) and characterised by Tonset, the lowest temperature where mass loss can be observed (see Fig. S2, ESI†). The major noticeable difference between the studied DILs and monovalent ILs that can be observed (Table 2 and Fig. S1, ESI†), is that the DILs are prone to degrade at lower temperatures. This is in stark contrast to reports claiming that DILs are inherently more thermally stable than similar monovalent ILs.30,31 This can clearly be seen from the Tonset values (Table 2) decreasing in the order [C10C1im]+ > [C10imC1imC1]2+ > [C10C1C1C1biim]2+. From these Tonset values, it appears these DILs decompose at temperatures approximately 40–60 °C lower than their related monovalent counterparts (Table 2). These Tonset values remain appreciable for organic compounds.
| IL | Cation charge | T onset /°C |
|---|---|---|
| a Determined from step tangent analysis with a heating rate of 10 °C min−1. | ||
| [C2C1im][NTf2] | 1+ | 450 |
| [C10C1im][NTf2] | 1+ | 441 |
| [(C3O)C1im][NTf2] | 1+ | 435 |
| [(C5O2)C1im][NTf2] | 1+ | 383 |
| [(C7O3)C1im][NTf2] | 1+ | 369 |
| [C10C1C1biim][NTf2] | 1+ | 368 |
| [C10C1C1C1biim][NTf2]2 | 2+ | 329 |
| [C10imC1imC1][NTf2]2 | 2+ | 400 |
| [(C7O3)imC1imC1][NTf2]2 | 2+ | 341 |
| [(C4im)2C1][NTf2]2 | 2+ | 411 |
| [((C3O)im)2C1][NTf2]2 | 2+ | 387 |
The rationale for the reduced Tonset of these DILs as compared to the more thermally stable DILs lies in their structure. Geminal divalent cations, i.e. those with m = 1 (Fig. 1A) and those with directly linked charged centres, are likely to display an increase in electrophilicity and/or α-hydrogen acidity, due to the decreased separation of the charged centres. These factors have been found to play a role in IL degradation pathways, and would account for the observed differences in thermal stability.30 The more stable DILs have m > 3, and hence the charge centres were separated by a much greater distance, reducing the electrophilicity of the cation. The observed increase in thermal stability of these might also arise from their substantially increased viscosity, through an effect on the onset temperatures. The results obtained here indicate that any viscosity effects are offset by the increased electrophilicity of cations that contain charge centres in close proximity.
As might be expected, ILs with heteroatom-rich functional groups degrade more easily than their unfunctionalised counterparts. This is the case for monovalent and divalent ether-functionalised ILs where the Tonset observed for [(C7O3)C1im][NTf2] and [(C7O3)imC1imC1][NTf2]2 were lower than for [C10C1im][NTf2] and [C10imC1imC1][NTf2]2, respectively. At high temperatures these functional groups could behave as nucleophiles or bases, which accelerate the rate of IL decomposition. Patil et al. proposed that these C–O bonds break after an intramolecular oxygen attack on β-carbon atoms in DILs with ether spacers.50 This transformation should be even more sterically accessible within mono-substituted polyethylene glycol chains. The monovalent [(C7O3)C1im][NTf2] shows a multi-step decomposition pattern (Fig. S1A, ESI†). Indeed, repeated degradation experiments for this IL at a lower heating rate show the first distinct mass loss to be approximately 13%, comparable with the expected 15% for a loss of ethylene glycol monomethyl ether (Fig. S3 in ESI†). This is in good agreement with the fact that non-functionalised ILs such as [C2C1im][NTf2] have significantly higher Tonset, as the [NTf2]− anion itself is remarkably stable and non-nucleophilic.
Physical properties of ionic liquid mixtures
Mixing the original ILs prepared herein allows the tuning of their physical properties without losing their structural features. Moreover, it will give additional insight into the relative importance of the divalency of the cation and the side-chain functionality. In order to understand how these ILs mix, we explored their thermodynamic properties of mixing. Firstly, the excess molar volume (VE) was calculated from the measured densities of the simple ILs and their mixtures, elucidating the free volume changes when the salts are mixed. Secondly, the enthalpy of mixing, or molar excess enthalpy (HE), was measured, providing direct experimental information about the energetic changes linked to the formation of these mixtures.The experimental partial molar enthalpies of mixing extrapolated towards infinite dilution (HE,∞i) can be regarded as a direct measurement of the interactions between the two components of the mixture.13 These experimental data are essential to better understand the resulting properties from mixing these complex salts. Liquid phase data alone, however, do not allow the calculation of the excess Gibbs energy (GE). The description of GE would permit a full comprehension of how these salts mix. Usually, vapour–liquid equilibrium measurements are used to access GE, which is practically unfeasible at temperatures close to ambient for mixtures of ILs, due their remarkably low vapour pressures.
As shown in Fig. 6 and 7, the studied IL mixtures have VE values close to zero, although they show significant trends. The magnitude of VE is considerably lower than the molar volume of the pure salts (Fig. 3 and Table S7, ESI†), which is in line with other IL mixtures studied in the literature.51
 | ||
| Fig. 6 Excess molar volume (VE, left) and excess molar enthalpy of mixing (HE, right) as a function of composition (x2, mole fraction of component 2) for the studied mixtures of ionic liquids (ILs) at 313.15 K. A visual overview of these mixtures—including symbols and line styles—is given in Fig. 7. Solid symbols and solid/dash-dotted lines correspond to mixtures containing a divalent and monovalent cation. Open symbols and dashed lines correspond to mixtures containing monovalent cations only. The red horizontal line at 0 represents ideal mixing behaviour. The symbols in the right panel were added for readability purposes and indicate the compositions at which the calorimetric injections were made. They do not correspond to raw data, as HE can only be obtained indirectly using isothermal titration calorimetry. See ESI,† Tables S7 and S8 for raw data and fitting parameters. | ||
 | ||
| Fig. 7 Partial molar excess enthalpy at infinite dilution (HE,∞i), the excess enthalpy of mixing at equimolar composition (HE0.5), and the excess molar volume at equimolar composition (VE0.5) of the mixture for the studied binary mixtures of ILs with common [NTf2]-anions. The symbols, colours, and line styles correspond to Fig. 6, a table-like representation is available in the ESI† (Table S9). | ||
The studied ILs exhibited some general trends upon mixing. When ILs with the same side-chain functionalisation (ether–ether and aliphatic–aliphatic) were mixed, a nearly ideal mixture in terms of the molar volume or a mixture with negative deviations from the ideal molar volume was formed (VE ≤ 0). This corresponds to a mixture molar volume proportional to that of the pure components’ or to a contraction of the molar volume upon mixing, respectively. On the other hand, IL mixtures with different functionalities (ether-aliphatic and vice-versa) showed quasi-ideal behaviour or positive deviations from ideality in terms of the molar volume, corresponding to an expansion upon mixing (VE ≥ 0). The most extreme VE values in each direction (−0.19 cm3 mol−1, and +1.15 cm3 mol−1) were found for the mixtures where a DIL with an aliphatic side-chain ([C10imC1imC1]2+) was mixed with a monovalent IL with the same aliphatic side-chain ([C10C1im]+); and where an ether functionalised DIL ([(C3Oim)2C1]2+) was mixed with a monovalent IL with an aliphatic side-chain ([C10C1im]+), respectively. These represent relatively small VE values, nonetheless several trends can be observed when comparing mixtures with a common IL.
The impact of side-chain functionality on IL mixing is highlighted by mixtures where [(C3Oim)2C1]2+ is combined with a series of monovalent ILs (6th row Fig. 7, circles in Fig. 6). When this DIL which is symmetrically functionalised with short ether side-chains is mixed with a monovalent cation with the same side-chain, the mixture behaves as quasi-ideal. Both VE and HE are close to zero. This shows that the presence of a divalent cation in a binary IL mixture does not necessarily lead to deviations from ideal mixing.
When the length of the ether side-chain of the monovalent IL2 is increased (R = C3O → C5O2 → C7O3), both VE and HE become increasingly negative. Hence, an increasing mismatch in the ether chain-length leads to a (slightly) more negative VE and HE, corresponding to a volume contraction and energetically more favourable mixing. For IL mixtures with a common anion negative HE is exceptional,13 as is a negative HE for side-chains with similar functionality. The few other mixtures that are known to have a negative HE are 1-butyl-2,3-dimethylimidazolium tetrafluoroborate with 1-ethyl-3-methylimidazolium tetrafluoroborate as well as acetate based ILs mixed with phosphonium and imidazolium cations.8
When the longest ether side-chain is substituted by an aliphatic side-chain of equivalent length ([(C7O3)C1im]+ → [C10C1im]+), the mixture with [(C3Oim)2C1]2+ shows positive VE and HE. This is in line with the behaviour of molecular binary n-alkane–ether mixtures that always show thermodynamically unfavourable mixing energies and volume expansion upon mixing.52
Five binary IL mixtures contained sets of cations with differently functionalised side-chains. Their excess properties were compared to those of molecular n-alkane–polyether mixtures at equimolar composition (Fig. 8).52 IL mixtures comprised of monovalent cations with different short side-chains mixed almost ideally (+0.04 kJ mol−1 and +0.05 cm3 mol−1 for HE and VE, respectively) and were therefore not included in Fig. 8. The order of magnitude of the IL mixture excess properties is similar to that of n-alkane–ethyl ether mixtures, although it seems that the ionic nature of the ILs leads to slightly suppressed excess properties as compared to those of the molecular mixtures. It is therefore notable that IL mixtures containing divalent ether-functionalised cations yielded larger volume expansions (VE) and energetically less favourable mixing (HE) than mixtures with monovalent cations, even when their side-chain was equivalent.
 | ||
| Fig. 8 The excess enthalpy of mixing at equimolar composition (HE0.5, top), and the excess molar volume at equimolar composition (VE0.5, bottom) for n-alkane–polyether mixtures52 (half-filled symbols) and studied IL mixtures (filled and open symbols). nC corresponds to the number of carbon atoms in the n-alkane or the alkyl side-chain of IL2 [C10C1im][NTf2], respectively. Structural formulas of the ethers and IL cations are given for clarity, lines are a guide to the eye. | ||
Divalent cations with high charge density, such as the ones studied herein, show enhanced excess properties when compared with those of mixtures of monovalent ions with similar side-chains. For example, VE becomes more negative and HE more positive when increasing the cation charge of IL1 for IL mixtures where both cations have aliphatic side-chains ([C10C1C1biim]+–[C10C1im]+vs. [C10imC1imC1]2+–[C10C1im]+).
The quasi-ideality of the [C10C1C1biim]+–[C10C1im]+ mixture shows that this is not an effect of the cationic head group size. Mixing the divalent [C10imC1imC1]2+ with its monovalent equivalent [C10C1im]+ yields small, but significant, excess properties (Fig. 7). For mixtures containing divalent ILs, the excess properties may be due to the increased local association of anions with 2 [NTf2]− anions required per divalent cation compared to one for the monovalent species. The interactions between this additional anion and the side-chain may be responsible for the enhanced excess properties, rather than (or in addition to) an increased charge density. To determine which mechanism is responsible for the observed effects, additional IL mixtures could be explored. Possible systems of interest are mixtures where both ILs are divalent, or mixtures containing symmetrically functionalised monovalent ILs such as [(C3O)(C3O)im][NTf2] as well as ILs with longer linker lengths to distinguish the effect of charge density from formal charge.
From Fig. 7 and 8 it can be observed that a mismatch in side-chain length has a stronger effect on the excess property magnitude than a mismatch in side-chain functionality. The short-chained mixture [(C3O)C1im]+–[C2C1im]+ behaves nearly ideally, while the short–long system [(C3O)C1im]+–[C10C1im]+ shows significant (positive) deviations from ideality. This trend is corroborated by the mixture [(C7O3)C1im]+–[C10C1im]+, which shows smaller deviations for a longer but comparable length ether side-chain. The same trend has been found for the VE of molecular n-alkane–ethyl ether mixtures52 (Fig. 8). However, the trend for the HE of IL mixtures is opposite to that of the molecular mixtures, showing that the ionic nature of ILs does affect the resulting mixing behaviour. It also confirms that VE and HE are decoupled. This is better illustrated by the [C10imC1imC1]+–[C10C1im]+ mixture, which shows opposite signs for VE and HE.
In addition to the HE at equimolar composition, partial excess enthalpies at infinite dilution (HE,∞i) are reported in Fig. 7. For three of the studied mixtures there is a significant difference between the HE,∞i of each IL, meaning that the energetics of mixing are different on each side of the equimolar composition. In particular, the addition of a limiting amount of [C10C1im]+ to the other IL, namely those based on [((C3O)im)2C1]2+, [(C7O3)imC1imC1]2+, or [(C3O)C1im]+ cations and to a lesser extent [(C7O3)C1im]+, is less favourable enthalpically than if a limiting addition is done the other way around. This behaviour is a sign of large perturbations of the structure and molecular interactions of the ether functionalised ILs by [C10C1im]+.
Finally, it should be noted that all values in Fig. 6 were interpolated using a zero-order Redlich–Kister function, yielding symmetrical curves. It is clear, however, that the excess volume of the [(C3Oim)2C1]2+–[C10C1im]+ mixture is asymmetric with composition. Hence, a small quantity of the monovalent cation disrupts the packing of the DIL, leading to a stronger expansion than in the reverse case. Higher-order Redlich–Kister expansions would be able to accurately describe this phenomenon. We specifically chose not to do so, because most systems were not evaluated at a sufficient number of different compositions (owing to limited sample quantities) to allow such accurate interpolation. Including higher-order descriptions only for mixtures for which three or more compositions were studied could lead to the false conclusion that the mixtures with fewer data exhibit a symmetrical behaviour of VE around equimolar composition.
Conclusions
We have prepared several novel DILs based on imidazolium cations and on the [NTf2]− anion, namely [((C3O)im)2C1][NTf2]2, [(C7O3)imC1imC1][NTf2]2, [C10C1C1C1biim][NTf]2 and [C10imC1imC1][NTf2]2. The cations of these DILs possess formal charges in closer proximity than similar DILs that have been previously reported. Many changes in the physical properties of these DILs, as compared to their monovalent counterparts, were consistent with those that have been observed for other DILs, such as a larger molar volumes and higher viscosities. However, the more localised positive charge of these DILs led to reduced thermal stability as compared to their monovalent equivalents, rather than the increase in thermal stability that has been observed for other classes of DILs. This reduction in thermal stability was attributed to increased electrophilicity when both formally charged groups are directly bonded or separated only by a single atom linker.The effect of oligoether side-chains was compared with aliphatic side-chains in the cations. Oligoether containing ILs were found to be generally denser and less thermally stable compared to their aliphatic counterparts. The viscosity of monovalent ILs with oligoether side-chains was lower than the corresponding ILs with aliphatic side-chains. The converse was true for DILs and the viscosity was found to increase for DILs with oligoether sidechains. This suggests that for DILs, terminal aliphatic chains rather than oligoether chains may lead to relatively lower viscosities.
Upon mixing the studied ILs, the sign of the excess properties is determined by the functionality of the side-chains. Both VE and HE are negative for mixtures of ILs that both have an ether-functionalised side-chain, and positive for mixtures of ILs where one contains ether side-chains and the other alkyl side-chains. The extent to which these mixtures showed deviations from ideality is governed by the mismatch in side-chain length, with DILs significantly enhancing this effect. The exact mechanism behind the differences observed between divalent–monovalent mixtures and solely monovalent-based mixtures is yet unknown and probably difficult to explain using simple descriptors. It should be kept in mind, however, that both the molar volume and the excess enthalpy of mixing are relatively low. This is especially true for the excess volume, which is typically two to four orders of magnitude lower than the molar volume of the mixture.
Collectively, these results suggest some potential design principles when developing DILs or their mixtures for various applications. When thermal stability is desired, it appears that the length of the spacer between cations should be greater than one carbon atom, despite the corresponding reduction in cation charge density. For DILs, the use of terminal aliphatic chains rather than oligoether chains appears to be a more successful route to reducing their viscosity. Since mixing DILs with less viscous monovalent ILs with the same side-chains yield virtually ideal mixtures, this could be an interesting route to predictably lowering the viscosity of these ILs similarly to asymmetric DILs with linkers longer than a single atom.23 Alternatively, combining mismatched sidechains of different functionality appears to be a route to generating slightly positive excess molar volumes, which often indicate an increase in free volume within the mixture. This effect is nevertheless difficult to predict using group contribution methods as they are not yet accurate enough for DILs. The origin of this inaccuracy remains to be determined, but can probably be solved by a re-parametrisation that explicitly accounts for the difference between CH2 and CH3 in the side-chain or for the divalent nature of studied cations.
This work shows that even when mixing ILs with very different structures, including significant variations in the size of polar domains, they behave as (almost) ideal mixtures from a thermodynamic standpoint. This is not always the case even for less complex ionic liquid mixtures which can show large deviations for ideality.8,39 Since we could quantify the small deviations from ideality very accurately, trends can still be identified depending on the functionalisation of the salts. These trends can then guide the design of new ILs and their mixtures for specific applications where DILs with increased charge density have been found beneficial.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
EB acknowledges UK Engineering and Physical Sciences Research Council for funding of doctoral studies, MCG and LP thank the Institute of Chemistry at Clermont-Ferrand for the use of the densimeter and the viscometer. MCG and AvdB acknowledge the financial support of the project IDEXLYON of the University of Lyon (ANR-16-IDEX-0005). TW acknowledges an ERC Advanced Investigator Grant for funding.Notes and references
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- M. T. Clough, C. R. Crick, J. Gräsvik, P. A. Hunt, H. Niedermeyer, T. Welton and O. P. Whitaker, Chem. Sci., 2015, 6, 1101–1114 RSC.
- R. P. Matthews, I. J. Villar-Garcia, C. C. Weber, J. Griffith, F. Cameron, J. P. Hallett, P. A. Hunt and T. Welton, Phys. Chem. Chem. Phys., 2016, 18, 8608–8624 RSC.
- N. J. Brooks, F. Castiglione, C. M. Doherty, A. Dolan, A. J. Hill, P. A. Hunt, R. P. Matthews, M. Mauri, A. Mele, R. Simonutti, I. J. Villar-Garcia, C. C. Weber and T. Welton, Chem. Sci., 2017, 8, 6359–6374 RSC.
- C. C. Weber, N. J. Brooks, F. Castiglione, M. Mauri, R. Simonutti, A. Mele and T. Welton, Phys. Chem. Chem. Phys., 2019, 21, 5999–6010 RSC.
- J. N. Canongia Lopes, T. C. Cordeiro, J. M. S. S. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, J. Phys. Chem. B, 2005, 109, 3519–3525 CrossRef.
- H. F. D. Almeida, J. N. C. Lopes, L. P. N. Rebelo, J. A. P. Coutinho, M. G. Freire and I. M. Marrucho, J. Chem. Eng. Data, 2016, 61, 2828–2843 CrossRef CAS.
- I. Otero, L. F. Lepre, A. Dequidt, P. Husson and M. Costa Gomes, J. Phys. Chem. B, 2017, 121, 9725–9736 CrossRef CAS.
- R. M. Lynden-Bell, Phys. Chem. Chem. Phys., 2010, 12, 1733–1740 RSC.
- K. Goloviznina, J. N. Canongia Lopes, M. Costa Gomes and A. A. H. Pádua, J. Chem. Theory Comput., 2019, 15, 5858–5871 CrossRef CAS.
- K. Shimizu, M. Tariq, M. C. Gomes, L. P. N. Rebelo and J. N. C. Lopes, J. Phys. Chem. B, 2010, 114, 5831–5834 CrossRef CAS.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- A. Podgoršek, J. Jacquemin, A. A. H. Pádua and M. Costa Gomes, Chem. Rev., 2016, 116, 6075–6106 CrossRef.
- L. F. Lepre, M. Costa Gomes, A. A. H. Pádua, R. A. Ando and M. C. C. Ribeiro, J. Phys. Chem. B, 2019, 123, 6579–6587 CrossRef CAS.
- M. Wang, L. Zhang, L. Gao, K. Pi, J. Zhang and C. Zheng, Energy Fuels, 2013, 27, 461–466 CrossRef CAS.
- A. M. Pinto, H. Rodríguez, Y. J. Colón, A. Arce Jr., A. Arce and A. Soto, Ind. Eng. Chem. Res., 2013, 52, 5975–5984 CrossRef CAS.
- L. F. Lepre, J. Szala-Bilnik, L. Pison, M. Traïkia, A. A. H. Padua, R. A. Ando and M. F. Costa Gomes, Phys. Chem. Chem. Phys., 2017, 19, 12431–12440 RSC.
- W. Y. Tsai, R. Lin, S. Murali, L. Li Zhang, J. K. McDonough, R. S. Ruoff, P. L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS.
- R. Lin, P.-L. Taberna, S. Fantini, V. Presser, C. R. Pérez, F. Malbosc, N. L. Rupesinghe, K. B. K. Teo, Y. Gogotsi and P. Simon, J. Phys. Chem. Lett., 2011, 2, 2396–2401 CrossRef CAS.
- X. Mao, P. Brown, C. Červinka, G. Hazell, H. Li, Y. Ren, D. Chen, R. Atkin, J. Eastoe, I. Grillo, A. A. H. Padua, M. F. Costa Gomes and T. A. Hatton, Nat. Mater., 2019, 18, 1350–1357 CrossRef CAS.
- N. Winterton, in Ionic Liquids Completely UnCOILed: Critical Expert Overviews, ed. N. V. Plechkova and K. R. Seddon, John Wiley & Sons, Inc., Hoboken, New Jersey, 2015, pp. 231–534 Search PubMed.
- F. D’Anna and R. Noto, Eur. J. Org. Chem., 2014, 4201–4223 CrossRef.
- S. I. Lall-Ramnarine, S. N. Suarez, E. D. Fernandez, C. Rodriguez, S. Wei, M. Gobet, J. R. P. Jayakody, S. B. Dhiman and J. F. Wishart, J. Electrochem. Soc., 2017, 164, H5150–H5159 CrossRef CAS.
- L. Qiao, H. Li, Y. Shan, S. Wang, X. Shi, X. Lu and G. Xu, J. Chromatogr. A, 2014, 1330, 40–50 CrossRef CAS.
- S. Li, K. L. Van Aken, J. K. McDonough, G. Feng, Y. Gogotsi and P. T. Cummings, J. Phys. Chem. C, 2014, 118, 3901–3909 CrossRef CAS.
- M. Matsumoto, S. Shimizu, R. Sotoike, M. Watanabe, Y. Iwasa, Y. Itoh and T. Aida, J. Am. Chem. Soc., 2017, 139, 16072–16075 CrossRef CAS.
- L. C. Branco, J. N. Rosa, J. J. Moura Ramos and C. A. M. Afonso, Chemistry, 2002, 8, 3671–3677 CrossRef CAS.
- E. Kuhlmann, S. Himmler, H. Giebelhaus and P. Wasserscheid, Green Chem., 2007, 9, 233 RSC.
- H. Shirota, H. Fukazawa, T. Fujisawa and J. F. Wishart, J. Phys. Chem. B, 2010, 114, 9400–9412 CrossRef CAS.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- H. Shirota, T. Mandai, H. Fukazawa and T. Kato, J. Chem. Eng. Data, 2011, 56, 2453–2459 CrossRef CAS.
- J. L. Anderson, R. Ding, A. Ellern and D. W. Armstrong, J. Am. Chem. Soc., 2005, 127, 593–604 CrossRef CAS.
- C. F. Poole and S. K. Poole, J. Sep. Sci., 2011, 34, 888–900 CrossRef CAS.
- C. F. Poole and N. Lenca, J. Chromatogr. A, 2014, 1357, 87–109 CrossRef CAS.
- T. V. Hoogerstraete, N. R. Brooks, B. Norberg, J. Wouters, K. V. Hecke, L. V. Meervelt and K. Binnemans, CrystEngComm, 2012, 14, 4902–4911 RSC.
- S. Yeganegi, A. Soltanabadi and D. Farmanzadeh, J. Phys. Chem. B, 2012, 116, 11517–11526 CrossRef CAS.
- J.-C. Xiao and J. M. Shreeve, J. Org. Chem., 2005, 70, 3072–3078 CrossRef CAS.
- J. Jacquemin, R. Ge, P. Nancarrow, D. W. Rooney, M. F. Costa Gomes, A. A. H. Pádua and C. Hardacre, J. Chem. Eng. Data, 2008, 53, 716–726 CrossRef CAS.
- L. F. Lepre, J. Szala-Bilnik, L. Pison, M. Traïkia, A. A. H. Pádua, R. A. Ando and M. F. Costa Gomes, Phys. Chem. Chem. Phys., 2017, 19, 12431–12440 RSC.
- D. T. Weiss, S. Haslinger, C. Jandl, A. Pöthig, M. Cokoja and F. E. Kühn, Inorg. Chem., 2015, 54, 415–417 CrossRef CAS.
- M. Blesic, M. Swadźba-Kwaśny, T. Belhocine, H. Q. N. Gunaratne, J. N. C. Lopes, M. F. C. Gomes, A. A. H. Pádua, K. R. Seddon and L. P. N. Rebelo, Phys. Chem. Chem. Phys., 2009, 11, 8939–8948 RSC.
- Y. Zhang and E. J. Maginn, J. Chem. Theory Comput., 2013, 9, 1592–1599 CrossRef CAS.
- Y. Deng, S. Morrissey, N. Gathergood, A.-M. Delort, P. Husson and M. F. Costa Gomes, ChemSusChem, 2010, 3, 377–385 CrossRef CAS.
- A. V. Plyasunov, N. V. Plyasunova and E. L. Shock, J. Chem. Eng. Data, 2004, 49, 1152–1167 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–647 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- S. I. Lall-Ramnarine, M. Zhao, C. Rodriguez, R. Fernandez, N. Zmich, E. D. Fernandez, S. B. Dhiman, E. W. Castner and J. F. Wishart, J. Electrochem. Soc., 2017, 164, H5247–H5262 CrossRef CAS.
- K. Shimizu, C. E. S. Bernardes, A. Triolo and J. N. Canongia Lopes, Phys. Chem. Chem. Phys., 2013, 15, 16256–16262 RSC.
- R. A. Patil, M. Talebi, A. Berthod and D. W. Armstrong, Anal. Bioanal. Chem., 2018, 410, 4645–4655 CrossRef CAS.
- M. T. Clough, C. R. Crick, J. Gräsvik, P. A. Hunt, H. Niedermeyer, T. Welton and O. P. Whitaker, Chem. Sci., 2015, 6, 1101–1114 RSC.
- R. Burgdorf, A. Zocholl, W. Arlt and H. Knapp, Fluid Phase Equilib., 1999, 164, 225–255 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp00208b |
| ‡ Present address: Laboratoire Magmas et Volcans, CNRS and Clermont Auvergne University, 6 Avenue Blaise Pascal, TSA 60026 – CS 60026, 63178 Aubière, France. |
| This journal is © the Owner Societies 2021 |