
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein–ligand free energies of binding from full-protein DFT calculations: convergence and choice of exchange–correlation functional†
Lennart
Gundelach
 a,
Thomas
Fox
b,
Christofer S.
Tautermann
b and
Chris-Kriton
Skylaris
*a
a,
Thomas
Fox
b,
Christofer S.
Tautermann
b and
Chris-Kriton
Skylaris
*a
aUniversity of Southampton Faculty of Engineering Science and Mathematics, Chemistry, University Road, Southampton, UK SO17 1BJ, UK. E-mail: c.skylaris@soton.ac.uk; Tel: +44 (023) 8059 9381
bBoehringer Ingelheim Pharma GmbH & Co KG, Medicinal Chemistry, Birkendorfer Str 65, 88397 Biberach an der Riss, Germany
First published on 30th March 2021
Abstract
The accurate prediction of protein–ligand binding free energies with tractable computational methods has the potential to revolutionize drug discovery. Modeling the protein–ligand interaction at a quantum mechanical level, instead of relying on empirical classical-mechanics methods, is an important step toward this goal. In this study, we explore the QM-PBSA method to calculate the free energies of binding of seven ligands to the T4-lysozyme L99A/M102Q mutant using linear-scaling density functional theory on the whole protein–ligand complex. By leveraging modern high-performance computing we perform over 2900 full-protein (2600 atoms) DFT calculations providing new insights into the convergence, precision and reproducibility of the QM-PBSA method. We find that even at moderate sampling over 50 snapshots, the convergence of QM-PBSA is similar to traditional MM-PBSA and that the DFT-based energy evaluations are very reproducible. We show that in the QM-PBSA framework, the physically-motivated GGA exchange–correlation functional PBE outperforms the more modern, dispersion-including non-local and meta-GGA-nonlocal functionals VV10 and B97M-rV. Different empirical dispersion corrections perform similarly well but the three-body dispersion term, as included in Grimme's D3 dispersion, is significant and improves results slightly. Inclusion of an entropy correction term sampled over less than 25 snapshots is detrimental while an entropy correction sampled over the same 50 or 100 snapshots as the enthalpies improves the accuracy of the QM-PBSA method. As full-protein DFT calculations can now be performed on modest computational resources our study demonstrates that they can be a useful addition to the toolbox of free energy calculations.
1 Introduction
Due to improved methodologies and greater access to computing resources, computational approaches are becoming increasingly valuable in drug discovery and development,1–4 and the ongoing COVID-19 pandemic illustrates how simulations on supercomputers can rapidly lead to valuable insights into a novel disease.5 The ability of a pharmaceutical drug to bind to and interact with a target protein is of central importance and thus, the accurate prediction of protein–ligand binding free energies is one of the grand challenges of computational chemistry. In modeling protein–ligand interactions, the two key challenges are the size and complexity of protein–ligand systems. As a result, protein–ligand binding free energy predictions have traditionally relied on low-cost approximate methods due to limited computing power. Classical mechanics docking and molecular-dynamics based approaches still dominate this field of research. While much progress has been made in addressing challenges like sampling a flexible protein or accurately describing the solvent, the fundamental limitation of force-field based approaches is their inability to explicitly describe important physical effects that influence protein–ligand binding. To account for electron polarization, charge transfer, halogen bonding and many-body effects a quantum mechanical description is essential.6,7 Force-fields that attempt to incorporate these effects are under development like the polarizable AMOEBA force-field for proteins8 as well as charge-transfer including force-fields.9–15 However, with increasing access to high-performance computing the use of accurate quantum mechanical methods, which innately describe all of the important physical interactions, is becoming viable.Most proteins consist of many thousands of atoms and thus cheap semi-empirical quantum mechanical (SEQM) methods like AM1 or SCC-DFTB are commonly applied. Despite using these cheaper, more approximate methods, often only the ligand16–19 or the ligand and surrounding protein-residues20–23 are treated at a QM level. Merz23 and Anisimov24,25 used linear-scaling SEQM methods on the whole protein. Ryde et al.26 used the SEQM methods AM1, RM1 and PM6 to calculated binding energies in three protein–ligand systems using a SEQM-GBSA approach. Most studies using more expensive ab initio density functional theory (DFT) only treat the ligand and surrounding protein sites at this level of theory.27–32 Fragmentation based approaches like PMISP33,34 or the fragment molecular orbital (FMO)35 method using QM calculations have been applied to various protein–ligand systems.36–39 These studies feature either no sampling or very low sampling from molecular-dynamics. Approaches based on sampling at a hybrid QM/MM level have also been developed.40
Even on supercomputers, it is prohibitive to perform conventional DFT calculations on entire proteins with thousands of atoms as the computational effort of DFT scales with the third power in the number of atoms. However, such calculations are enabled by the use of linear-scaling DFT approaches.41 In 2010, Cole et al.42 used linear-scaling DFT energy-evaluations with classical molecular-dynamics (MM) sampling on an entire protein–protein complex. In 2012 and 2014, Fox et al.43,44 extended this approach and evaluated the binding free energies of a full protein–ligand system using ab initio linearly-scaling DFT. The QM-PBSA method combines MM sampling with implicit-solvent full-QM (DFT) energy evaluations. The predicted binding free energies outperformed traditional, classical mechanics based MM-PBSA.44
Our goal is to improve the accuracy, transferability and reproducibility of binding free energy calculations in protein–ligand systems by using high-accuracy ab initio DFT. In this study, we push the boundary of DFT-based binding free energy calculations by drastically increasing the number of full-protein DFT calculations to over 2900. This allows us to assess, in-depth, the convergence of the QM-PBSA method. We compare the performance of dispersion-including non-local and meta-GGA-nonlocal exchange–correlation functionals VV1045 and B97M-rV46,47 with the popular GGA functional PBE. In addition to exploring these modern dispersion-including DFT functionals, we compare different empirical dispersion-corrections to the PBE functional and assess the significance of the three-body dispersion term. We demonstrate exceptional reproducibility of DFT energies, determine statistical errors at different levels of sampling and study the entropy correction term. The new general-purpose semi-empirical QM method GFN2-XTB48 is also tested. Overall, we lay the foundation for large-scale applications of the QM-PBSA method, comment on best practice and demonstrate that with modern computing capabilities, DFT binding free energy calculations are viable and are a promising avenue of research and industry application.
Section 2.1 outlines the theory of the MM- and QM-PBSA method and some specific aspects of our linear-scaling DFT based approach. Section 2.2 describes the design of this computational study and computational details are summarized in Section 2.3. The results are presented in Section 3, discussing reproducibility (Section 3.1), convergence (Section 3.2), the entropy correction term (Section 3.3), statistical errors (Section 3.4), a comparison of DFT functionals (Section 3.5) and a comparison of different MM-, SEQM- and QM-PBSA methods. Section 4 contains the discussion of the results and we present our conclusions in Section 5.
2 Methods
2.1 The MM- and QM-PBSA methods
MM-PBSA was first proposed by Kollman et al. in 200049 and has become a popular method for estimating binding free energies. The two key assumptions in MM-PBSA are (1) sampling only from the endpoints of the binding process, and (2) treating the solvent implicitly. By sampling only the endpoints of the binding process, the computational cost is reduced greatly. Sampling is usually implemented using molecular-dynamics (MD) or Monte Carlo (MC) methods with an explicit solvent model. A representative ensemble of snapshots is extracted to estimate binding free energies. The binding free energy of a ligand B to a receptor protein A is the difference of the average free energy of the complex and its constituents,| ΔGbind = 〈GAB〉 − 〈GA〉 − 〈GB〉. | (1) |
In MM-PBSA the mean free energies of the complex, protein and ligand are deconstructed into the following terms,
| 〈G〉 = 〈E〉 + 〈Gsolvation〉 − T〈S〉, | (2) |
Substituting eqn (2) into (1) gives,
| ΔGbind = 〈ΔE〉 + 〈ΔGsolvation〉 − T〈ΔS〉 = 〈ΔHbind〉 − T〈ΔS〉, | (3) |
The MM-PBSA method is used actively in prospective drug design and lead identification. Recent examples include efforts to identify potential treatments of Covid-1953,54 and Alszheimer's55,56 and to better understand Down Syndrome.57 A variety of improvements to the original formulation by Kollman et al.49 have also been suggested. Duan et al.58 have proposed an alternative method, called interaction entropy, of estimating the entropy correction term in MM-PBSA. More involved definitions of the solvent accessible surface area (SASA) like the weighted-SASA54 approach have also been developed as well as volume based estimates of the cavitation energy in the non-polar solvation term.59 For more background and applications of the MM-PBSA method we recommend the reviews by Genheden,60 Wang61 and Poli.62
In the QM-PBSA method, the gas-phase energies, 〈E〉, and solvation energies, 〈Gsolvation〉, are calculated at a QM level. In our implementation of QM-PBSA, we use linear-scaling DFT to calculate QM gas-phase and solvation energies.
While MM-PBSA is a very cheap and approximate method that is outclassed by more sophisticated, thermodynamically rigorous and computationally expensive MM approaches like free energy perturbation (FEP) or thermodynamic integration (TI), its extension to QM is straight forward. The MM- and QM-PBSA methods differ only in how the gas-phase and solvation energy are calculated and thus, a direct comparison between MM and QM is possible. Because of this, we choose QM-PBSA as a stepping stone method, through which we may study the tractability, convergence, errors and other aspects of full-QM protein–ligand binding free energies. We expect, that our findings will aid future developments of QM-variants of more rigorous and involved MM methods.
2.2 Linear-scaling DFT
Due to the cubic scaling of conventional density functional theory, full-protein calculations on many thousands of atoms are not feasible. To study larger systems, linearly-scaling versions of DFT have been developed.41 The ONETEP code63 is one such linear-scaling DFT implementation and exploits hybrid MPI-OMP parallelization64 for efficient and scaleable calculations. The unique characteristic of ONETEP is that even though it is linear-scaling, it is able to retain large basis set accuracy as in conventional cubic-scaling DFT calculations. The implicit solvation model is a minimal-parameter Poisson–Boltzmann (PB) based model which is implemented self-consistently as part of the DFT calculation65,66 and uses the smeared-ion formalism and electron-density iso-surfaces to construct solute cavities.2.3 Design of computational study
 | ||
| Fig. 1 Phenol bound in the buried binding site of the T4-lysozyme double mutant (L99A/M102Q).67 PDB Code: 1LI2. | ||
We re-use the identical 1000 MD snapshots for 7 ligands (shown in Fig. 2) in T4-lysozyme in this study. Fox et al. applied the QM-PBSA method to a subset of 50 snapshots, equally spaced within the 1000 extracted from the MD trajectories. Ligand 8, a non-binder, from the 2014 ligand set was excluded from this study as due to its larger size, it is more prone to inducing sidechain motion in the protein upon binding73 which are unlikely to be captured in 20 ns of MD.
 | ||
| Fig. 2 An overview of sampling and methods for each ligand. Ligand set A consists of the first 5 ligands and 50 snapshots of sampling with DFT functionals PBE, VV10 and B97M-rV. Ligand set B consists of all 7 ligands and 100 snapshots for PBE and GFN2-XTB. Structures drawn using Marvin JS on chem-space.com. | ||
Hydroxyaniline is a non-binder and thus does not have a well defined experimental binding free energy. The experimental assay used in ref. 74 could identify measurable binders up to a dissociation constant of 10 mMol which corresponds to a binding free energy of −2.7 kcal mol−1 at 300 K. Thus, the lower limit of the non-binder's free energy of binding is −2.7 kcal mol−1 while the theoretical upper limit is 0 kcal mol−1. Going forward, all metrics applied to relative binding energies are calculated for the lower limit, upper limit and under the exclusion of the non-binder.
PBE is a generalized gradient approximation (GGA) functional based on exact constraints and minimal empiricism. Because PBE cannot describe long-range correlation effects, an empirical force-field-like dispersion-correction is added. In this study, we test ONETEP's default dispersion-correction63 and variants of Grimme's D275 and D376–78 empirical dispersion-corrections. We chose PBE because it is highly popular and is based on physical considerations with only moderate empiricism.
VV10 was selected because it is a non-local dispersion-including GGA functional. It combines rPW86 exchange, PBE correlation and VV10 dispersion-correlation.45 In 2016, Womack et al.47 implemented a more numerically efficient version, rVV10, into ONETEP.
Going beyond GGA functionals, meta-GGAs (mGGA) incorporate the electron kinetic energy density as well as the density gradient. This allows for more flexibility in the functional form and in general, mGGAs outperform GGAs but are more computationally demanding. In a benchmarking study by Head-Gordon et al.79 the most accurate mGGA was the relatively new empirically-parameterized functional, B97M-rV46 which incorporates rVV10 non-local dispersion.
2.4 Computational details
3 Results
3.1 Reproducibility
This study provides a valuable opportunity to demonstrate the reproducibility of DFT-based binding free energy calculations. The calculations by Fox et al.44 were performed with a 2012 version (3.1.15.2) of the ONETEP63 code while this study uses version 5.3.2.6 from late 2019. As shown in Table 1, despite 7 years of active code development the average absolute difference between the new and old results using the same structures and functional is 0.1 kcal mol−1. This underlines the robustness and precision of the DFT methodology and the ONETEP code in particular.| Ligands | PBE | PBE-2014 | Delta |
|---|---|---|---|
| Catechol | −8.9 | −9.0 | 0.14 |
| Fluoroaniline | −6.0 | −5.9 | −0.12 |
| Hydroxyaniline | −6.2 | −6.2 | 0.00 |
| Methylphenol | −8.7 | −8.5 | −0.16 |
| Toluene | −5.0 | −4.8 | −0.18 |
| Chlorophenol | −6.9 | −6.9 | 0.01 |
| Absolute mean | 0.10 | ||
3.2 Convergence
Fig. 3 shows the SEM convergence, calculated by bootstrapping, of each enthalpy component and the total enthalpy for catechol in T4-lysozyme. The enthalpies shown are net enthalpies, as defined in eqn (3). The SEM of the net gas-phase enthalpy, 〈ΔE〉, and the solvation energy, 〈ΔGsolvation〉, in catechol is only slightly higher for PBE than for the other methods. However, the SEM of PBE in the cavity-corrected solvation energy, 〈ΔGsolvation–cav–cor〉, is significantly higher. At 100 snapshots the SEM of the cavity-corrected solvation energy is 0.385 kcal mol−1 while the other methods have SEMs below 0.184 kcal mol−1. This leads to the overall higher SEM in the total enthalpy change upon binding, 〈ΔHbind〉, for the PBE method. The higher SEM of PBE is also reflected in the SEM of functionals VV10 and B97M-rV over ligand set A.
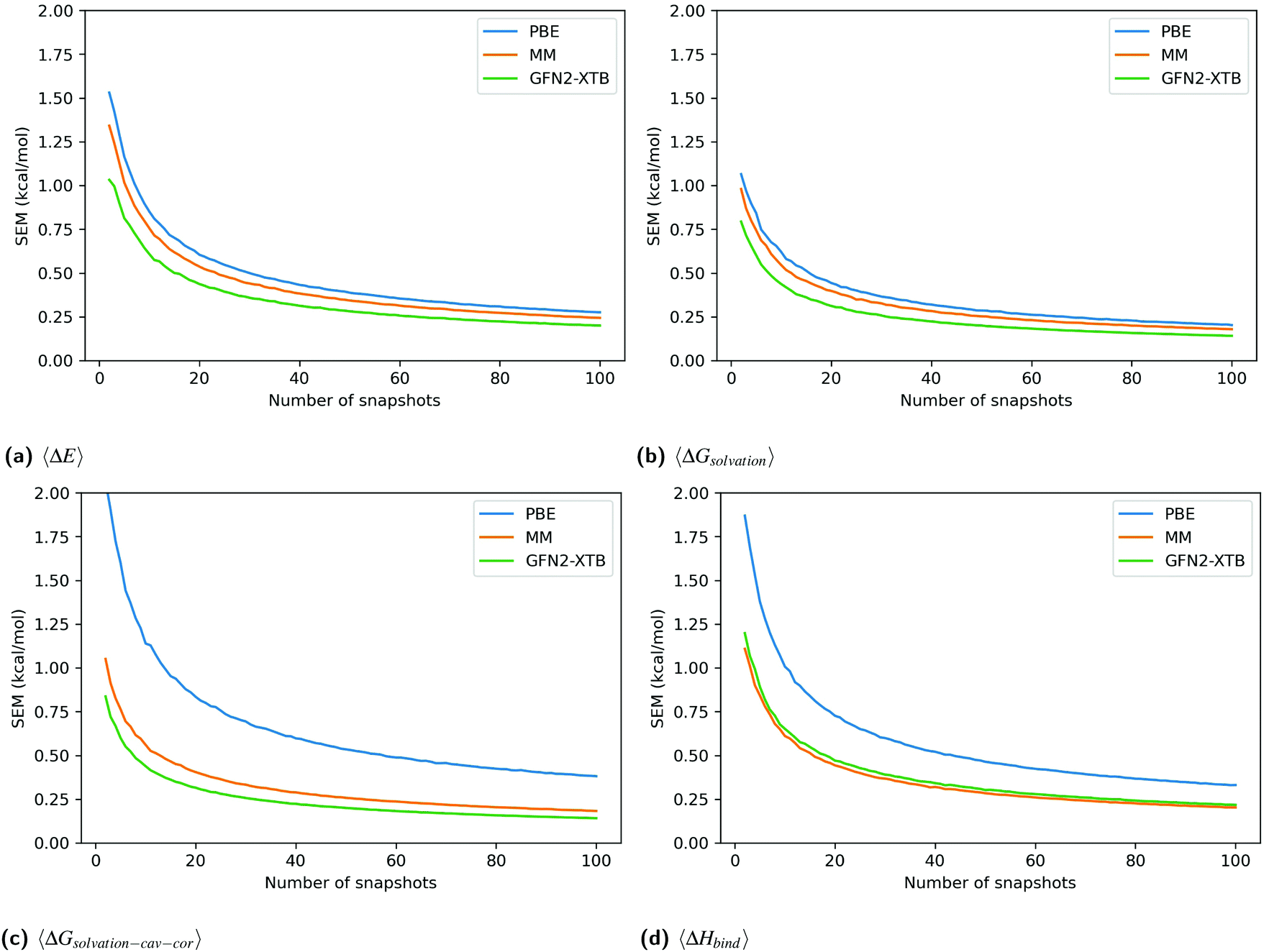 | ||
| Fig. 3 The standard error of the mean (SEM) calculated by bootstrapping (1000 re-samples) of the change upon binding in the gas-phase energy, 〈ΔE〉, solvation energy, 〈ΔGsolvation〉, cavity-corrected solvation energy, 〈ΔGsolvation–cav–cor〉, and total enthalpy, 〈ΔHbind〉 = 〈ΔE〉 + 〈ΔGsolvation–cav–cor〉, for catechol up to 100 snapshots for MM, the DFT functional PBE and the SEQM method GFN2-XTB. | ||
Unlike for PBE, the MM-style cavity-correction term applied to the MM-PBSA results (labeled MM) only minimally increases the solvation energy SEM. GFN2-XTB is shown without cavity-correction and has a similar SEM to MM. The above observations are consistent for all ligands (figures in Section 2.1 of ESI†).
 | ||
| Fig. 4 Left: Mean change in total enthalpy upon binding, 〈ΔHbind〉, of each ligand at different numbers of equally spaced snapshots. Right: Absolute deviation of 〈ΔHbind〉 at different numbers of equally spaced snapshots from the ‘converged’ mean over 100 snapshots. Methods: MM (a and b), DFT(PBE) (c and d). | ||
3.3 Entropy correction
The entropy term in QM- and MM-PBSA is calculated by normal mode analysis as detailed in the methods section. The maximum SEM at low numbers of snapshots is lower than for the enthalpic components, especially the DFT cavity-corrected solvation energies. However, the rate of convergence is also slower. The entropy SEM at 100 snapshots is larger than that of the total enthalpy change upon binding calculated with MM and GFN2-XTB, and is comparable to that of PBE.Fig. 5 shows a similar analysis for entropy as done for the enthalpic terms. Panels 5a and b show the convergence of the mean net-entropy term and absolute deviation from the mean net-entropy term at 100 snapshots. There are significant fluctuations below 50 snapshots (>1 kcal mol−1). Fluctuations of about 0.5 kcal mol−1 remain even beyond 50 snapshots and, compared to the enthalpic terms, the entropy term appears qualitatively slower in convergence.
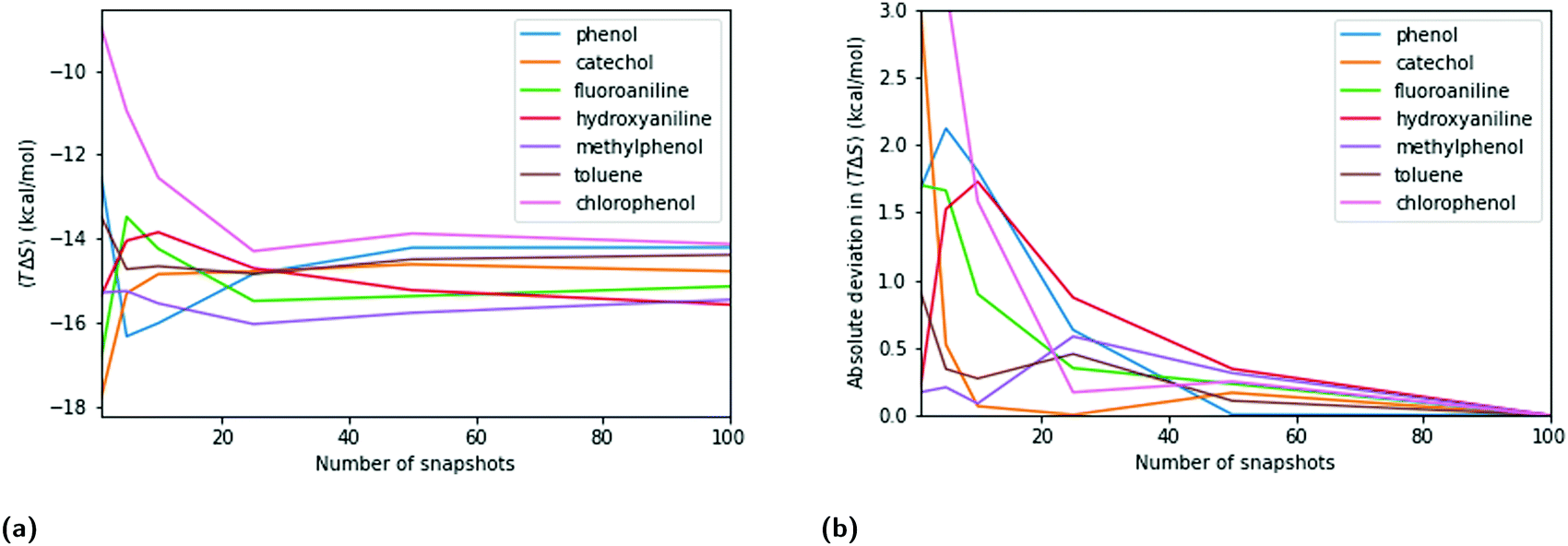 | ||
| Fig. 5 (a) Convergence of mean entropy change upon binding, TΔ〈S〉, over 100 equally spaced snapshots. (b) Absolute deviation of TΔ〈S〉 from “converged” value at 100 snapshots at different numbers of equally spaced snapshots. | ||
The degree of entropy sampling significantly changes the RMSDtr of calculated against experimental relative binding free energies. Fig. 6 shows the RMSDtr over ligand set B at 100 enthalpy snapshots and increasing levels of entropy sampling. Including a small number of entropy snapshots (5, 10, 25) increases the RMSDtr by up to 1.3 kcal mol−1 for MM-PBSA and 0.4 kcal mol−1 for QM-PBSA. At 50 entropy snapshots and beyond the RMSDtr decreases compared to no entropy correction. The lowest RMSDtr is reached at 100 snapshots of entropy, i.e. the same level of sampling as for the enthalpy terms. Beyond this, the sampling of snapshots not included for calculating the enthalpy terms does not further improve accuracy vs. experiment. All three treatments of the non-binder exhibit the increased RMSDtr at low levels of entropy sampling (Figures in ESI†).
 | ||
| Fig. 6 Root mean square deviation from experiment after removal of mean signed error (kcal mol−1) of calculated binding free energies for ligand set B, at different levels of entropy sampling. Enthalpy sampled over 100 snapshots. RMSDtr calculated with upper limit for non-binder (lower limit and binders only in ESI†). | ||
3.4 Statistical error due to incomplete sampling
The calculated absolute binding free energies are the sum of two separate means, the mean enthalpy and mean entropy, sampled over a selection of protein–ligand conformations, i.e. snapshots. By propagation of errors, the SEM of the entropy and enthalpy terms of each ligand and the chosen reference ligand are combined to estimate the total statistical error due to imperfect sampling in the relative binding free energies.Table 2 shows the maximum statistical error due to incomplete sampling across all choices of reference ligands for each method at different levels of sampling. Enthalpy and entropy terms are evaluated over the same 10, 25, 50 and 100 equally spaced snapshots. The maximum statistical errors for PBE are almost identical to those for VV10 and B97M-rV and hence only PBE is shown. We use these values as estimates of the uncertainty in our calculated binding free energies going forward. This statistical error or uncertainty, due to imperfect sampling, should not be confused with the RMSDtr of calculated against experimental binding free energies, used to quantify the closeness of predicted to experimental results.
| Snapshots/methods | Max statistical errors (kcal mol−1) | |||
|---|---|---|---|---|
| 10 | 25 | 50 | 100 | |
| PBE | 1.88 | 1.22 | 0.87 | 0.62 |
| MM | 1.63 | 1.05 | 0.76 | 0.54 |
| GFN2-XTB | 1.57 | 1.02 | 0.73 | 0.52 |
3.5 Ligand set A: comparing DFT functionals
Table 3 shows the root mean square deviation after removal of the mean signed error of the calculated relative binding free energies with respect to experimental binding free energies68,74 of ligand set A using 50 enthalpy and 50 entropy snapshots. The RMSDtr is shown for all treatments of the non-binder and the average standard error (SE) is estimated by bootstrapping with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 resamples.
000 resamples)
000 resamples.
000 resamples)
| Method | RMSD after removal of systematic error (MSE) | ||
|---|---|---|---|
| All ligands [*] | All ligands [‡] | Binders only | |
| B97M-rV | 2.49 | 1.86 | 1.93 |
| VV10 | 2.68 | 2.11 | 2.21 |
| PBE + ONETEP Disp | 2.26 | 1.76 | 1.93 |
| PBE + D2 | 2.25 | 1.71 | 1.84 |
| PBE + D3(BJ) | 2.25 | 1.79 | 1.97 |
| PBE + D3(BJ) + ABC | 2.17 | 1.73 | 1.91 |
| PBE + D3(BJM) | 2.22 | 1.77 | 1.95 |
| PBE + D3(BJM) + ABC | 2.14 | 1.71 | 1.90 |
| Average SE | 0.26 | 0.27 | 0.30 |
Overall, VV10 is the worst performing exchange–correlation functional and has a consistently higher RMSDtr, irrespective of the treatment of the non-binder. The PBE + dispersion methods have slightly lower RMSDtr than B97M-rV when the non-binder is included, either via the upper or lower bound, but given the estimated standard error, this difference is likely not significant. For the subset of binders only, B97M-rV and PBE + dispersion methods achieve the same RMSDtr. All the empirical dispersion corrections to PBE perform well but are indistinguishable given the standard error. The inclusion of the three-body dispersion term (ABC) always slightly reduces RMSDtr but the change is much smaller than the standard error.
3.6 Ligand set B: PBE, GFN2-XTB and MM
We now compare the accuracy vs. experiment of MM, PBE + dispersion and GFN2-XTB on ligand set B at 100 snapshots of enthalpy and entropy. Table 4 shows the RMSDtr of the calculated relative binding free energies against experiment.68,74 The RMSDtr is shown for all treatments of the non-binder and the average standard error (SE) is estimated by bootstrapping with 10000 resamples.
000 resamples)
| Method | RMSD after removal of systematic error (MSE) | ||
|---|---|---|---|
| All ligands [*] | All ligands [‡] | Binders only | |
| MM | 2.21 | 1.61 | 1.55 |
| PBE + ONETEP Disp | 2.01 | 1.57 | 1.65 |
| PBE + D2 | 2.09 | 1.63 | 1.69 |
| PBE + D3(BJ) | 2.11 | 1.73 | 1.84 |
| PBE + D3(BJ) + ABC | 2.03 | 1.66 | 1.77 |
| PBE + D3(BJM) | 2.11 | 1.74 | 1.85 |
| PBE + D3(BJM) + ABC | 2.02 | 1.67 | 1.79 |
| GFN2-XTB | 3.65 | 3.16 | 3.12 |
| Average SE | 0.15 | 0.16 | 0.17 |
Overall, the accuracy against experiment as described by the RMSDtr is comparable for the MM- and QM-PBSA approach. Only the SEQM-PBSA approach using the GFN2-XTB energy method performs significantly worse. The different empirical dispersion corrections are indistinguishable given the standard error. As in ligand set A, the three-body dispersion term does slightly reduce the RMSDtr of both the PBE + D3(BJ) and PBE + D3(BJM) methods but this change is within the estimates standard error.
3.7 Comment on correlation
Correlation to experiment is not included as a quality metric for two main reasons. First, the ligand set is very small. Second, the range of experimental binding free energies is only 1.4 kcal mol−1 and multiple ligands have identical, or near identical, experimental energies. In comparison, the estimated statistical error in the computed relative binding free energies due to incomplete sampling is 0.87 kcal mol−1 at 50 snapshots and 0.62 kcal mol−1 at 100 snapshots for PBE. As a result, the correlation values obtained vary greatly depending on both the choice of reference ligand and the treatment of the non-binder. Furthermore, the 90% confidence intervals calculated by bootstrapping for Pearson r-values exhibit very large ranges of r-value, often above 0.5. Thus, no meaningful comparison between methods is possible. We conclude from this, that both a larger number of ligands and a larger range of experimental binding free energies are key requirements for future protein–ligand system selection.4 Discussion
4.1 Computational cost
Gathering the results for this study posed a serious computational challenge. Excluding initial testing and exploratory work, 3600 ab initio DFT calculations were completed, 2900 of which were on the entire 2600-atom T4-lysozyme. This was made possible by (1) the linear-scaling of the ONETEP DFT code, (2) the efficient hybrid MPI-OMP parallelization of the ONETEP code and (3) access to three different HPC centers. Running calculations concurrently on three HPC facilities for 6 months, the DFT calculations alone required more than 1 million core-hours. Taking into account the 21000 normal mode calculations, 15000 empirical dispersion calculations and 2100 SEQM calculations we estimate a total wall-time of about 30000 hours or 1250 days.
With full access to the 5632 compute nodes of the tier-0 EU HPC facility HAWK, the entirety of the calculations for this study could be completed in less than 24 hours.
With this study, we have shown that a benchmarking study on the QM-PBSA method comprising of multiple protein systems with up to or beyond 25 ligands is feasible.
4.2 Convergence and errors
One criticism of QM-PBSA and related methods is that sampling and energy evaluation are performed using different energy functions.7 We expected this to lead to poor convergence of the QM energy terms in comparison to the MM energies. While the higher SEM for DFT methods, as compared to MM, initially indicated this to be true, the source of the higher SEM is predominantly the QM cavity-correction. Further investigation showed that the QM non-polar solvation terms calculated in ONETEP have larger variance than the MM non-polar terms. This is exacerbated by the functional form of the QM cavity-correction which combines the host and complex non-polar terms and then scales the result by a factor of 7.116.44 This magnifies the effect of the larger variance in the QM non-polar terms on the total binding free energies, leading to a larger overall SEM. The standard deviation of the cavity-corrected solvation energy for PBE ranges from 2.7 to 3.9 kcal mol−1 in ligand set B while the range for MM is 1.3 to 1.9 kcal mol−1. One possible reason for the larger variance in the DFT non-polar term is the more complex definition of the binding cavity via electron-density iso-surfaces. GN2-XTB has similar or lower standard deviations than MM.Detailed analysis of the convergence of the total enthalpy change upon binding of each ligand showed that beyond 25 snapshots, the QM results appear equally converged as the MM results. Analysis of the absolute deviations from the ‘converged’ enthalpy change upon binding at 100 snapshots using different numbers of equally spaced and randomly selected snapshots also confirmed this. At low numbers of snapshots (<25) the DFT energies fluctuated significantly more than those from MM, reflected by the higher SEM of the QM methods. The SEQM method GFN2-XTB showed similar convergence to MM, even below 25 snapshots. This is likely because the method does not suffer the increased SEM due to the QM cavity-correction.
In terms of precision, the maximum estimated statistical error for PBE in Table 2 is only 0.08 kcal mol−1 higher than for MM at 100 snapshots and 0.11 kcal mol−1 higher at 50 snapshots. This further suggests, that at and beyond 50 snapshots the convergence and precision of MM and DFT methods are comparable. The key finding is that in this system, the QM-PBSA method (irrespective of choice of functional) does not suffer from poorer convergence compared to MM-PBSA.
In this study, snapshots generated from a single MD simulation were used. Sampling snapshots from independent MD simulations may result in large standard errors of the mean and may impact the rate of convergence of both the MM and QM methods.
These results indicate that the MM force-fields (GAFF and ff99SB) used for sampling are well parameterized for this system and produce configurations that overlap well with the true QM ensemble. As a result, the QM calculations converge quickly as no high QM-energy configurations are present in the MM ensemble. In terms of the potential energy landscape, this would mean that the position of energy minima in the MM and QM representation are very similar. The difference in calculated binding free energies is then a result of the different depths and shapes of these energy minima for the different energy functions.
4.3 QM-PBSA: improvements and recommendations
In the same study, Risthaus et al.88 found the D3 three-body dispersion term to contribute 2.3% to 14.6% in large, dispersion-dominated systems. We have confirmed that the three-body dispersion term is significant (about 10% of total dispersion) in protein–ligand systems of this size and tends to improve RMSDtr slightly, however well within the estimated standard error. Given the size of the three-body dispersion term and its tendency to reduce RMDStr, we recommend the use of the D3 empirical dispersion correction due to its ability to include the three-body dispersion term.
Why do the newer and more computationally expensive VV10 and B97M-rV, which explicitly account for dispersion, not improve upon the PBE functional with empirical dispersion in this QM-PBSA study of a large dispersion-dominated system? Application of the non-parametric Kolmogorov–Smirnov test for equality of two one-dimensional distributions showed that the distributions of non-cavity-corrected absolute solvation energies of PBE, VV10 and B97M-rV are very similar. This echoes our past experience using the ONETEP solvent model that showed the solvation energies to be independent of the choice of DFT functional. Based on the Kolmogorov–Smirnov test, the gas-phase energy distributions, which include the dispersion energy, are dissimilar. Both VV10 and B97M-rV use the non-local rVV10 dispersion term. While the dispersion term rVV10 results in different gas-phase energies than the empirical dispersion corrections to PBE, this does not translate to improved accuracy vs. experiment compared to PBE + dispersion in this QM-PBSA study.
Furthermore, the rVV10 non-local dispersion term cannot describe three-body effects, which we found to be significant using the empirical D3-ABC method. Risthaus et al.88 have suggested that the three-body dispersion term from D3 could be added, in a post hoc fashion, to dispersion including functionals, however, this was not tested here.
B97M-rV is a meta-GGA functional and thus almost twice as computationally intensive as the GGA functionals PBE and VV10. The data-set used to design and test B97M-rV consisted almost entirely of small molecules.46 The only protein–ligand system was a 1686 atom HIV II-protease/indinavir complex split into 21 interaction fragment pair structures. In the 2017 benchmarking study by Head-Gordon et al.,79 B97M-rV was the most accurate meta-GGA, however, of the almost 5000 data points tested, there were only 21 protein–ligand fragments (same as above) and 12 protein–DNA complexes with a maximum size of 58 atoms. While we can only comment on the suitability of the functionals to the QM-PBSA method, and not their accuracy on the whole, it is interesting that B97M-rV produced worse or comparable results to PBE + dispersion. A potential explanation is that the accuracy of exchange–correlation functionals on small molecule test sets is not indicative of their applicability to much larger systems. This study serves as an example that moving up the “Jacob's ladder” of functional complexity does not guarantee improved results.
Given the small range of binding energies in the ligand set, the null hypothesis of assigning each ligand the same binding energy yields relatively low errors against experiment. The null hypothesis has a RMSDtr of 0.57 kcal mol−1 if the non-binder is excluded, and 1.00 kcal mol−1 and 1.87 kcal mol−1 when the non-binder is included via the lower and upper bound, respectively. Fundamentally, more ligands with a wider range of binding free energies are needed allow for a comparison between MM- and QM-PBSA and we are actively working on achieving this.
In this study on the T4-lysozyme double mutant (L99A/M102Q) our linear-scaling DFT-based QM-PBSA method achieves a RMSDtr of about 1.7 kcal mol−1 across the 6 binders and MM-PBSA achieves a RMSDtr of 1.6 kcal mol−1. To place our results into context, we briefly outline the results of some other QM- and SEQM-PB(GB)SA studies. For a more in-depth review of QM based biding free energy calculations we recommend the review by Ryde and Söderhjelm.7 In 2011, Anisimov et al.24,25 applied a SEQM-PBSA style method using the PM3 Hamiltonian and a COSMO solvation model to 5 ligands binding to the LcK SH2 domain and 4 binders to BRCA1. They achieved MAD of 0.7 kcal mol−1 and 1.7 kcal mol−1, respectively. In both cases the SEQM approach was more accurate than MM-PBSA. In 2012, Mikulskis et al.26 tested a SEQM-GBSA approach with the AMI, RM1 and PM6 Hamiltonians on three protein–ligand systems. The overall best performing energy function, AM1, achieved an MADtr of 1.8–12.0 kcal mol−1 in avidin, 1.1–1.3 kcal mol−1 in fXa and 0.3–4.9 kcal mol−1 in ferritin, depending on the details of the hydrogen bond correction and choice of dispersion correction. Only in the ferritin system was the best SEQM-GBSA method able to convincingly outperform MM-GBSA and MM-PBSA. In 2010, Söderhjelm et al.34 used the PMISP approach on 7 biotin analogues binding to avidin. They achieved a MADtr of 4.5 kcal mol−1 and the QM approach performed worse than MM-PBSA (3.3 kcal mol−1).
One of the key motivations to extend binding free energy calculations to the quantum mechanical level is that the QM energy evaluations can in principle describe a wider range of physics. In this ligand set and binding site however, the MM force-field is not challenged by high charges, large polarization, charge transfer or similar phenomena that are not well described in traditional empirical force-fields. This may in part explain the similar accuracy of MM- and QM-PBSA in this system. Going forward, we will focus our efforts on protein–ligand systems which explicitly challenge traditional force-fields and where the more involved, quantum mechanical description may be necessary.
5 Conclusions
In this study, we have shown that in the context of protein–ligand binding studies for drug design applications, thousands of ab initio DFT calculations of full protein–ligand systems are feasible with modest computational effort. In testing the exchange–correlation functionals PBE, VV10 and B97M-rV we find that the computationally cheapest functional, PBE, is the most promising candidate for the application of the QM-PBSA method. Our findings highlight that benchmarking studies focused almost entirely on small systems may not be representative of the performance of the functionals in a QM-PBSA approach applied to much larger systems (2600 atoms in our case). Different empirical dispersion corrections to PBE all perform well but their accuracy against experiment are all within the estimated standard error. The D3 three-body dispersion term is significant in size (≈10%) and tends to improve results slightly. By expanding the QM calculations to 100 snapshots for the PBE functional, we can show that sampling at 50 snapshots is likely sufficient for convergence. While going beyond 50 snapshots reduces statistical error, no improvement in predicted against experimental binding energies is observed. Furthermore, the QM-PBSA and MM-PBSA methods exhibit near indistinguishable convergence beyond 25 snapshots of sampling. This is shown by the similar statistical errors and the convergence of the mean binding energies. In this system, the inclusion of an entropy correction term is only beneficial when sampled over at least 25 snapshots. Entropy terms with less sampling increase RMSDtr. Sampling entropy beyond 100 snapshots does not improve results.Our study demonstrates that QM-PBSA with full protein calculations is now feasible and can be a useful addition to the toolbox of free energy calculations, especially in cases where force field parameterization may not be sufficiently able to capture effects such as charge transfer and polarisation which are included by default in quantum descriptions. Looking to the future, we believe that the extension of more rigorous classical mechanical binding free energy methods to full-QM, using linear-scaling density functional theory, has significant potential and that the QM-PBSA method is an important stepping stone.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the use of the IRIDIS 5 High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work. We are grateful for computational support from the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/P020194 and EP/T022213/1). We are also grateful for access to the ARCHER national supercomputer which was obtained via the UKCP consortium, funded by EPSRC grant ref EP/P022030/1. L. G. would also like to thank the CDT for Theory and Modelling in the Chemical Sciences and Boehringer Ingelheim for financial support in the form of a PhD studentship.Notes and references
- B. Kuhn, M. Tichý, L. Wang, S. Robinson, R. E. Martin, A. Kuglstatter, J. Benz, M. Giroud, T. Schirmeister, R. Abel, F. Diederich and J. Hert, J. Med. Chem., 2017, 60, 2485–2497 CrossRef CAS PubMed.
- Z. Li, Y. Huang, Y. Wu, J. Chen, D. Wu, C. G. Zhan and H. B. Luo, J. Med. Chem., 2019, 62, 2099–2111 CrossRef CAS PubMed.
- M. Kuhn, S. Firth-Clark, P. Tosco, A. S. Mey, M. MacKey and J. Michel, J. Chem. Inf. Model., 2020, 60, 3120–3130 CrossRef CAS PubMed.
- L. F. Song and K. M. Merz, J. Chem. Inf. Model., 2020, 60, 5308–5318 CrossRef CAS PubMed.
- L. Casalino, A. Dommer, Z. Gaieb, E. P. Barros, T. Sztain, A. Trifan, A. Brace, A. Bogetti, H. Ma, H. Lee, S. Khalid, L. Chong, C. Simmerling, D. J. Hardy, J. D. C. Maia, J. C. Phillips, T. Kurth, A. Stern, L. Huang, J. Mccalpin, T. Gibbs, J. E. Stone, S. Jha, A. Ramanathan and R. E. Amaro, biorxiv, 2020 DOI:10.1101/2020.11.19.390187.
- C. N. Cavasotto, N. S. Adler and M. G. Aucar, Front. Chem., 2018, 6, 188 CrossRef PubMed.
- U. Ryde and P. Söderhjelm, Chem. Rev., 2016, 116, 5520–5566 CrossRef CAS PubMed.
- Y. Shi, Z. Xia, J. Zhang, R. Best, C. Wu, J. W. Ponder and P. Ren, J. Chem. Theory Comput., 2013, 9, 4046–4063 CrossRef CAS PubMed.
- D. V. Sakharov and C. Lim, J. Am. Chem. Soc., 2005, 127, 4921–4929 CrossRef CAS PubMed.
- M. Soniat and S. W. Rick, J. Chem. Phys., 2014, 140, 184703 CrossRef PubMed.
- M. Soniat and S. W. Rick, J. Chem. Phys., 2012, 137, 6146–6952 CrossRef PubMed.
- M. Soniat, G. Pool, L. Franklin and S. W. Rick, Fluid Phase Equilib., 2015, 407, 31–38 CrossRef.
- M. Soniat, R. Kumar and S. W. Rick, J. Chem. Phys., 2015, 143, 044702 CrossRef PubMed.
- M. Soniat, L. Hartman and S. W. Rick, J. Chem. Theory Comput., 2015, 11, 1658–1667 CrossRef CAS PubMed.
- A. J. Lee and S. W. Rick, J. Chem. Phys., 2011, 134, 184507 CrossRef PubMed.
- F. Gräter, S. M. Schwarzl, A. Dejaegere, S. Fischer and J. C. Smith, J. Phys. Chem. B, 2005, 109, 10474–10483 CrossRef PubMed.
- M. A. Ibrahim, J. Chem. Inf. Model., 2011, 51, 2549–2559 CrossRef CAS PubMed.
- M. Retegan, A. Milet and H. Jamet, J. Chem. Inf. Model., 2009, 49, 963–971 CrossRef CAS PubMed.
- K. D. Dubey and R. P. Ojha, J. Biol. Phys., 2011, 37, 69–78 CrossRef CAS PubMed.
- Y. T. Wang and Y. C. Chen, Mol. Inf., 2014, 33, 240–249 CrossRef CAS PubMed.
- F. Barbault and F. Maurel, J. Comput. Chem., 2012, 33, 607–616 CrossRef CAS PubMed.
- K. Wichapong, A. Rohe, C. Platzer, I. Slynko, F. Erdmann, M. Schmidt and W. Sippl, J. Chem. Inf. Model., 2014, 54, 881–893 CrossRef CAS PubMed.
- N. Díaz, D. Suárez, K. M. Merz and T. L. Sordo, J. Med. Chem., 2005, 48, 780–791 CrossRef PubMed.
- V. M. Anisimov and C. N. Cavasotto, J. Comput. Chem., 2011, 32, 2254–2263 CrossRef CAS PubMed.
- V. M. Anisimov, A. Ziemys, S. Kizhake, Z. Yuan, A. Natarajan and C. N. Cavasotto, J. Comput.-Aided Mol. Des., 2011, 25, 1071–1084 CrossRef CAS PubMed.
- P. Mikulskis, S. Genheden, K. Wichmann and U. Ryde, J. Comput. Chem., 2012, 33, 1179–1189 CAS.
- M. Kaukonen, P. Söderhjelm, J. Heimdal and U. Ryde, J. Phys. Chem. B, 2008, 112, 12537–12548 CrossRef CAS PubMed.
- X. Chen, X. Zhao, Y. Xiong, J. Liu and C. G. Zhan, J. Phys. Chem. B, 2011, 115, 12208–12219 CrossRef CAS PubMed.
- H. Lu, X. Huang, M. D. M. Abdulhameed and C. G. Zhan, Bioorg. Med. Chem., 2014, 22, 2149–2156 CrossRef CAS PubMed.
- M. Wang and C. F. Wong, J. Chem. Phys., 2007, 126, 026101 CrossRef PubMed.
- S. Manta, A. Xipnitou, C. Kiritsis, A. L. Kantsadi, J. M. Hayes, V. T. Skamnaki, C. Lamprakis, M. Kontou, P. Zoumpoulakis, S. E. Zographos, D. D. Leonidas and D. Komiotis, Chem. Biol. Drug Des., 2012, 79, 663–673 CrossRef CAS PubMed.
- K. E. Tsitsanou, J. M. Hayes, M. Keramioti, M. Mamais, N. G. Oikonomakos, A. Kato, D. D. Leonidas and S. E. Zographos, Food Chem. Toxicol., 2013, 61, 14–27 CrossRef CAS PubMed.
- P. Söderhjelm, J. Kongsted, S. Genheden and U. Ryde, Interdiscip. Sci.: Comput. Life Sci., 2010, 2, 21–37 CrossRef PubMed.
- P. Söderhjelm, J. Kongsted and U. Ryde, J. Chem. Theory Comput., 2010, 6, 1726–1737 CrossRef PubMed.
- D. G. Fedorov, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1322 Search PubMed.
- T. Sawada, D. G. Fedorov and K. Kitaura, J. Am. Chem. Soc., 2010, 132, 16862–16872 CrossRef CAS PubMed.
- R. Kurauchi, C. Watanabe, K. Fukuzawa and S. Tanaka, Comput. Theor. Chem., 2015, 1061, 12–22 CrossRef CAS.
- U. Tagami, K. Takahashi, S. Igarashi, C. Ejima, T. Yoshida, S. Takeshita, W. Miyanaga, M. Sugiki, M. Tokumasu, T. Hatanaka, T. Kashiwagi, K. Ishikawa, H. Miyano and T. Mizukoshi, ACS Med. Chem. Lett., 2016, 7, 435–439 CrossRef CAS PubMed.
- A. Heifetz, E. I. Chudyk, L. Gleave, M. Aldeghi, V. Cherezov, D. G. Fedorov, P. C. Biggin and M. J. Bodkin, J. Chem. Inf. Model., 2016, 56, 159–172 CrossRef CAS PubMed.
- T. J. Giese and D. M. York, J. Chem. Theory Comput., 2019, 15, 5543–5562 CrossRef CAS PubMed.
- D. R. Bowler and T. Miyazaki, Rep. Prog. Phys., 2012, 75, 036503 CrossRef CAS PubMed.
- D. J. Cole, C. K. Skylaris, E. Rajendra, A. R. Venkitaraman and M. C. Payne, EPL, 2010, 91, 37004 CrossRef.
- S. J. Fox, PhD thesis, University of Southampton, 2012.
- S. J. Fox, J. Dziedzic, T. Fox, C. S. Tautermann and C. K. Skylaris, Proteins: Struct., Funct., Bioinf., 2014, 82, 3335–3346 CrossRef CAS PubMed.
- O. A. Vydrov and T. Van Voorhis, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2015, 142, 074111 CrossRef PubMed.
- J. C. Womack, N. Mardirossian, M. Head-Gordon and C. K. Skylaris, J. Chem. Phys., 2016, 145, 204114 CrossRef PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS.
- C. Wang, D. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2018, 4, 87 CrossRef PubMed.
- J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 1998, 120, 9401–9409 CrossRef CAS.
- V. K. Bhardwaj, R. Singh, J. Sharma, V. Rajendran, R. Purohit and S. Kumar, J. Biomol. Struct. Dyn., 2020, 1–10 CrossRef PubMed.
- J. Wang, J. Chem. Inf. Model., 2020, 60, 3277–3286 CrossRef CAS PubMed.
- R. Singh, V. Bhardwaj, P. Das and R. Purohit, J. Biomol. Struct. Dyn., 2020, 38, 5126–5135 CrossRef CAS PubMed.
- A. Kaur, S. Shuaib, D. Goyal and B. Goyal, Phys. Chem. Chem. Phys., 2020, 22, 1543–1556 RSC.
- V. K. Bhardwaj, R. Singh, J. Sharma, P. Das and R. Purohit, Comput. Methods Progr. Biomed., 2020, 194, 105494 CrossRef PubMed.
- K. Huang, S. Luo, Y. Cong, S. Zhong, J. Z. Zhang and L. Duan, Nanoscale, 2020, 12, 10737–10750 RSC.
- C. Tan, Y. H. Tan and R. Luo, J. Phys. Chem. B, 2007, 111, 12263–12274 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed.
- G. Poli, C. Granchi, F. Rizzolio and T. Tuccinardi, Molecules, 2020, 25, 1971 CrossRef CAS PubMed.
- J. C. Prentice, J. Aarons, J. C. Womack, A. E. Allen, L. Andrinopoulos, L. Anton, R. A. Bell, A. Bhandari, G. A. Bramley, R. J. Charlton, R. J. Clements, D. J. Cole, G. Constantinescu, F. Corsetti, S. M. Dubois, K. K. Duff, J. M. Escartín, A. Greco, Q. Hill, L. P. Lee, E. Linscott, D. D. O'Regan, M. J. Phipps, L. E. Ratcliff, Á. R. Serrano, E. W. Tait, G. Teobaldi, V. Vitale, N. Yeung, T. J. Zuehlsdorff, J. Dziedzic, P. D. Haynes, N. D. Hine, A. A. Mostofi, M. C. Payne and C. K. Skylaris, J. Chem. Phys., 2020, 152, 174111 CrossRef CAS PubMed.
- K. A. Wilkinson, N. D. Hine and C. K. Skylaris, J. Chem. Theory Comput., 2014, 10, 4782–4794 CrossRef CAS PubMed.
- J. Dziedzic, H. H. Helal, C. K. Skylaris, A. A. Mostofi and M. C. Payne, EPL, 2011, 95, 43001 CrossRef.
- J. C. Womack, L. Anton, J. Dziedzic, P. J. Hasnip, M. I. Probert and C. K. Skylaris, J. Chem. Theory Comput., 2018, 14, 1412–1432 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- B. Q. Wei, W. A. Baase, L. H. Weaver, B. W. Matthews and B. K. Shoichet, J. Mol. Biol., 2002, 322, 339–355 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Genet., 2006, 65, 712–725 CrossRef CAS PubMed.
- D. Case, T. Darden, T. Cheatham, C. Simmerling, J. Wang, R. Duke, R. Luo, M. Crowley, R. Walker, W. Zhang, K. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. Wong, F. Paesani, J. Vanicek, X. Wu, S. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. Mathews, M. Seetin, C. Sagui, V. Babin and P. Kollman, AMBER 10, 2008 Search PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- J. W. Ponder and D. A. Case, Adv. Protein Chem., 2003, 66, 27–85 CrossRef CAS PubMed.
- D. L. Mobley and M. K. Gilson, Annu. Rev. Biophys., 2017, 46, 531–558 CrossRef CAS PubMed.
- S. E. Boyce, D. L. Mobley, G. J. Rocklin, A. P. Graves, K. A. Dill and B. K. Shoichet, J. Mol. Biol., 2009, 394, 747–763 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. G. Smith, L. A. Burns, K. Patkowski and C. D. Sherrill, J. Phys. Chem. Lett., 2016, 7, 2197–2203 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. Sun, Y. Li, S. Tian, L. Xu and T. Hou, Phys. Chem. Chem. Phys., 2014, 16, 16719–16729 RSC.
- M. Elstner, P. Hobza, T. Frauenheim, S. Suhai and E. Kaxiras, J. Chem. Phys., 2001, 114, 5149–5155 CrossRef CAS.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- P. Pracht, E. Caldeweyher, S. Ehlert and S. Grimme, ChemRxiv, 2019, 1–19 Search PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- D. Case, R. Betz, D. Cerutti, T. Cheatham, T. Darden, R. Duke, T. Giese, H. Gohlke, A. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. Merz, G. Monard, H. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, D. Roe, A. Roitberg, C. Sagui, C. Simmerling, W. Botello-Smith, J. Swails, R. Walker, J. Wang, R. Wolf, X. Wu, L. Xiao and P. Kollman, Amber 16, 2016 Search PubMed.
- S. S. Shapiro and M. B. Wilk, Biometrika, 1965, 52, 591–611 CrossRef.
- T. Risthaus and S. Grimme, J. Chem. Theory Comput., 2013, 9, 1580–1591 CrossRef CAS PubMed.
- S. Zhong, K. Huang, Z. Xiao, X. Sheng, Y. Li and L. Duan, J. Phys. Chem. B, 2019, 123, 8704–8716 CrossRef CAS.
- L. Duan, X. Liu and J. Z. Zhang, J. Am. Chem. Soc., 2016, 138, 5722–5728 CrossRef CAS PubMed.
- P.-C. Su, C.-C. Tsai, S. Mehboob, K. E. Hevener and M. E. Johnson, J. Comput. Chem., 2015, 36, 1859–1873 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details, supplementary figures and directions to all input and output files. See DOI: 10.1039/d1cp00206f |
| This journal is © the Owner Societies 2021 |