
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the population of triplet states of 2-seleno-thymine†
Danillo
Valverde
 a,
Sebastian
Mai
*bc,
Adalberto Vasconcelos
Sanches de Araújo
d,
Sylvio
Canuto
a,
Leticia
González
c and
Antonio Carlos
Borin
*d
a,
Sebastian
Mai
*bc,
Adalberto Vasconcelos
Sanches de Araújo
d,
Sylvio
Canuto
a,
Leticia
González
c and
Antonio Carlos
Borin
*d
aInstitute of Physics, University of São Paulo, Rua do Matão 1371. 05508-090, São Paulo, SP, Brazil
bPhotonics Institute, Vienna University of Technology, Gußhausstraße 27-29, 1040 Vienna, Austria
cInstitute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Straße 17, 1090 Vienna, Austria. E-mail: sebastian.mai@univie.ac.at
dDepartment of Fundamental Chemistry, Institute of Chemistry, University of São Paulo, Av. Prof. Lineu Prestes 748. 05508-000, São Paulo, SP, Brazil. E-mail: ancborin@iq.usp.br
First published on 23rd February 2021
Abstract
The population and depopulation mechanisms leading to the lowest-lying triplet states of 2-Se-Thymine were studied at the MS-CASPT2/cc-pVDZ level of theory. Several critical points on different potential energy hypersurfaces were optimized, including minima, conical intersections, and singlet–triplet crossings. The accessibility of all relevant regions on the potential energy hypersurfaces was investigated by means of minimum energy paths and linear interpolation in internal coordinates techniques. Our analysis indicates that, after the population of the bright S2 state in the Franck–Condon region, the first photochemical event is a barrierless evolution towards one of its two minima. After that, three viable photophysical deactivation paths can take place. In one of them, the population in the S2 state is transferred to the T2 state via intersystem crossing and subsequently to the T1 state by internal conversion. Alternatively, the S1 state could be accessed by internal conversion through two distinct conical intersections with S2 state followed by singlet–triplet crossing with the T2 state. The absence of a second minimum on the T1 state and a small energy barrier on pathway along the potential energy surface towards the ground state from the lowest triplet state are attributed as potential reasons to explain why the lifetime of the triplet state of 2-Se-Thymine might be reduced in comparison with its thio-analogue.
1 Introduction
In DNA and RNA, the genetic information1,2 is encoded by the sequence of the canonical nucleobases, which are purine and pyrimidine derivatives. These canonical nucleobases absorb in the ultra-violet (UV) region, meaning that they could in principle undergo harmful photochemical reactions. Fortunately, these canonical nucleobases have a native and very efficient self-protection mechanism that brings them back to the ground state nonradiatively on an ultrafast timescale after photoexcitation.3,4 Here, conical intersections act as the photophysical decay funnels that enable the efficient population transfer among different electronic states.5,6Replacement of a carbonyl oxygen atom by a sulfur transforms a canonical nucleobase into a so-called thiobase. It has been shown that this strongly boosts intersystem crossing (ISC), leading to an increased triplet yield after excitation.7,8 Triplet states are often very reactive, which can be harmful but can also be taken advantage of in photodynamical therapy9—a medicinal technique widely employed in the treatment of several diseases.10–14 Hence, thiobases have been extensively studied in the last years—both experimentally and theoretically15–20—with focus on their prospective suitability as photosensitizers for photodynamical therapy.
Recent experimental21 and theoretical investigations22–26 have shown that selenium-substituted nucleobases (selenobases) could be even more effective photosensitizers. This is due to their near-unity triplet yields and their red-shifted into the visible absorption spectra (relative to the thiobases), thus increasing the possible light penetration into biological tissue.27 Accordingly, a comprehensive understanding of the population pathways after light irradiation is dearly needed, if selenobases are to be exploited in practical application as photosensitizers. Recently, some of us carried out a comprehensive analysis of the unusual photophysical properties of 2-Se-Uracil (2SeUra), showing that it has some potential as a photosensitizer.24 Here we aim at investigating the photodeactivation mechanisms of 2-Se-Thymine (2SeThy).
Uracil and thymine (5-methyl-uracil) differ only in the presence of a methyl group at the C5 position in thymine. It has been shown in detail that the presence of the methyl group (and other chemical modifications) on C5 position, significantly affects the excited-state decay due to subtle changes in the potential energy surfaces that in turn affect the nuclear motion, leading to slower deactivation and in general to a more complex relaxation mechanism for thymine as compared to uracil.6,28–31 This pushed us to investigate the effects of methylation on the triplet population mechanisms of 2-Se-Uracil.24
Some preliminary aspects of the 2SeThy ISC mechanisms were already investigated theoretically by Pirillo and coworkers,22 but it focused only on the Franck–Condon region. In this paper we carry out a comprehensive analysis of the global potential energy surfaces (PESs) of 2SeThy, mapping state minima, minimum energy paths and minimum energy crossing regions, allowing us to derive photophysical relaxation mechanisms. To this aim, we employ accurate multiconfigurational methods that include both static and dynamic electron correlation.
2 Computational details
Both the computation of vertical excitation energies (VEEs) and the optimization of critical points of the PESs were carried out using the multi-state complete active space perturbation theory of second order32 (MS-CASPT2) method. The VEEs were calculated with the cc-pVDZ33 basis set and without relativistic effects, as well as with the relativistic ANO-RCC-VDZP34 basis set and considering relativistic effects following the Douglas–Kroll–Hess formalism.35–37 Since the results of both basis sets are very similar (see Tables S1 and S2 in ESI†), all subsequent steps were performed with the less expensive cc-pVDZ basis set. Two-electron integrals were computed with the Cholesky decomposition method38 to speed up the calculations.Geometries were pre-optimized with the complete active space self-consistent field39 (CASSCF) method, employing an active space of 12 electrons in 9 orbitals (denoted as CAS(12,9)), comprising 8 π/π* orbitals plus the lone pair of the selenium atom, as shown in Fig. 1 (green background). The final optimization of minima and crossing points (and associated energy calculations) was carried out at the MS-CASPT2 level of theory with the same active space. Additionally, VEEs were computed with a larger CAS(14,11) active space, by augmenting the CAS(12,9) active space with the σ and σ* orbitals localized on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se group, see Fig. 1 (blue background). Test calculations indicated that the nO lone pair localized on the oxygen atom remains virtually doubly occupied in all cases and hence was excluded from the active space (further details in Table S3 in ESI†).
Se group, see Fig. 1 (blue background). Test calculations indicated that the nO lone pair localized on the oxygen atom remains virtually doubly occupied in all cases and hence was excluded from the active space (further details in Table S3 in ESI†).
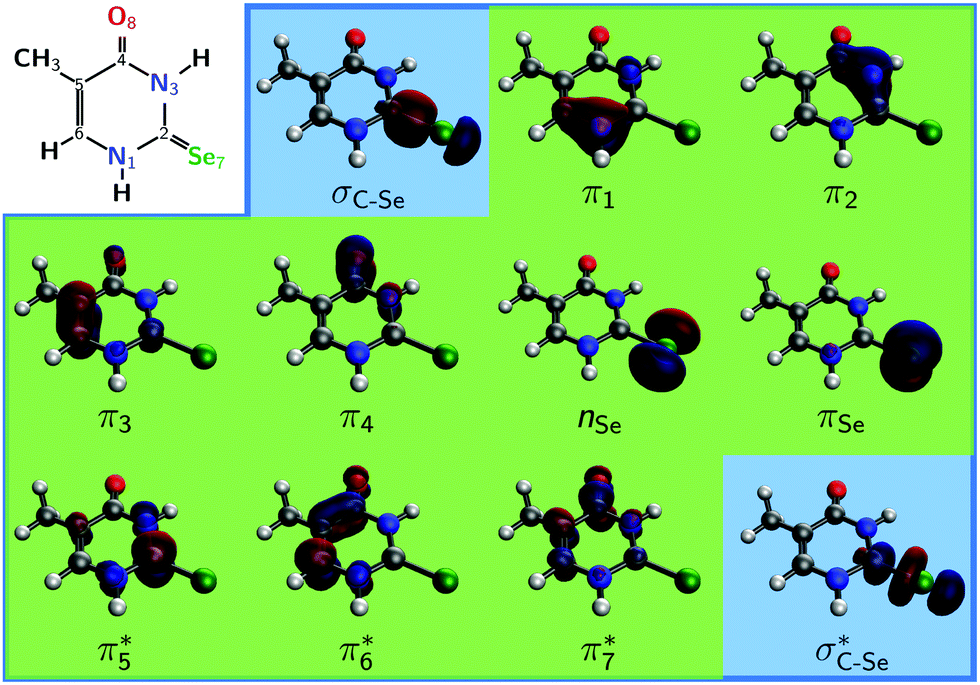 | ||
| Fig. 1 Schematic structure of 2SeThy (with atom numbering) together to the active space orbitals employed in the calculations. The smaller CAS(12,9) is shown with green background. The larger CAS(14,11) additionally contains the two orbitals with blue background. Orbitals are shown for the ground state equilibrium geometry. | ||
All calculations were carried out with the OpenMolcas40 package. As OpenMolcas does not average density matrices over states of different multiplicities, singlet and triplet states were computed separately, state-averaging over 4 singlet or 3 triplet states, respectively. An imaginary level-shift correction41 of 0.2 a.u. was used to prevent intruder states problems. Based on recommendations from literature,42 the IPEA shift43 was set to zero. Spin–orbit coupling (SOC) elements were computed through the AMFI (Atomic Mean Field Integrals) approach.44
Minimum energy crossing points (MECPs) were optimized with the restricted Lagrange multipliers technique.45 If the two states involved in a MECP structure have the same spin multiplicity, it is a conical intersection (CI) and if they belong to different spin symmetries (singlet and triplet, in our case), is a singlet–triplet crossing (STC). The initial relaxation pathway starting from the ground state minimum geometry was scanned with a minimum energy path (MEP) calculation (sphere of radius of 0.025 a.u.), carried out at the MS-CASPT2(12,9)/cc-pVDZ level of theory. Subsequent segments of the relaxation pathway were obtained by connecting the optimized structures with the linear interpolation in internal coordinates (LIIC) technique, computing the energies at each interpolated point at the same level of theory. One should note that the PES profiles obtained through LIIC scans only provide an upper limit for any energy barrier (as curved pathways with lower barriers could exist). However, the absence of any barrier from a LIIC scan segment can be taken as an unambiguously proof that no barrier is present.
3 Results and discussion
3.1 Vertical excitation energies
The computed VEEs and associated oscillator strengths of 2SeThy are shown in Table 1, together with other results available from the literature. All calculations predict that the lowest-lying excited state S1 has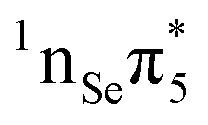 character, i.e. it derives from a single excitation from the non-bonding orbital (nSe) situated on the Se atom to the
character, i.e. it derives from a single excitation from the non-bonding orbital (nSe) situated on the Se atom to the  molecular orbital (Fig. 1). The low oscillator strength identifies it as a dark state. It is interesting to note that our results agree nicely with those reported by Pirillo et al.22 at the M062X/6-31+G* level of theory, whereas those obtained by Manae and Hazra46 with MS-CASPT2(14,10)/cc-pVTZ (IPEA = 0.25 a.u.) are overestimated by about 0.2 eV, probably due to the different IPEA shift and basis set employed.
molecular orbital (Fig. 1). The low oscillator strength identifies it as a dark state. It is interesting to note that our results agree nicely with those reported by Pirillo et al.22 at the M062X/6-31+G* level of theory, whereas those obtained by Manae and Hazra46 with MS-CASPT2(14,10)/cc-pVTZ (IPEA = 0.25 a.u.) are overestimated by about 0.2 eV, probably due to the different IPEA shift and basis set employed.
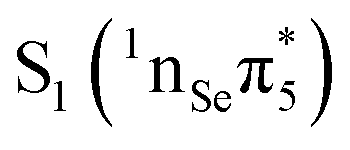
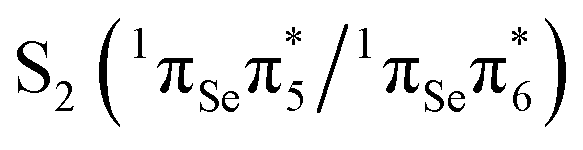
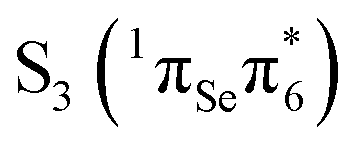
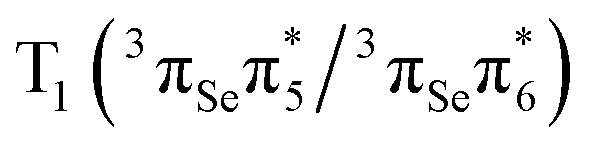
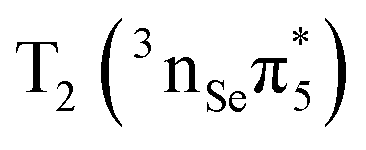
The second excited state (S2) is predicted at 3.91 eV at our highest level of theory (MS-CASPT2(14,11)) and is a bright state. Its character is a linear combination of two single excitations, 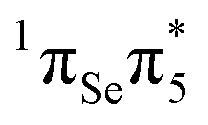 (44%) and
(44%) and 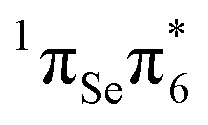 (45%). As it is associated with the largest computed oscillator strength (0.265), the S2 is designated the initially excited state in our relaxation mechanism. The VEE of S2 is in line with experimental results obtained in ethanol,47 which report an intense absorption band peaking at 3.95 eV (314 nm). Our results are also consistent with the observation that 2SeThy shows an absorption red-shifted by about 0.5 eV relative to the corresponding thiobase 2-thio-thymine.48
(45%). As it is associated with the largest computed oscillator strength (0.265), the S2 is designated the initially excited state in our relaxation mechanism. The VEE of S2 is in line with experimental results obtained in ethanol,47 which report an intense absorption band peaking at 3.95 eV (314 nm). Our results are also consistent with the observation that 2SeThy shows an absorption red-shifted by about 0.5 eV relative to the corresponding thiobase 2-thio-thymine.48
The S3 state is calculated 4.30 eV vertically above the ground state, is also bright, and can be described as being of 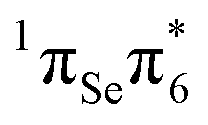 character. While MS-CASPT2(14,11) predicts similar oscillator strength for the S2 and S3 (0.265 and 0.241), other levels of theory calculate a considerable smaller oscillator strength for S3. This can be attributed to the fact that both states derive from contributions of the
character. While MS-CASPT2(14,11) predicts similar oscillator strength for the S2 and S3 (0.265 and 0.241), other levels of theory calculate a considerable smaller oscillator strength for S3. This can be attributed to the fact that both states derive from contributions of the 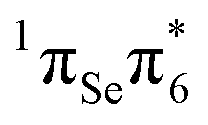 and
and 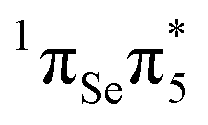 transitions, which show very strong mixing (and hence intensity exchange) that depends sensitively on the geometry and level of theory. The S3 state could be populated by excitation energies higher than that employed experimentally47 (3.95 eV), although this is not the focus of the present work. Nonetheless, even if S3 is populated, it is likely that this state decays quickly to the S2 state, due to the small energy gap between these states that makes the presence of a conical intersection probable. Hence, the most relevant photochemical events would be governed by the S2 potential energy hypersuface. Henceforth we shall focus only on the bright S2 state to discuss the photophysical deactivation pathways of 2SeThy.
transitions, which show very strong mixing (and hence intensity exchange) that depends sensitively on the geometry and level of theory. The S3 state could be populated by excitation energies higher than that employed experimentally47 (3.95 eV), although this is not the focus of the present work. Nonetheless, even if S3 is populated, it is likely that this state decays quickly to the S2 state, due to the small energy gap between these states that makes the presence of a conical intersection probable. Hence, the most relevant photochemical events would be governed by the S2 potential energy hypersuface. Henceforth we shall focus only on the bright S2 state to discuss the photophysical deactivation pathways of 2SeThy.
The lowest T1 and T2 triplet states are computed at 2.85 eV and 3.29 eV, respectively. The T1 is dominated by a 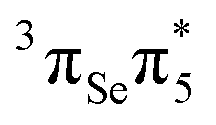 configuration, whereas the T2 state has
configuration, whereas the T2 state has 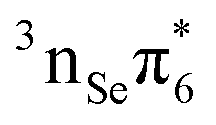 character. The energy of the T1 is in reasonable agreement regardless the level of theory employed. However, the excitation energy of the T2 state is lower at the M062X/6-31+G* level of theory22 than that predicted by our multiconfigurational calculations. We also note that the inclusion of the σ and σ* orbitals in the active space affects the T1 and T2, stabilizing them by 0.12 and 0.15 eV, respectively.
character. The energy of the T1 is in reasonable agreement regardless the level of theory employed. However, the excitation energy of the T2 state is lower at the M062X/6-31+G* level of theory22 than that predicted by our multiconfigurational calculations. We also note that the inclusion of the σ and σ* orbitals in the active space affects the T1 and T2, stabilizing them by 0.12 and 0.15 eV, respectively.
3.2 Excited states and crossing regions
Fig. 2 shows the geometry of the calculated critical points of 2SeThy. Key geometrical parameters of minima and MECPs with adiabatic excitation energies (AEEs) are displayed in Table 2. Ring deformations were analyzed with Cremer–Pople parameters49 and the Boeyens classification scheme.50 The out-of-plane movement of the selenium atom was described by the SeC2N1N4 pyramidalization angle, defined as 90 degrees minus the angle between the Se–C bond and the normal vector of C2–N1–N4.51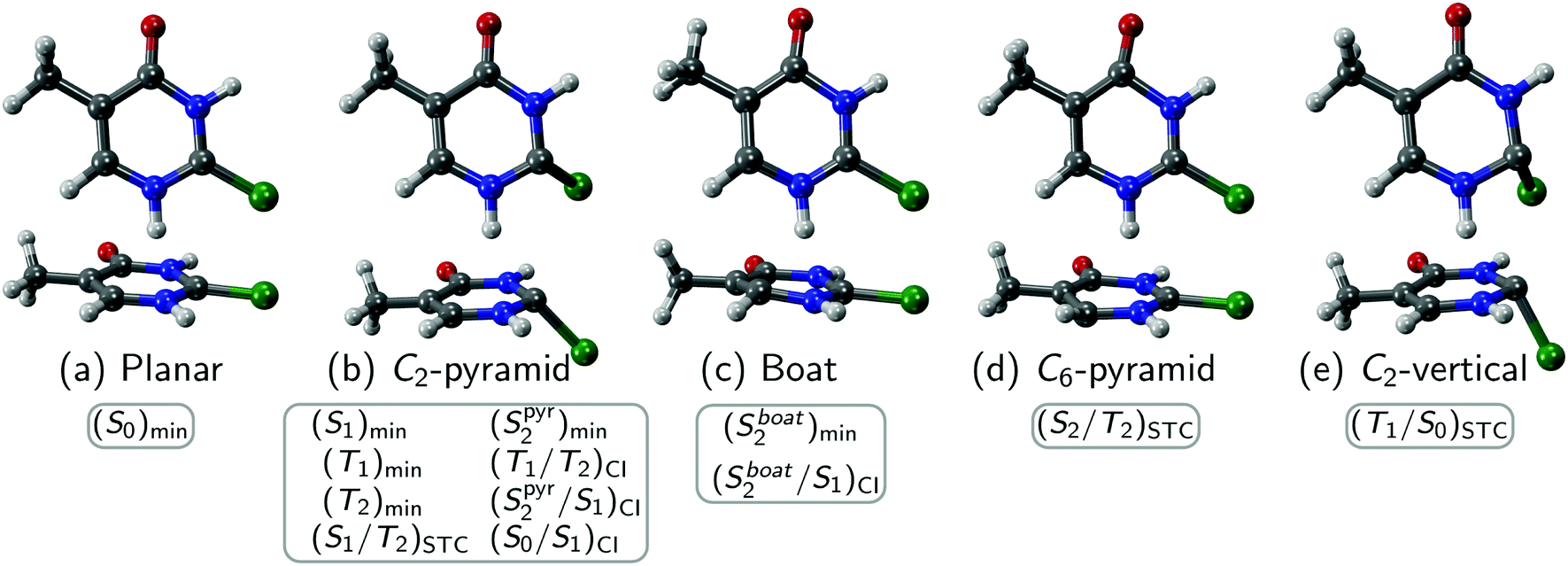 | ||
| Fig. 2 Geometries of 2SeThy critical points in gas phase at the MS-CASPT2(12,9)/cc-pVDZ level of theory. | ||
| Parameters | Minima | |||||
|---|---|---|---|---|---|---|
| (S0)min | (S1)min | (Sboat2)min | (Spyr2)min | (T1)min | (T2)min | |
| ΔE (eV) | 0.00 | 2.89 | 3.37 | 3.16 | 2.53 | 2.67 |
| r(N1C2) | 1.374 | 1.394 | 1.330 | 1.372 | 1.395 | 1.383 |
| r(C2N3) | 1.371 | 1.400 | 1.344 | 1.374 | 1.403 | 1.385 |
| r(N3C4) | 1.415 | 1.402 | 1.496 | 1.410 | 1.398 | 1.406 |
| r(C4C5) | 1.465 | 1.471 | 1.420 | 1.469 | 1.472 | 1.471 |
| r(C5C6) | 1.368 | 1.369 | 1.433 | 1.369 | 1.372 | 1.366 |
| r(N1C6) | 1.383 | 1.376 | 1.423 | 1.379 | 1.369 | 1.380 |
| r(C2Se) | 1.823 | 1.987 | 1.920 | 2.120 | 1.988 | 2.074 |
| r(C5C7) | 1.502 | 1.502 | 1.504 | 1.500 | 1.501 | 1.501 |
| r(C5O8) | 1.225 | 1.228 | 1.239 | 1.225 | 1.229 | 1.226 |
| p(SeC2N1N3) | 0.1 | 40.0 | 2.1 | 48.9 | 44.2 | 42.1 |
| p(HC6N1C5) | 0.2 | 1.8 | −7.4 | −0.2 | 0.2 | 1.5 |
| Q | 0.00 | 0.07 | 0.12 | 0.04 | 0.05 | 0.09 |
| φ | — | 72.5 | 115.3 | 128.9 | 70.2 | 135.3 |
| θ | — | 73.8 | 99.9 | 65.0 | 64.7 | 57.9 |
| Boeyens | — | 3S2 | B 3,6 | E 2 | E 2 | E 2 |
| Minimum energy crossing points | |||||||
|---|---|---|---|---|---|---|---|
| (Sboat2/S1)CI | (Spyr2/S1)CI | (S0/S1)CI | (S2/T2)STC | (S1/T2)STC | (T1/T2)CI | (T1/S0)STC | |
| ΔE (eV) | 3.47 | 3.23 | 3.53 | 3.44 | 2.92 | 2.77 | 2.73 |
| r(N1C2) | 1.308 | 1.366 | 1.436 | 1.331 | 1.403 | 1.390 | 1.441 |
| r(C2N3) | 1.306 | 1.367 | 1.414 | 1.326 | 1.397 | 1.389 | 1.413 |
| r(N3C4) | 1.467 | 1.413 | 1.396 | 1.563 | 1.398 | 1.403 | 1.388 |
| r(C4C5) | 1.398 | 1.470 | 1.475 | 1.397 | 1.477 | 1.476 | 1.486 |
| r(C5C6) | 1.425 | 1.370 | 1.363 | 1.441 | 1.367 | 1.364 | 1.355 |
| r(N1C6) | 1.434 | 1.379 | 1.370 | 1.421 | 1.381 | 1.391 | 1.372 |
| r(C2Se) | 1.920 | 2.157 | 2.299 | 1.922 | 1.965 | 2.020 | 2.043 |
| r(C5C7) | 1.504 | 1.507 | 1.507 | 1.510 | 1.501 | 1.501 | 1.502 |
| r(C5O8) | 1.223 | 1.224 | 1.229 | 1.229 | 1.227 | 1.226 | 1.228 |
| p(SeC2N1N3) | 0.5 | 32.3 | 35.9 | 2.5 | 42.0 | 38.9 | 57.0 |
| p(HC6N1C5) | 10.7 | −0.3 | −3.9 | 9.3 | 1.7 | 2.5 | 0.9 |
| Q | 0.06 | 0.05 | 0.14 | 0.08 | 0.10 | 0.28 | 0.07 |
| φ | 78.4 | 126.8 | 116.1 | 64.5 | 70.7 | 32.3 | 52.5 |
| θ | 55.0 | 64.8 | 134.8 | 56.7 | 63.5 | 75.2 | 66.2 |
| Boeyens | 5 H 6 | E 2 | E 2 | E 6 | E 2 | 1S2 | E 2 |
The 2SeThy ground state optimized geometry ((S0)min, Fig. 2a) is planar, with only the CH3 hydrogen atoms out of the molecular plane. The CSe bond length is 1.82 Å, significantly larger than observed for the analogous CO (about 1.21 Å) or CS (about 1.56 Å) double bonds.52
The minimum of the first excited state, denoted as (S1)min, is placed adiabatically 2.89 eV above the ground state minimum. Its ring remains planar as in the ground state, but the selenium atom leaves the molecular plane by 40 degrees (Fig. 2b). Also noticeable is the stretching of the CSe bond by 0.16 Å with respect to the equilibrium geometry.
At MS-CASPT2(12,9)/cc-pVDZ level, the S2 excited state PES exhibits two minima, as it was also observed for 2TUra53 and 2SeUra.24 The first minimum, with an AEE of 3.37 eV, has a boat conformation (B3,6); thus, we labeled it (Sboat2)min (Fig. 2c). This geometry shows a slight pyramidalization of the hydrogen atom on the 6-position (about 7 degrees) and a moderate increase on the CSe bond (about 0.1 Å) with respect to the ground state minimum. The second minimum is located at a lower energy (3.16 eV) and exhibits strong out-of-plane displacement of the Se atom, as given by the pyramidalization angle of C2 of about 50 degrees. Thus, we label this minimum as (Spyr2)min (Fig. 2b).
The T1 optimized geometry (T1)min is also characterized by a large pyramidalization (44.2 degrees) on the selenium atom (Fig. 2b). We failed to locate a second minimum on the T1 PES—as was reported for 2-thiothymine54,55 and other thiobases56—but consistent with the findings for 2SeUra.24 This is of high relevance, as the presence and energy of the T1 minima is decisive for the T1 life time and the decay back to the ground state. Indeed, the presence of only one T1 minimum in 2SeThy and 2SeUra could be a reason for their strongly decreased triplet life time as compared to their thio-analogues.21
We also optimized the MECPs involving the S1 and S2 states, obtaining two structures. For one of them, denoted (Spyr2/S1)CI (Fig. 2b), we noted a large pyramidalization on the selenium atom, but not so pronounced as in the (S1)min and (Spyr2)min structures. The second structure, (Sboat2/S1)CI, is characterized by a boat conformation, resembling the (Sboat2)min optimized minimum (Fig. 2c). The latter crossing is a bit different than that found for 2SeUra,24 because the bond distances in the ring are smaller for 2SeThy (by about 0.02 Å) as well as the pyramidalization angle (10 degrees versus 25 degrees). In addition, the optimized crossing structure between the S0 and S1 states ((S0/S1)CI) (Fig. 2b) exhibits the largest CSe bond among all optimized structures, with a pyramidalization on the selenium atom of 36°.
We have also located three STC structures. The first one involves the S2 and T2 states. This (Spyr2/T2)STC structure is slightly non-planar, which an envelope (E6) deformation and minor pyramidalization of the C2 (2.5 degrees) and C6 (9.3 degrees) atoms (Fig. 2d). The second STC (S1/T2)STC involves the S1 and T2 states, with an envelope deformation (E2), out-of-plane position of the Se atom (pyramidalization angle of 42 degrees), and a C–Se bond length of 1.922 Å (Fig. 2b). Thus, its geometry is very similar to (S1)min. The third STC structure involves the T1 and S0 states, thus is denoted the (T1/S0)STC, and features the largest pyramidalization on the selenium atom among all the optimized structures (Fig. 2e).
For the three STCs, we computed effective spin–orbit couplings (SOCs) as 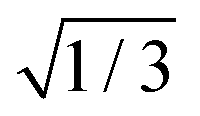 times the norm of the vector containing all spin–orbit matrix elements involving the multiplet components of the two involved states (see the ESI,† for the corresponding equation). We note that this definition differs by the
times the norm of the vector containing all spin–orbit matrix elements involving the multiplet components of the two involved states (see the ESI,† for the corresponding equation). We note that this definition differs by the 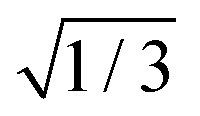 factor from our preceding paper on 2SeUra.24 At the (Spyr2/T2)STC geometry, we obtained a SOC of about 477 cm−1 between S2 and T2. Likewise, for the (S1/T2)STC STC a SOC of 239 cm−1 was obtained. Both values are in qualitative agreement with the Franck–Condon results reported by Pirillo et al.22 (B3LYP/6-31G*) as 433 cm−1 (the paper lists it as 750 cm−1, but this does not include the
factor from our preceding paper on 2SeUra.24 At the (Spyr2/T2)STC geometry, we obtained a SOC of about 477 cm−1 between S2 and T2. Likewise, for the (S1/T2)STC STC a SOC of 239 cm−1 was obtained. Both values are in qualitative agreement with the Franck–Condon results reported by Pirillo et al.22 (B3LYP/6-31G*) as 433 cm−1 (the paper lists it as 750 cm−1, but this does not include the 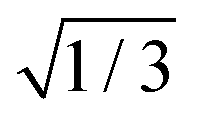 factor we use). At the (S0/T1)STC geometry the SOC is computed to be about 450 cm−1 (Fig. 3).
factor we use). At the (S0/T1)STC geometry the SOC is computed to be about 450 cm−1 (Fig. 3).
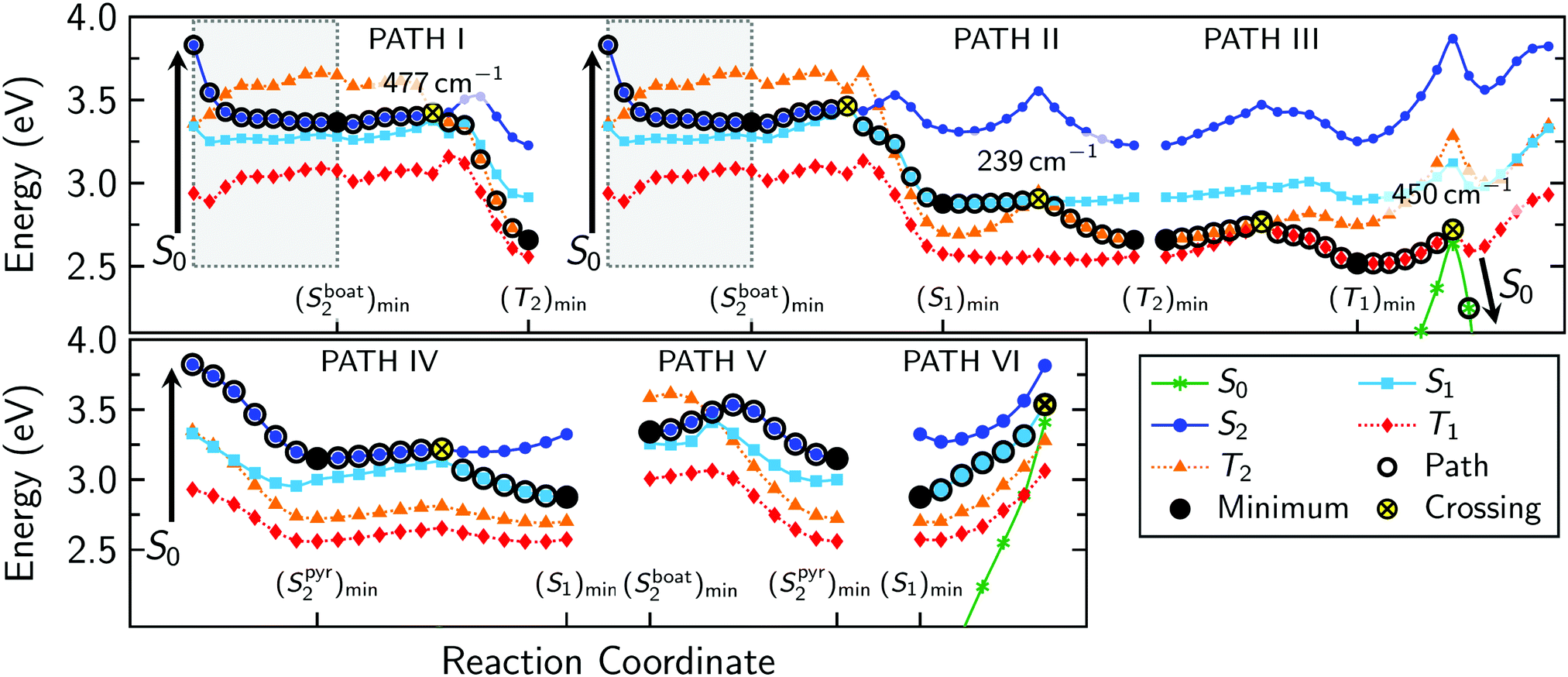 | ||
| Fig. 3 Photophysical deactivation pathways of 2SeThy. At each structure obtained by MEP (gray background) and LIIC approaches, energies were computed at the MS-CASPT2(14,11)/cc-pVDZ level. Spin–orbit couplings (in cm−1) are reported for the crossing seam involving singlet and triplet states. Solid circles indicate the expected evolution of the system after light irradiation. | ||
3.3 Mapping the potential energy surface
In order to obtain plausible deactivation pathways, the optimized minima and MECPs were connected either by MEPs or LIICs. Fig. 3 shows all the discussed deactivation pathways. We note here that in all LIIC scans the S3 state stayed always above 4.0 eV relative to the S0 minimum, and thus was not included in Fig. 3.PATH I begins with a barrierless MEP from the Franck–Condon region along the S2 PES to the (Sboat2)min structure at 3.37 eV. The path proceeds by linear interpolation to the (S2/T2)STC crossing point, 0.07 eV above the (Sboat2)min structure. At the crossing point, the system might undergo ISC to the T2 state and subsequently relaxes to the (T2)min that is 0.77 eV below the crossing.
PATH II also starts with the MEP from the Franck–Condon region to (Sboat2)min, from where the system can access the (Sboat2/S1)CI conical intersection that is located 0.10 eV above the S2 minimum. After internal conversion to the S1 state, the system relaxes to the (S1)min, from where ISC to the T2 is easily possible at the (S1/T2)STC, which is only 0.03 eV above the S1 minimum. After ISC, the system will first continue to the (T2)min. A direct ISC channel from S1 to T1—as hypothesized by Pirillo et al.22 based on the large SOC between these states at the Franck–Condon point—is very unlikely based on our computed relaxation pathways and intermediates.
It is interesting to note that an early (S2/T2)STC can be observed in Paths I and II (Fig. 3). However, as this crossing is located on the steepest region of the path, there is not enough time for an efficient population transfer to take place. On the other hand, the early S2/T2 crossing is likely on the same seam as the later S2/T2 crossing in PATH I, which leads us to conclude that they would share the same decay path.
PATH III continues at the common endpoint of PATH I and PATH II, which is the (T2)min. Internal conversion to the T1 is possible at the (T2/T1)CI conical intersection, 0.10 eV above the T2 minimum. Subsequently, the molecule can access the (T1)min region, at an energy of 2.53 eV above the ground state minimum. As the (T1)min is the lowest-energy excited-state minimum, it is likely decisive for the excited state life time of 2SeThy. However, the T1 can nonradiatively decay back to the ground state at the (T1/S0)STC crossing, placed 0.20 eV above the (T1)min. This energy difference is in good agreement with the value reported for 2SeUra at the MS-CASPT2/cc-pVDZ level of theory.24
Although the MEP from the Franck–Condon region should be the most appropriate pathway to describe the initial relaxation, it is in principle possible that the momentum of the initial wave packet leads part of it along other pathways. For 2SeThy, PATH IV in Fig. 3 shows that from the Franck–Condon point also the second S2 minimum, ((Sboat2)min), could be accessed in a barrierless fashion along a linearly interpolated pathway. Hence, a bifurcation of the initial wave packet into PATH I/II and PATH IV is rather likely. On the contrary, once either of the S2 minima is reached, conversion to the other minimum is hampered by the energy barrier (0.17 eV above (Sboat2)min) shown in PATH V. In general, nonadiabatic molecular dynamics simulations would be necessary to judge which of these two directions is more relevant in the initial relaxation step. However, ultimately we do not expect a significantly different behaviour between PATH I/II and PATH IV, because also PATH IV eventually leads to (S1)min. This is possible due to the presence of the (S2Spyr2/S1)CI conical intersection, at an energy of 0.07 eV above the (S2py)min.
Finally, we analyzed the possibility of the system returning to the ground state via an internal conversion process, as shown in PATH-VI. As it can be seen, the LIIC path shows that this process is very unlikely because the (S1/S0)CI is placed 0.42 eV above the S1 state minimum structure. This finding is in agreement with previous theoretical works about thio- and seleno-nucleobases.18,57
In summary, after 2SeThy absorbs UV radiation, our calculations predict three possible deactivation mechanisms (I–III, II–III and IV–(II)–III in Fig. 3) that reach the lowest triplet state, see Fig. 4. Subsequently, a decay from the T1 minimum to the ground state is possible via reverse ISC. This deactivation mechanism can be summarized as (S2)FC → Sboat2/Spyr2 → S1 → T2 → T1 → S0. Our calculations do not allow clarifying which of the two S2 minima constitutes the primary deactivation pathway, as this would require explicit simulation of the nonadiabatic dynamics of the molecule. Lacking such simulations, we can only estimate the S2 minimum preference from the results published for 2SeUra24 at the ADC(2) level of theory, which suggest that the (Sboat2)min region is only populated for a few femtoseconds before the trajectories turn towards the lower-energy (Spyr2)min region.
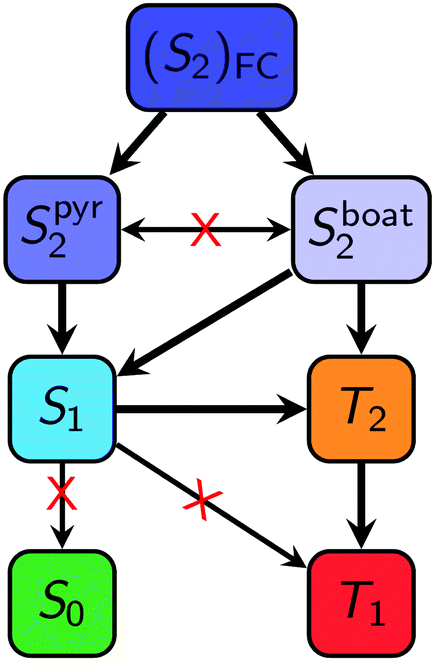 | ||
| Fig. 4 Possible deactivation mechanisms of 2SeThy in gas phase from the S2, calculated at the MS-CASPT2(14,11)/cc-pVDZ//MS-CASPT2(12,9)/cc-pVDZ level of theory. | ||
In comparison to 2SeUra,24 we can conclude that in this case methylation leads to minor effects on the excited-state PES of 2SeThy, as it only slightly reduces the energy barrier between the (T1)min and (T1/S0)STC structures. This energy barrier affects the life time of the triplet state, which we will discuss in more detail in the next section.
3.4 Life time of the triplet state and dependence on the level of theory
A final open issue concerns the life time of the 2SeThy triplet state in relation to that of 2SeUra, both in gas phase and in solution. Here, we will scrutinize the dependence of the relevant energy barrier on the level of theory, in order to obtain a more comprehensive view on this issue. To this end, we have optimized the (T1)min and (S0/T1)STC structures for both molecules in water using different levels of theory, which comprises different active spaces, basis set, and imaginary level shift, including the effects of the environment by means of the Polarizable Continuum Model (PCM).58,59 These results are presented in Table 3.| Level of theory | 2SeThy | 2SeUra | |
|---|---|---|---|
| Gas | MS(12,9)//MS(12,9)/cc-pVDZ, imag = 0.2 a.u. | 0.18 | 0.2324 |
| MS(14,11)//MS(12,9)/cc-pVDZ, imag = 0.2 a.u. | 0.20 | 0.25 | |
| Water | MS(12,9)//MS(12,9)/cc-pVDZ, imag = 0.2 a.u. | 0.14 | |
| MS(12,9)//MS(12,9)/cc-pVDZ, imag = 0.3 a.u. | 0.18 | 0.0624 | |
| MS(14,11)//MS(12,9)/cc-pVDZ, imag = 0.2 a.u. | 0.24 | 0.20 | |
| MS(14,11)//MS(12,9)/cc-pVDZ, imag = 0.3 a.u. | 0.20 | ||
| MS(14,11)//MS(12,9)/ANO-RCC-VDZP, imag = 0.2 a.u. | 0.18 | ||
In gas phase, the energy barrier for 2SeThy has a value smaller than that obtained for 2SeUra, although the difference is small. Therefore, the triplet states life time of 2SeThy in gas phase should be approximately equal or slightly shorter than the one of 2SeUra.
Our initial computation of the energy barrier between (T1)min and (S0/T1)STC regions in solution at the MS-CASPT2(14,11)//MS-CASPT2(12,9)/cc-pVDZ level of theory yielded a value of 0.24 eV. As this value is significantly larger than the previously reported barrier of 0.06 eV in 2SeUra,24 we recomputed the 2SeUra barrier at our present level of theory and found 0.20 eV. In order to rule out a strong dependence of the barrier on the level of theory, we performed further optimizations for 2SeThy (see Table 3), which showed that the barrier depends slightly on the level of theory, varying between 0.14 and 0.24 eV. Importantly, the height of the barrier seems to be little affected by solvation in our new computations, as the different solution-phase results are generally consistent with the gas-phase results. Hence, we can deduce that the triplet states life time should be on the same order of magnitude in both environments. Similarly, we expect that 2SeThy and 2SeUra will have similar triplet life times in solution. Thereby, the conclusion of the previous work24 about the 2SeUra should be revised.
Similar to the gas-phase results presented above, the optimized structures in water always lead to a single minimum in the T1 state. As discussed previously,24 this finding is at odds with that predicted for 2-thiouracil54 and 2-thiothymine.55 According to MS-CASPT2 calculations, both of these thiobases have two different minima on the T1 hypersurface, a boat-like one and a pyramidalized one. The presence of these two minima structures and the higher barriers to access the T1/S0 crossing point (0.36 eV54 in 2-thiouracil and 0.30 eV55 for 2-thiothymine) could enhance the trapping of the triplet population in the thiobases. Based on these findings, we conclude that the triplet state life time should be significantly shorter in 2SeThy than in 2-thiothymine.
4 Conclusion
In this contribution the photophysical deactivation pathways of 2SeThy was investigated at the MS-CASPT2/cc-pVDZ level of theory. Several excited-state minima and crossing points were found, establishing relaxation pathways that should lead to the ultrafast population of the lowest triplet state after the absorption of light. Two mimina on the S2 PES were found, an (Sboat2) structure and an (Spyr2) structure. Even though our calculations cannot accurately predict which of those structures will be more relevant in the dynamics, both of them would allow for an efficient decay to the S1 state or the triplet states. In summary, three feasible photophysical paths can be followed to populate the triplet state: (i) (S2)FC → Sboat2 → T2 → T1; (ii) (S2)FC → Sboat2 → S1 → T2 → T1; and (iii) (S2)FC → Spyr2 → S1 → T2 → T1. Based on the presented energy profiles, we suggest that the latter is the preferential photochemical channel, although nonadiabatic dynamics simulations are required to confirm this prediction.According to our results, the photochemical deactivation mechanisms of 2SeThy are very close to that determined for 2SeUra,24 which implies that the effects due to methylation on the C5 position is rather small. Hence, we expect that both molecules will exhibit very similar triplet life times after excitation—both in gas phase and in solution—although 2SeThy might show slightly shorter life times due to its slightly lower activation barrier.
Compared to its thiobase analogue 2-thiothymine, in 2SeThy the absorption spectrum is red-shifted by about 0.5 eV, due to the weaker bound electrons in the nSe and πSe orbitals. The general relaxation mechanism after excitation should be very similar for 2SeThy, 2SeUra, 2-thiothymine, and 2-thiouracil. However, the two thiobases exhibit two rather deep minima53,54 on the T1 potential energy surface, that can more effectively trap population in the triplet state, compared to the single T1 minimum in the selenobases. Furthermore, the SOCs in the selenobases are approximately four times larger than in the thiobases.24,53,54 These two facts suggest that ISC from T1 to S0 should be significantly faster in the selenobases than in the thiobases. Such shorter triplet life times for selenobases were already reported for 6-selenoguanine,21 who also suggested that for this reason, selenobases might be preferentially employed as short-lived Type I photosensitizer that only affects its immediate vicinity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. V. acknowledges financial support from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), under grants 2017/02612-4 and 2019/04413-4. S. C. thanks CAPES for the BioMol project 23038.004630/2014-35 and the National Institute of Science and Technology Complex Fluids (INCT-FCx) with the CNPq grant 141260/2017-3 and FAPESP grant 2014/50983-3. A. C. B. and A. V. S. A. thank the CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for research fellowships (project numbers 302318/2017-8 and 153104/2015-5, respectively). A. C. B. thanks FAPESP for the research grant 2018/19454-5. Computations were performed on resources provided by HPC through Superintendência de Tecnologia da Informação da Universidade de São Paulo. The authors acknowledge the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing HPC resources of the SDumont supercomputer (http://sdumont.lncc.br), which have contributed to the research results reported within this paper. S. M. and L. G. gratefully acknowledge the Vienna Scientific Cluster (VSC3) for allocation of computational resources and the University of Vienna for continuous support.References
- A. Rich, Q. Rev. Biophys., 2009, 42, 117–137 CrossRef CAS.
- A. Travers and G. Muskhelishvili, FEBS J., 2015, 282, 2279–2295 CrossRef CAS.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Chem. Rev., 2004, 104, 1977–2020 CrossRef.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122–127 CrossRef CAS.
- A. Giussani, J. Segarra-Martí, D. Roca-Sanjuán and M. Merchán, in Excitation of Nucleobases from a Computational Perspective I: Reaction Paths, ed. M. Barbatti, A. C. Borin and S. Ullrich, Springer International Publishing, Cham, 2015, pp. 57–97 Search PubMed.
- S. Mai, M. Richter, P. Marquetand and L. González, in Excitation of Nucleobases from a Computational Perspective II: Dynamics, ed. M. Barbatti, A. C. Borin and S. Ullrich, Springer International Publishing, Cham, 2015, pp. 99–153 Search PubMed.
- B. Ashwood, M. Pollum and C. E. Crespo-Hernández, Photochem. Photobiol., 2019, 95, 33–58 CrossRef CAS.
- R. Brem, F. Li and P. Karran, Nucleic Acids Res., 2009, 37, 1951–1961 CrossRef CAS.
- R. Darlenski and J. W. Fluhr, J. Biomed. Opt., 2012, 18, 061208 CrossRef.
- L. P. Rosa and F. C. da Silva, J. Med. Microbiol., 2014, 3, 1 Search PubMed.
- M. Kim, H. Y. Jung and H. J. Park, Int. J. Mol. Sci., 2015, 16, 23259–23278 CrossRef CAS.
- A. Oniszczuk, K. A. Wojtunik-Kulesza, T. Oniszczuk and K. Kasprzak, Biomed. Pharmarcother., 2016, 83, 912–929 CrossRef CAS.
- S. B. Brown, E. A. Brown and I. Walker, Lancet Oncol., 2004, 8, 497–508 CrossRef.
- S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S. I. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, J. Photochem. Photobiol., C, 2011, 12, 46–67 CrossRef CAS.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS.
- Y. Harada, C. Okabe, T. Kobayashi, T. Suzuki, T. Ichimura, N. Nishi and Y.-Z. Xu, J. Phys. Chem. Lett., 2010, 1, 480–484 CrossRef CAS.
- Y. Harada, T. Suzuki, T. Ichimura and Y.-Z. Xu, J. Phys. Chem. B, 2007, 111, 5518–5524 CrossRef CAS.
- S. Mai, M. Pollum, L. Martínez-Fernández, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernández and L. González, Nat. Commun., 2016, 7, 13077 CrossRef CAS.
- A. Mohamadzade, S. M. Bai, M. Barbatti and S. Ullrich, Chem. Phys., 2018, 515, 572–579 CrossRef CAS.
- H. Kuramochi, T. Kobayashi, T. Suzuki and T. Ichimura, J. Phys. Chem. B, 2010, 114, 8782–8789 CrossRef CAS.
- K. M. Farrell, M. M. Brister, M. Pittelkow, T. I. Solling and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2018, 140, 11214–11218 CrossRef CAS.
- J. Pirillo, B. C. De Simone and N. Russo, Theor. Chem. Acc., 2016, 135, 8 Search PubMed.
- J. Pirillo, G. Mazzone, N. Russo and L. Bertini, J. Chem. Inf. Model., 2017, 57, 234–242 CrossRef CAS.
- S. Mai, A.-P. Wolf and L. González, J. Chem. Theory Comput., 2019, 15, 3730–3742 CrossRef CAS.
- Y.-G. Fang, Q. Peng, Q. Fang, W. Fang and G. Cui, ACS Omega, 2019, 4, 9769–9777 CrossRef CAS.
- Q. Peng, Y.-H. Zhu, T.-S. Zhang, X.-Y. Liu, W.-H. Fang and G. Cui, Phys. Chem. Chem. Phys., 2020, 22, 12120–12128 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- A. Sharonov, T. Gustavsson, S. Marguet and D. Markovitsi, Photochem. Photobiol. Sci., 2003, 2, 362–364 CrossRef CAS.
- R. J. Malone, A. M. Miller and B. Kohler, Photochem. Photobiol., 2003, 77, 158–164 CrossRef CAS.
- T. Gustavsson, A. Bányász, E. Lazzarotto, D. Markovitsi, G. Scalmani, M. J. Frisch, V. Barone and R. Improta, J. Am. Chem. Soc., 2006, 128, 607–619 CrossRef CAS.
- S. Yamazaki and T. Taketsugu, J. Phys. Chem. A, 2012, 116, 491–503 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- P.-O. Widmark, P.-Å. Malmqvist and B. O. Roos, Theor. Chim. Acta, 1990, 77, 291–306 CrossRef CAS.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742–3748 CrossRef CAS.
- M. Reiher, Wiley Interdiscip. Rev.: Dev. Biol., 2012, 2, 139–149 CAS.
- F. Aquilante, R. Lindh and T. Bondo Pedersen, J. Chem. Phys., 2007, 127, 114107 CrossRef.
- B. O. Roos, The Complete Active Space Self-Consistent Field Method and its Applications in Electronic Structure Calculations, John Wiley & Sons, Ltd, 2007, pp. 399–445 Search PubMed.
- I. F. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov and R. K. Carlson, et al. , J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef.
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- J. P. Zobel, J. J. Nogueira and L. González, Chem. Sci., 2017, 8, 1482–1499 RSC.
- G. Ghigo, B. O. Roos and P.-Å. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- B. O. Roos and P.-Å. Malmqvist, Adv. Quantum Chem., 2004, 47, 37–49 CrossRef CAS.
- L. De Vico, M. Olivucci and R. Lindh, J. Chem. Theory Comput., 2005, 1, 1029–1037 CrossRef CAS.
- M. A. Manae and A. Hazra, J. Phys. Chem., 2017, 121, 8147–8153 CrossRef CAS.
- H. G. Mautner, J. Am. Chem. Soc., 1956, 78, 5292–5294 CrossRef CAS.
- G. Cui and W.-H. Fang, J. Chem. Phys., 2013, 138, 044315 CrossRef.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- J. Boeyens, J. Cryst. Mol. Struct., 1978, 8, 317 CrossRef.
- S. Mai, M. Richter, M. Heindl, M. F. S. J. Menger, A. Atkins, M. Ruckenbauer, F. Plasser, L. M. Ibele, S. Kropf and M. Oppel, et al., SHARC2.1: Surface Hopping Including Arbitrary Couplings—Program Package for Non-Adiabatic Dynamics, sharc-md.org, 2019.
- D. R. Lide, CRC handbook of chemistry and physics, CRC press, 2004, vol. 85 Search PubMed.
- S. Mai, P. Marquetand and L. González, J. Phys. Chem., 2015, 119, 9524–9533 CrossRef CAS.
- J. A. Sánchez-Rodríguez, A. Mohamadzade, S. Mai, B. Ashwood, M. Pollum, P. Marquetand, L. González, C. E. Crespo-Hernández and S. Ullrich, Phys. Chem. Chem. Phys., 2017, 19, 19756–19766 RSC.
- S. Bai and M. Barbatti, Phys. Chem. Chem. Phys., 2017, 19, 12674–12682 RSC.
- L. Martnez-Fernández, I. Corral, G. Granucci and M. Persico, Chem. Sci., 2014, 5, 1336–1347 RSC.
- Y.-G. Fang, D. Valverde, S. Mai, S. Canuto, A. C. Borin, G. Cui and L. González, J. Phys. Chem. B, 2021, 125, 1778–1789 CrossRef CAS.
- L. Serrano-Andrés, M. P. Fülscher and G. Karlström, Int. J. Quantum Chem., 1997, 65, 167–181 CrossRef.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The following files are available free of charge: (i) comparison of vertical excitation energies calculations with different active spaces and atomic basis set; (ii) comparison of the ground-state geometry using two atomic basis set; (iii) Cartesian coordinates of the optimized structures. See DOI: 10.1039/d1cp00041a |
| This journal is © the Owner Societies 2021 |