
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical, third order non-linear optical and electrochemical properties of dipolar, centrosymmetric and C2v organoimido polyoxometalate derivatives†
Ahmed
Al-Yasari
ab,
Hani
El Moll‡
a,
Ryan
Purdy
a,
Kevin B.
Vincent
a,
Philip
Spence
 a,
Jean-Pierre
Malval
c and
John
Fielden
*a
a,
Jean-Pierre
Malval
c and
John
Fielden
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: John.Fielden@uea.ac.uk
bDepartment of Chemistry, Faculty of Science, University of Kerbala, Kerbala, Iraq
cInstitut de Science des Matériaux de Mulhouse CNRS-UMR 7361, Université de Haute Alsace, 15 rue Jean Starcky, Mulhouse, 68057, France
First published on 7th May 2021
Abstract
A family comprising seven arylimido-polyoxometalate (POM) hybrid chromophores (three of which are new), with linear dipolar, C2v and linear centrosymmetric geometries have been synthesised and studied by electronic absorption spectroscopy, electrochemistry, Z-scans (two photon absorption, TPA) and computation (DFT/TD-DFT). These reveal that POM acceptor units are an effective basis for TPA materials: the centrosymmetric bis-POM chromophores produce significant cross sections (δ up to 82 GM) from a single aryl bridge, a similar performance to larger dipolar π-systems combining carbazole or diphenylamino donors with the imido-POM acceptor. DFT/TD-DFT calculations indicate strong communication between POM and organic components is responsible for the linear and non-linear optical behaviour of these compounds, while electrochemical measurements reveal class II mixed valence behaviour resulting from an interplay of through-bond and through-space effects.
Introduction
Polyoxometalates (POMs) – a vast family of well-defined, nano- or sub-nanosized molecular metal oxide clusters1 – are attractive functional building blocks for use in materials and devices.1,2 This is because of the wide range of properties obtained by incorporating transition metal heteroatoms or organic moieties into their structures, and the ability of many POMs to reversibly accept one or more electrons without degradation or structural change.3 These features have made POMs a focus of attention in oxidation catalysis for many years,4 and because of their oxygen-to-metal charge transfer bands, POMs are also strong UV chromophores and UV photocatalysts.5 More recently, there has been a growing interest in the interaction of POMs and POM hybrid materials with visible light,6 and the resulting optical, photophysical and photochemical properties.7Non-linear optical effects are a particular focus because of their relevance to many current, emerging and future technologies – for example terahertz generation, telecommunications (electro-optic modulators), imaging, optical limiting, and optical and electro-optical switching and computing.8 We recently demonstrated that in donor-functionalised arylimido-polyoxometalates, strong electronic coupling between the organic and POM sub-units results in strong charge transfer transitions and strong second order optical nonlinearities (relevant to laser frequency doubling).9 In many examples, static first hyperpolarizabilities β0 significantly exceed those obtained for comparable structures with organic acceptors. Meanwhile, several other groups have demonstrated that POMs can offer third order NLO properties (e.g. laser frequency tripling, two-photon absorption (TPA) and reverse saturable absorption (RSA)), finding that incorporation of POMs in films of organic chromophores (such as porphyrins) can enhance third order NLO susceptibility10 and adjust saturable/reverse saturable absorption. Coordination of Keggin ([XM12O40]n−, X = B, P, Si; M = Mo, W) POMs and their lacunary derivatives to first row transition metal complexes of pyridines and other conjugated organic ligands has produced materials with high TPA cross sections,11 while covalent molecular POM-porphyrin materials show similar enhancements of non-linear absorption (vs. porphyrins alone) to those found in films.12 However, so far third order NLO properties have mostly only been studied in POM hybrid systems where POM-organic electronic communication is intrinsically weak, as there is either no covalent linkage, or an aliphatic bridge. The only prior study of an arylimido-POM investigated overall non-linear absorption of a porphyrin-derivatised arylimido-Lindqvist ([ArNMo6O18]2−) system with a nanosecond laser source,12b thus including longer time-domain effects such as excited state absorption as well as simultaneous TPA.
Here, we present the synthesis and TPA cross sections of several centrosymmetric and C2v aryl bis-[imido Lindqvist] hexamolybdate derivatives, along with those of previously published dipolar derivatives featuring carbazole and diphenylamino groups. These first TPA measurements on arylimido-polyoxometalates show that the imido-POM acceptor unit is effective for use in 3rd order, as well as second order NLO, yielding π-electron normalised TPA cross sections in dipolar D–π–A and centrosymmetric A–π–A structures that are comparable to efficient organic materials. DFT/TD-DFT calculations suggest this is consequence of strong electronic communication between the POMs and organic groups, which also leads to electrochemically-observed Class II mixed valence behaviour, while methylation ortho to the imido-groups reveals a subtle interplay of through-space and through-bond communication effects.
Experimental
Materials and procedures
Acetonitrile (MeCN) was freshly distilled under nitrogen from calcium hydride.13 Dry (sure seal) dimethyl sulfoxide (DMSO) was purchased from Sigma Aldrich. All other reagents and solvents were obtained as ACS grade from Sigma Aldrich, Alfa Aesar or Fisher Scientific and used as supplied. Tetrabutylammonium hexamolybdate14 was synthesized according to previously published methods, as were the dipolar hexamolybdate derivatives 1 to 3.9b,c Other organoimido hexamolybdate derivatives were synthesised using an adapted literature procedure,15 under an atmosphere of dry nitrogen using standard Schlenk techniques, full details are given in the ESI.†General physical measurements
FT-IR spectra were measured using PerkinElmer FT-IR spectrum BX and Bruker FT-IR XSA spectrometers. NMR spectra were acquired using a Bruker Ascend 500 (500 MHz) spectrometer and all shifts are quoted with respect to TMS using the solvent signals as a secondary standard (s = singlet, d = doublet, t = triplet, q = quartet, sex = sextet, dt = doublet of triplets, m = multiplet). Elemental analyses and accurate mass spectrometry were outsourced to London Metropolitan University, and the UK National Mass Spectrometry Service at Swansea University respectively. UV-Vis spectra were obtained by using an Agilent Cary 60 UV-Vis spectrophotometer. Cyclic voltammetric measurements were carried out using Autolab PGStat 30 potentiostat/galvanostat. A single-compartment or a conventional three-electrode cell was used with a silver/silver chloride reference electrode (3M NaCl, saturated AgCl), glassy carbon or platinum working electrode and Pt wire auxiliary electrode. Acetonitrile was freshly distilled (from CaH2) and [N(C4H9-n)4]BF4,4 was used as the supporting electrolyte. Solutions containing ca. 10−3 M analyte (0.1 M electrolyte) were degassed by purging with nitrogen. All E1/2 values were calculated from (Epa + Epc)/2 at a scan rate of 100 mV s−1 and referenced to Fc/Fc+.Z-Scan measurements
The two-photon absorption cross sections were measured at 806 nm by using open-aperture Z-scan method16 with a femtosecond mode-locked Ti:sapphire laser (Coherent, Chameleon Ultra II: pulse duration: 140 fs; repetition rate: 80 MHz; wavelength range: 680–1080 nm). After passing through a beam expander (×4), the laser beam was focused using an f = 15 cm lens and passed through a quartz cell (1 mm optical path length). The position of the sample cell was varied along the laser-beam direction (z-axis) using a Z-step motorized stage controlled by a computer. At constant incident excitation, the local power density within the sample is changed, and the corresponding transmitted laser beam, T(z), recorded with a silicon photodetector (Ophir PD300), is monitored in connection with the z-position of the cell. The on-axis peak intensity of the incident pulses at the focal point, I0, ranged from 2 to 5 GW cm−2. If we assume that the linear absorption of the sample is negligible at the working wavelength and that the laser exhibits a Gaussian beam profile in space and time, the nonlinear absorption coefficient β can be calculated from the curve fitting to the experimental transmittance with the following equation: | (1) |
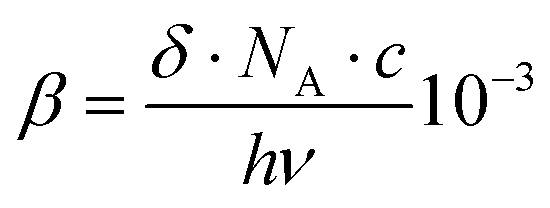 | (2) |
X-ray crystallography
Crystals of 4·2MeCN were obtained by room temperature vapour diffusion of diethyl ether into a solution of 4 in acetonitrile. Structures of 1 to 3 have been previously published.9b,c Data were collected on an Oxford Diffraction XCalibur 3 diffractometer, data reduction, cell refinement and absorption correction were carried out using Agilent Technologies CrysAlisPro,18 and the structure was solved using SHELXS-201419via WinGX.20 Refinement was achieved by full-matrix least-squares on all F02 data using SHELXL-201421 and molecular graphics were prepared using Mercury 3.8.22 None of the bis-polyoxometalate compounds obtained crystallise well, multiple attempts were made with all four compounds and dataset presented for 4·2MeCN used was best obtained. Evidence of twinning was seen in the images obtained, but twin data reduction and twin refinement were not successful, resulting in significant excess electron density on the Mo atoms in the final structure. Support provided by other data (mass spectra, elemental analysis) means that the identity of these atoms is not in question and the obtained structure provides a useful confirmation of connectivity. Restraints were applied to the thermal parameters of atoms in phenyl group, plus several positions on the {Mo6} units and tetrabutylammonium cations. Full crystallographic data and refinement details are presented in Table S1 (ESI†) and an ORTEP representation of the asymmetric unit is provided in Fig. S1 (ESI†).Computational methods
DFT and TD-DFT calculations were carried out using the Gaussian16 suite23 on the UEA high performance cluster for geometry optimisations and linear light absorption properties, and GAMESS24 on a dedicated 8-core workstation for TPA cross sections. Geometry optimisations were performed using the range separated, Grimme D2 dispersion corrected hybrid functional ωB97XD with the 6-311+G(d,p) basis set for C, H, N, O, and the LANL2DZ basis set for Mo atoms, as this approach was found to best reproduce experiment (Table S1, ESI†).25 These were carried out first in the gas phase, and subsequently with acetonitrile solvent introduced using Truhlar and coworkers’ SMD solvation model.26 TIGHT convergence thresholds on the residual forces on the atoms (1.5 × 10−5 Hartree/Bohr or Hartree/radian) were applied. For linear light absorption we used the same ωB97XD/6-311+G(d,p)/LANL2DZ level of theory, with the SMD model for acetonitrile solvent. The 200 lowest energy transitions were calculated using a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mix of singlet and triplet transitions, but only the singlet absorptions proved significant. For calculations of two-photon absorption properties we used GAMESS with the range-separated CAM-B3LYP functional and Huzinaga's 3 Gaussian minimal basis set (MINI) with addition of one polarization function on heavy elements. Solvation (acetonitrile) was again applied using the SMD model.
:50 mix of singlet and triplet transitions, but only the singlet absorptions proved significant. For calculations of two-photon absorption properties we used GAMESS with the range-separated CAM-B3LYP functional and Huzinaga's 3 Gaussian minimal basis set (MINI) with addition of one polarization function on heavy elements. Solvation (acetonitrile) was again applied using the SMD model.
Assuming linearly polarized light, the rotationally averaged TPA probabilities in atomic units can be obtained from the TPA transition moment S:
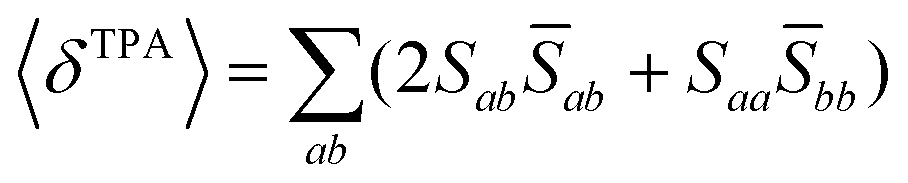 | (3) |
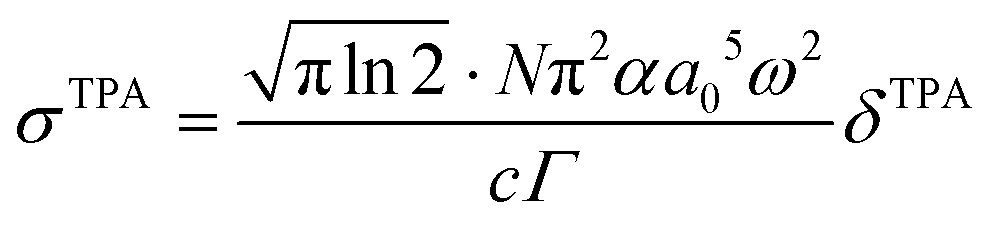 | (4) |
 are both applied when compared to Z-scan single beam experiments8,27 and Γ is the lifetime Gaussian broadening functions (= 0.1 eV), as previously employed28 for comparison with experiment. The macroscopic TPA cross section in GM (Göppert–Mayer units) units were obtained from the calculated values in cgs units from eqn (2) multiplied by 105 as previously reported.29
are both applied when compared to Z-scan single beam experiments8,27 and Γ is the lifetime Gaussian broadening functions (= 0.1 eV), as previously employed28 for comparison with experiment. The macroscopic TPA cross section in GM (Göppert–Mayer units) units were obtained from the calculated values in cgs units from eqn (2) multiplied by 105 as previously reported.29
Results and discussion
Molecular design, synthesis and structure
Our synthetic approach to hexamolybdate derivatives is based on the DCC-mediated coupling of anilines and other amines with (n-Bu4N)2[Mo6O19],30 although in our hands dimethylsulfoxide has proved a more reliable solvent for this chemistry than the commonly used acetonitrile.9 Of the compounds presented in Scheme 1, three are new (5 to 7). Of the others, 1 to 3 were previously published by us, and 4 by Wei and co-workers.31 The series is designed to test the effect of centrosymmetric, vs. C2v geometry, and the influence of electron donors, on the linear and two-photon absorption properties of the compounds. Moreover, we wished to test steric demand (provided by the methyl groups of 5 and 7, compared to protons in equivalent positions in 4 and 6) as a means to protect against hydrolysis of the imido-bond – a problem for some hexamolybdate imido derivatives, notably 2.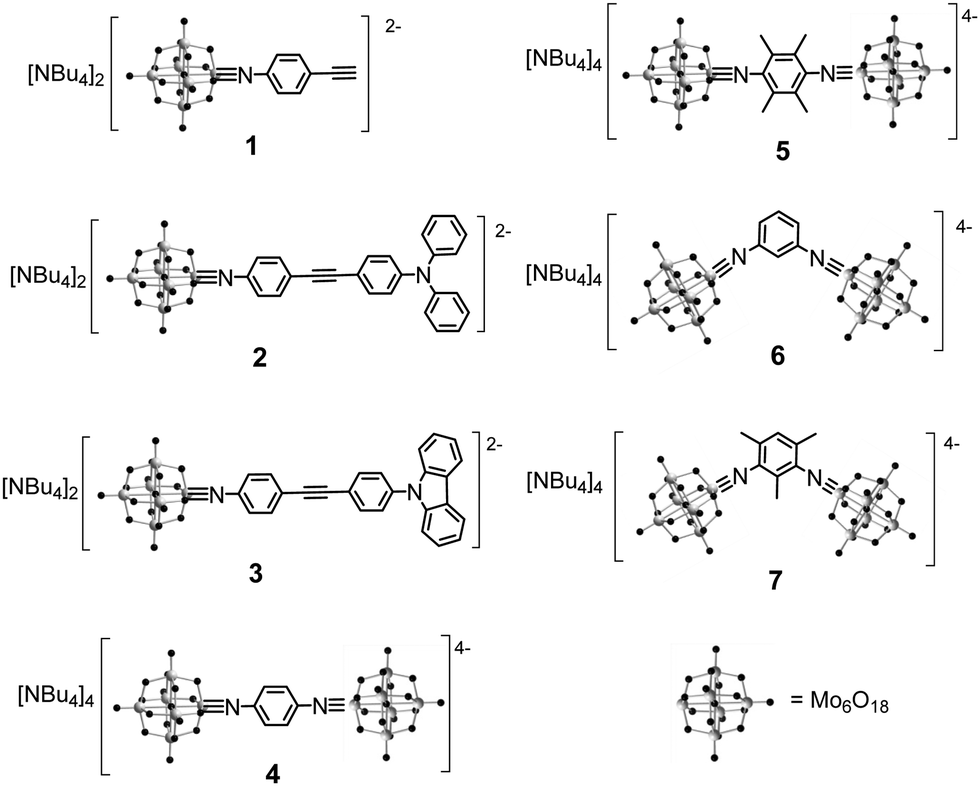 | ||
| Scheme 1 Hexamolybdate derivatives 1 to 7 included in this study. | ||
Crystallisation of 4 to 7 proved much more challenging than for mono derivatives 1 to 3,9 presumably because their higher charge leads to rapid precipitation from organic media. Nonetheless, crystals of 4 were obtained suitable for X-ray diffraction and the resulting structure is presented in Fig. 1 – notably, a structure of this known material was not published before. It reveals the expected molecular “dumbbell” structure with two POMs bridged by the di-imido phenyl ligand. The Mo–N bond distance (1.71(1) Å) is within the range typically observed for imido-molybdate species, and although the C–N–Mo bond angle (161(1)°) is a little further from 180° than we have typically observed in dipolar systems such as 1 to 3,9 it is still linear enough to indicate significant Mo–N triple bond character. Consistent with the centrosymmetric structure, the phenyl group shows no sign of polarisation (such as the quinoidal structure evident in the phenyl groups of 1), however the {Mo6} units do show the typical imido-Lindqvist pattern of a shortened bond length from the imido-Mo to the central oxygen, lengthened equatorial bond lengths from the imido-Mo to the bridging oxygens, and a lengthened axial bond length from the Mo trans to the imido to the central oxygen.32
 | ||
| Fig. 1 X-ray crystal structure of the molecular anion in 4·2MeCN. Thermal ellipsoids are at the 30% probability level; C atoms are grey; N, purple; Mo, cyan; O, red; H atoms are white spheres of arbitrary radii. | ||
Electronic absorption spectra
UV-vis spectra of the centrosymmetric and C2v compounds 4 to 7 are shown in Fig. 2, while those of the dipolar compounds 1 to 3 are displayed in Fig. S2 (ESI†) and the data are summarised in Table 1. Two main types of band are observed – high energy bands between 200 and 300 nm associated with ligand π → π* and O → Mo transitions, and a lower energy band (350 to 450 nm) we assign as an intra-hybrid charge transfer (IHCT) band.9b Depending on the nature of the organic group (e.g. presence of resonance donor or acceptor substituents) and geometry this band may have contributions from charge transfer both towards and away from the POM, and what are essentially π → π* transitions perturbed and lowered in energy by participation of the POM orbitals. Introduction of a strong organic donor, such as the diphenylamino unit in 3, leads to dominant ligand-to-POM charge transfer (LPCT) and the pronounced red shift that is observed between phenylethynyl derivative 1, and 3.9 TD-DFT calculations (vide infra) confirm that the lower energy bands of 4 to 7 all involve transitions from the imido-aryl groups, to orbitals that either have significant POM character or are entirely POM based, indicating that they are also LPCT in nature.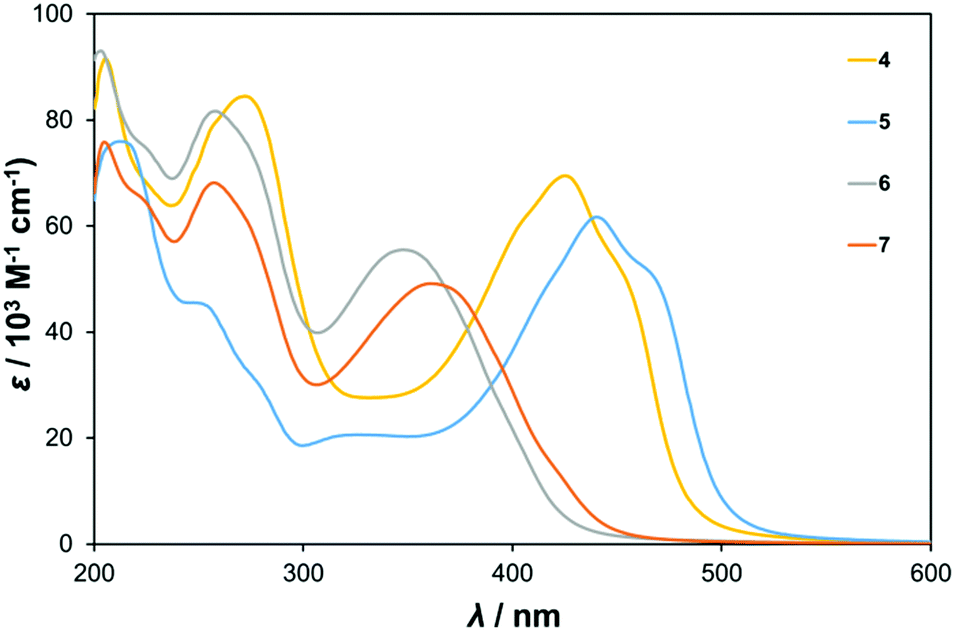 | ||
| Fig. 2 UV-vis absorption spectra of centrosymmetric bis-hexamolybdate derivatives 4 and 5, and C2v6 and 7. | ||
| λ max/nma (ε, 103 M−1 cm−1) | E max (eV) | Assignment | E 1/2/V (ΔE/mV)bvs. Ag/AgCl | |
|---|---|---|---|---|
| a Concentrations ca. 10−5 M in MeCN. b Solutions ca. 10−3 M in analyte and 0.1 M in [NBu4][BF4] at a glassy carbon working electrode with a scan rate of 100 mV s−1. c Wave consists of two closely spaced one-electron reductions, as shown by differential pulsed voltammetry. Ferrocene internal reference E1/2 = 0.46 V, ΔEp = 80 mV. Data for 1 are from ref. 9b and for 2 and 3 from ref. 9c. | ||||
| 1 | 264 (36.2) | 4.69 | O → Mo and π → π* | −0.499 (61) |
| 358 (27.0) | 3.46 | IHCT | ||
| 2 | 292 (41.2) | 4.28 | O → Mo and π → π* | −0.503 (64) |
| 327 (36.2) | 3.74 | O → Mo and π → π* | ||
| 414 (45.3) | 2.98 | IHCT | ||
| 3 | 227 (70.4) | 5.28 | O → Mo and π → π* | −0.503 (64) |
| 292 (48.4) | 4.25 | O → Mo and π → π* | ||
| 327 (31.3) | 3.78 | O → Mo and π → π* | ||
| 341 (33.7) | 3.63 | O → Mo and π → π* | ||
| 385 (43.4) | 3.20 | IHCT | ||
| 4 | 205 (91.6) | 6.05 | O → Mo and π → π* | −0.498 (117)c |
| 272 (84.6) | 4.56 | O → Mo and π → π* | ||
| 425 (69.5) | 2.92 | IHCT | ||
| 5 | 214 (75.7) | 5.79 | O → Mo and π → π* | −0.517 (132)c |
| 254 (45.0) | 4.88 | O → Mo and π → π* | ||
| 440 (61.7) | 2.82 | IHCT | ||
| 6 | 203 (93.0) | 6.11 | O → Mo and π → π* | −0.526 (134)c |
| 258 (81.6) | 4.81 | O → Mo and π → π* | ||
| 348 (55.5) | 3.56 | IHCT | ||
| 7 | 205 (75.8) | 6.05 | O → Mo and π → π* | −0.562 (123)c |
| 257 (68.0) | 4.82 | O → Mo and π → π* | ||
| 361 (49.1) | 3.43 | IHCT | ||
The spectra of the C2v compounds 6 and 7 are essentially very similar to that of 1. This is because all three are based on small π systems with no strong electron donor, and no electron acceptor other than the POMs. The IHCT λmax is found at 348 or 361 nm respectively, but is around twice as intense in the bis-POM systems 6 and 7 as in 1. This is consistent with involvement of the POM in the transition. The red shift observed on moving from 6, to the trimethylated analogue 7, is consistent with a HOMO level with strong participation from the phenyl group being raised in energy by the inductive electron donating methyls. Remarkably, the centrosymmetric “dumbell” derivatives 4 and 5 show by far the strongest, and longest wavelength absorption. The IHCT bands of these systems have λmax values of 425 and 440 nm respectively, over 50 nm to the red of 1 and their C2v analogues. In the absence of strong electron donors, this can be ascribed as consequence of better inter-acceptor (POM) communication in the 1,4-geometry, and their centrosymmetry, which leads to an intermediate 1B1u state with an allowed transition from the 1Ag ground state – of lower energy than the 1A → 2A transition of comparable dipolar systems.8d
UV-vis spectroscopy also enabled investigation of the effect of introducing steric bulk around the imido bond on its hydrolytic stability. The Mo–N bond can hydrolyse yielding the [Mo6O19]2− cluster and the parent amine, and many literature {Mo6} imido compounds feature methyl or isopropyl groups adjacent to the imido bond.30,32 Yet, neither quantitative nor qualitative information exists in the literature to indicate how such structural modifications can influence the vulnerability of organoimido-polyoxometalates to hydrolysis. Thus, Fig. S3 (ESI†) shows the absorbance decline of ca. 10−5 M solutions of compounds 4 (with protons in the ortho and meta positions) and 5 in kept in the dark in acetonitrile after deliberate addition of a large excess of water, monitored at the IHCT band λmax. As neither [Mo6O19]2− nor the resulting anilines have any significant absorption at the IHCT λmax, the absorbance loss at this wavelength is directly proportional to the extent of hydrolysis of the imido groups. For 4, around 25% of the absorbance is lost over the course of 10 hours, of which 10% is lost within the first hour. For 5, the loss is <3% over 10 hours, of which nearly all occurs in the first hour. This indicates a substantial stabilising influence, even for the relatively small methyl group, in return for only modest changes (red shift, decreased ε) to the optical properties.
Electrochemistry
The hexamolybdate anion, [Mo6O19]2− has a reversible one electron reduction at −0.32 V vs. Ag/AgCl under the conditions used here. The mono-{Mo6} species 1 to 3 all show a similar reduction,9 shifted negative by around 180 mV, as electron donation from the arylimido unit to the Mo d-orbitals makes the POM a weaker acceptor. The influence of arylacetylene donor ring systems connected to the 4-position of the imidophenyl group in these systems on the POM reductions is very weak, as 1 to 3 have reduction potentials within 5 mV of one another. At first glance, addition of a second, electron accepting imido-{Mo6} in the 3- or 4-position (i.e. in 4 to 7) also seems to have little effect on the reduction potential vs.1 to 3 (Table 1). However, this is inconsistent with prior observations that π-donor or acceptor substituents (–NMe2, –NO2) directly connected at the 4-position exert a large effect (±70 mV) on the POM reduction potential,9b and there is a noticeable shift to more negative potential in the 1,3-systems – particularly when comparing 5vs.7 (Fig. S4, ESI†). Close inspection reveals that all four bis-POM systems have a much larger ΔEp (>110 mV) than the near ideal one electron couples of 1 to 3 (ΔEp = 61–64 mV). Current responses with scan rate, and independence of the peak separation from scan rate, suggested that the observed waves consist of two overlapping, reversible one electron reductions at different potentials – that is reduction of one {Mo6} unit makes reduction of the second one more difficult. Differential pulsed voltammetry (DPV) indeed uncovers two processes for each of 4 to 7, separated by 73–82 mV from one another (Table 2 and Fig. S5–S8, ESI†). This shows that for 4 the first reduction (E1) shifts around 90 mV more positive than those of 1 to 3, a similar effect to that previously observed for a para-nitro substituent (values in Table 2 are Ep not E1/2). Introducing methyl groups in 5 produces the expected, small negative shift in E1 as the aryl group becomes more electron rich. The E1s of the 1,3 systems are less positively shifted vs. the mono-POM compounds, consistent with weaker through-bond communication in this geometry, but reveal a stronger effect for the methyl groups. This stronger effect of methylation in the 1,3 geometry may be explained through DFT-optimised geometries (vide infra) that suggest possible C–H⋯O hydrogen bonding interactions between methyl groups and POMs. These lead to a shortening of POM⋯POM distances in both methylated structures (Table 4), with the effect being much more substantial in 6/7 (0.15 Å centre-to-centre, 0.621 Å closest O⋯O) than in 4/5 (0.03 Å centre-to-centre, 0.03 Å closest O⋯O). This would increase repulsion between the added electron and the negative charge on the second POM group.As well as delineating trends in the first reduction potential, the DPV provides a direct means of assessing the level of communication between the POMs through the separation between the two reductive processes. This communication is quantified by comproportionation constant (Kcom) derived from the difference in the redox potentials (ΔE), such that Kcom = e(ΔE·F/RT) for one electron processes.33a,bKcom allows assignment of the mixed-valence system according to the Robin-Day classification, where non-communicating redox centres are Class I (Kcom < 4), complete delocalisation is Class III (Kcom > 106) and intermediate communication is Class II.33 While this classification is simple, it should be noted that other factors such as solvation, ion-pairing, and intra-molecular electrostatic effects can influence Kcom as well as through-bond communication.33c For 4 to 7, ΔE values fall into a narrow (<9 mV range), yielding Kcom values (17–25) consistent with weakly coupled Class II mixed valence behaviour,33 and similar to values obtained for the 1,4-dicyanobenzene bridge in bis-ruthenium pentammine complexes.34 Comparing 1,4 and 1,3 geometries (4vs.6, 5vs.7) shows that communication is stronger for 1,4-consistent with expectations and the observation that the bridge participates in the LUMO for this geometry, but not for the 1,3 geometry. However, the difference is very small, particularly when the methyl groups are present, such that Kcom for 7 slightly exceeds that of 4. This may be explained by increased electron–electron repulsion when the two POM acceptors are closer in the 1,3-geometry, which increases with methylation as the POMs are forced closer together. Moreover, short C–H⋯O contacts consistent with hydrogen bonding in the calculated structure of 7 may provide another means for charge on one POM to influence the bridge, and thus feed through to the second POM.
Two-photon absorption measurements
Measurement of third order NLO effects with nanosecond laser sources, for both electrostatic35 and aliphatically bridged12a POM hybrids have previously shown significant enhancements in non-linear absorption (1.1× to 2.8×) resulting from introduction of the POM. Due to a dependence on the LUMO level of the POM, these effects were ascribed to electron transfer to the POM – a longer time domain process consistent with non-simultaneous absorption of the second photon as permitted by ns laser pulses. Ultrafast (ps) through-space electron transfer from excited dyes to POMs, and similarly rapid electron transfer from reduced POMs to excited or oxidised dyes has been previously documented.36 Thus, the non-linear absorption in these systems likely results from a combination of excited state absorption, absorption by the dye oxidised state and POM reduced states (which often have broad absorptions between 500 nm and the near-IR), and potentially even dye regenerated or reduced by electron transfer from the reduced POM, rather than enhancement of simultaneous TPA. However, a greater enhancement of non-linear absorption (4.5×) measured by a ns source has been described for tetraphenylporphyrin derivatised with imido-Lindqvist units,12b potentially indicating that the strong electronic coupling provided by the imido linkage also enhances simultaneous (intrinsic) TPA.Thus, to investigate intrinsic TPA in 1 to 7, we have obtained TPA cross sections by Z-scan measurements with a femtosecond 806 nm source (Table 3). Fig. S9–S10 (ESI†) display some typical z-scan traces of POM compounds, the linear correlation between the variation of transmission (ΔT) and the incident power confirms the two-photon absorption regime.37 The values of TPA cross sections are modest compared to very strong two-photon absorbers (which can exceed 104 GM),8d but relatively small numbers of π-electrons are involved – and the cross sections normalized for the number of effective π-electrons38 (δ/Neff2) are broadly comparable to efficient organic and metallo-organic two-photon absorbers.39 Notably, the highest normalized values are obtained for the centrosymmetric A–π–A, bis-POM derivatives 4 and 5 – whose δ values approach those of dipolar 2 and 3 despite much smaller organic π-systems (lower Neff), while mono-POM 1 with the same Neff produces no appreciable TPA cross section (likely due to the relative lack of CT character in mono-POM derivatives with no resonance electron donor9b). This clearly implies that the POMs play a role in the third order NLO properties of these compounds, as should be expected from the role TD-DFT (vide infra) shows that they play in the electronic transitions, and indicates that they are effective acceptors for use in TPA. But, unlike our observations for second order NLO,9 it does not impute a general advantage to imido-POMs compared to efficient organic donor or acceptor moieties. This is because, based on known theory and structure/activity relationships for organic chromophores, the centrosymmetric geometry of 4 and 5 should be expected to yield more effective two photon absorbers than dipolar or C2v systems. Indeed, similar or higher δ/Neff2 values than those of 4 and 5 can be obtained for stilbene bridged organic D–π–D structures,38 and stilbene bridged dipolar organics have shown higher δ/Neff2 than 2 and 3.39b Moreover, this comparison does not take account of the steric bulk and mass of the POM, which would be relevant considerations for the efficiency of any bulk TPA material.
| Compound | ε 403/103 M−1 cm−1 | δ 806/GM | δ 806/Neff2 |
|---|---|---|---|
| a Non-linear absorbance too weak for accurate measurement. | |||
| 1 | 10.2 | — | — |
| 2 | 43.6 | 112 | 0.34 |
| 3 | 38.4 | 70 | 0.18 |
| 4 | 60.1 | 82 | 0.82 |
| 5 | 38.8 | 58 | 0.58 |
| 6 | 19.2 | 20 | 0.20 |
| 7 | 26.1 | 26 | 0.26 |
| Compound | Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–C N–C |
Mo–N | N–C | Mo–Oca | Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
POM⋯POMb (Oc⋯Oc) | POM⋯POMc (closest) | C(H)⋯Od | H⋯Oc | C–H⋯Oc |
|---|---|---|---|---|---|---|---|---|---|---|
| a Central oxygen. b Distance between central oxygens of the two POMs. c Shortest O⋯O distance between the two POMs. d Average of short (C⋯O <3.6 Å) C–H⋯O interactions in calculated structures. | ||||||||||
| 4 | 172 | 1.74 | 1.37 | 2.23 | 1.70 | 13.44 | 9.835 | — | — | — |
| 5 | 176.6 | 1.73 | 1.37 | 2.22 | 1.70 | 13.41 | 9.789 | 3.487 | 2.402 | 173.39 |
| 6 | 158.1 | 1.74 | 1.38 | 2.21 | 1.70 | 12.72 | 9.079 | 3.348 | 2.474 | 136.85 |
| 7 | 167.6 | 1.73 | 1.38 | 2.22 | 1.70 | 12.57 | 8.458 | 3.277 | 2.407 | 135.44 |
Within the bis-POM compounds, contrasting effects are seen for the methylated derivatives – a decrease in δ upon methylation in the centrosymmetric 4 and 5 and an increase in C2v6 and 7. This is likely a result of the effect of shifts in the TPA allowed resonance wavelengths relative to the 403 nm second harmonic (SH) wavelength, induced by the more electron rich phenyl rings – it is notable that the change in δ approximately tracks the change in the one photon extinction coefficient at 403 nm. The difference in TPA cross sections observed between C2v and centrosymmetric systems is likely also related to the position of the TPA allowed resonance wavelengths, vs. the SH: for C2v6 and 7 the S0 → S1 transition is TPA allowed, and thus the TPA resonance wavelength should correspond to the linear absorption spectrum λmax (ca. 50 nm to the blue of the second harmonic), whereas for centrosymmetric 4 and 5 S0 → S1 is TPA forbidden and the TPA allowed S0 → S2 resonance will be accessible to the blue of the one-photon λmax, and thus potentially quite close to the 403 nm SH wavelength.
Computational study
Accurate calculations of both linear light absorption, and non-linear optical properties can be challenging for polyoxometalate derivatives, but recent work from Champagne and co-workers has shown that excellent results can be achieved for linear absorption and second-order non-linear optical properties (frequency doubling, β) using range-separated hybrid functionals.25 Accordingly, testing a number of functional/basis set combinations (Table S2, ESI†) on dumbbell compound 4 revealed that ωB97XD/6-311+G(d,p)/LanL2DZ produced the best match to the crystallographically obtained bond lengths and angles. Replacing LanL2DZ with the larger LanL2TZ basis set was computationally expensive, and in fact slightly worsened the match on the MoN–C angle. Extending the approach to 2, 3 and 5 to 7 also produced geometries consistent with known imido-species (Fig. 3 and Fig. S11, Table 4 and Table S3, ESI†) with Mo–N bond lengths of 1.73 to 1.79 Å and Mo–N–C angles of between 154 and 177°. The lower (more bent) end of this range is occupied by the dipolar and C2v derivatives 2, 3, 6 and 7.
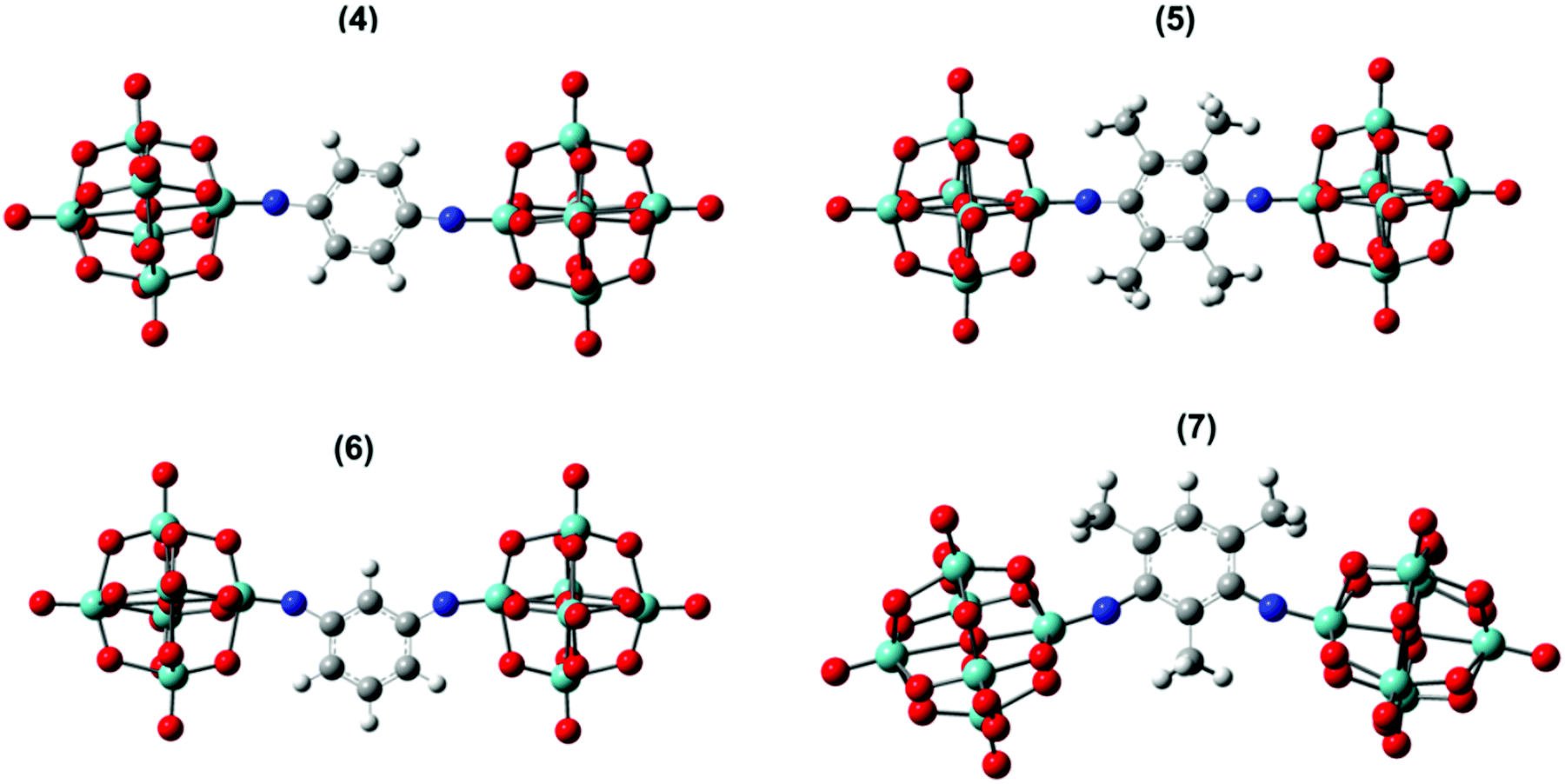 | ||
| Fig. 3 Solvent phase optimized geometry of 4–7 anions. Grey, bayside, white, blue, and red colours represent C, Mo, H, N, and O atoms respectively. | ||
Calculation of UV-vis absorption properties (Gaussian, ωB97X-D) produced a much better match to experimental spectra than prior calculations for dipolar 2 and 3 (Table S4, ESI†) using the SAOP model functional,9c but show a very similar picture of the MO-to-MO transitions (Fig. S12 and S13, ESI†). For the centrosymmetric and C2v systems, the match to experimental spectra is excellent (Table 5) – comparing the strongest calculated transition (based on oscillator strength) with the experimental λmax indicates an overestimate of the transition energy of only around 0.2 eV for the centrosymmetric dumbbells 4 and 5, and a 0.05 eV overestimate to 0.1 eV underestimate for the C2v systems 6 and 7. The overall pattern of the transition energies relative to one another is consistent with experiment, and expectations based on the geometries and the presence of the methyl groups in 5 and 7, which raise the level of the aryl-based LUMOs leading to a smaller HOMO–LUMO gap. For all four compounds, the HOMO/HOMO-1 involved in the transitions spreads across the entire arylimido unit, and also has some POM character (Fig. 4, 5 and Fig. S14, S15, ESI†). The LUMO and LUMO+x acceptor orbitals are more POM-based in all cases, although a significant number also involve the π-bridge. This clear involvement of the POM as an acceptor in the electronic transitions, and presence of hybrid delocalized orbitals with participation from both POM and organic group, implies that the origin of TPA by the POM derivatives is the introduction of charge transfer character and the extension of the CT transitions onto the POM. The lack of TPA activity in 1 most likely results from the relative lack of CT character in its transitions, as shown previously by computation and electroabsorption spectroscopy.9b
| Compound | Experimental | Calculated | |||||
|---|---|---|---|---|---|---|---|
| λ max/nm | E max/eV | λ max/nm | E max/eV | Oscillator strength (fos) | Transition | Contr. % | |
| 4 | 425 | 2.917 | 415 | 2.985 | 0.0144 | HOMO → LUMO+1 | 25 |
| HOMO → LUMO+2 | 14 | ||||||
| HOMO → LUMO+5 | 8 | ||||||
| HOMO → LUMO+8 | 7 | ||||||
| 414 | 2.996 | 0.0214 | HOMO → LUMO+1 | 22 | |||
| HOMO → LUMO+2 | 14 | ||||||
| HOMO → LUMO+5 | 5 | ||||||
| HOMO → LUMO+3 | 2 | ||||||
| 399 | 3.108 | 1.8896 | HOMO → LUMO | 76 | |||
| HOMO → LUMO+16 | 4 | ||||||
| HOMO → LUMO+8 | 3 | ||||||
| 5 | 440 | 2.818 | 446 | 2.778 | 0.0178 | HOMO → LUMO | 32 |
| HOMO → LUMO+4 | 14 | ||||||
| HOMO → LUMO+2 | 9 | ||||||
| 409 | 3.033 | 1.8028 | HOMO → LUMO+2 | 68 | |||
| HOMO → LUMO+4 | 7 | ||||||
| HOMO → LUMO+15 | 6 | ||||||
| HOMO → LUMO | 3 | ||||||
| 6 | 348 | 3.563 | 359 | 3.454 | 0.0429 | HOMO → LUMO+10 | 14 |
| HOMO → LUMO+3 | 10 | ||||||
| HOMO-1 → LUMO+2 | 5 | ||||||
| HOMO → LUMO | 2 | ||||||
| 343 | 3.620 | 1.2651 | HOMO → LUMO+9 | 24 | |||
| HOMO → LUMO+16 | 10 | ||||||
| HOMO-1 → LUMO+8 | 9 | ||||||
| HOMO → LUMO+10 | 5 | ||||||
| HOMO-1 → LUMO+2 | 4 | ||||||
| 7 | 361 | 3.434 | 367 | 3.383 | 1.3606 | HOMO → LUMO+17 | 14 |
| HOMO → LUMO+6 | 10 | ||||||
| HOMO → LUMO+7 | 8 | ||||||
| HOMO → LUMO+3 | 6 | ||||||
| HOMO-1 → LUMO+7 | 3 | ||||||
| 362 | 3.423 | 0.1185 | HOMO → LUMO+16 | 13 | |||
| HOMO → LUMO+7 | 12 | ||||||
| HOMO → LUMO+2 | 8 | ||||||
| HOMO → LUMO | 2 | ||||||
| HOMO → LUMO+3 | 2 | ||||||
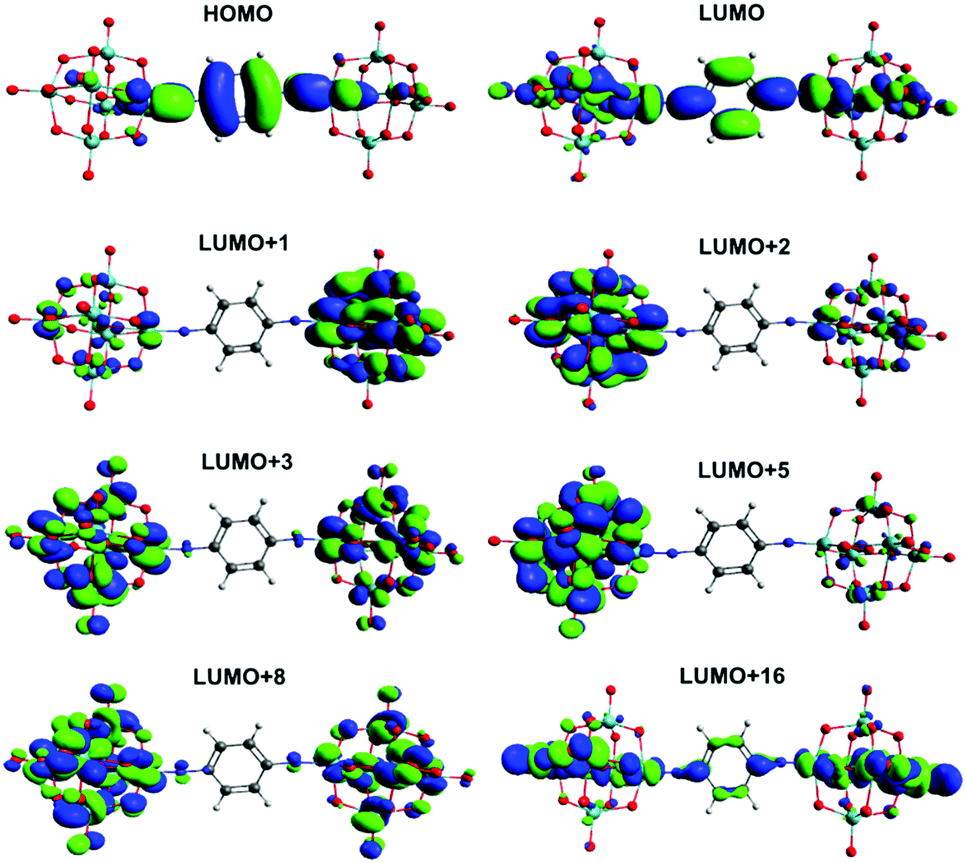 | ||
| Fig. 4 Solution phase frontier orbitals involved in significant UV-vis transitions of 4. | ||
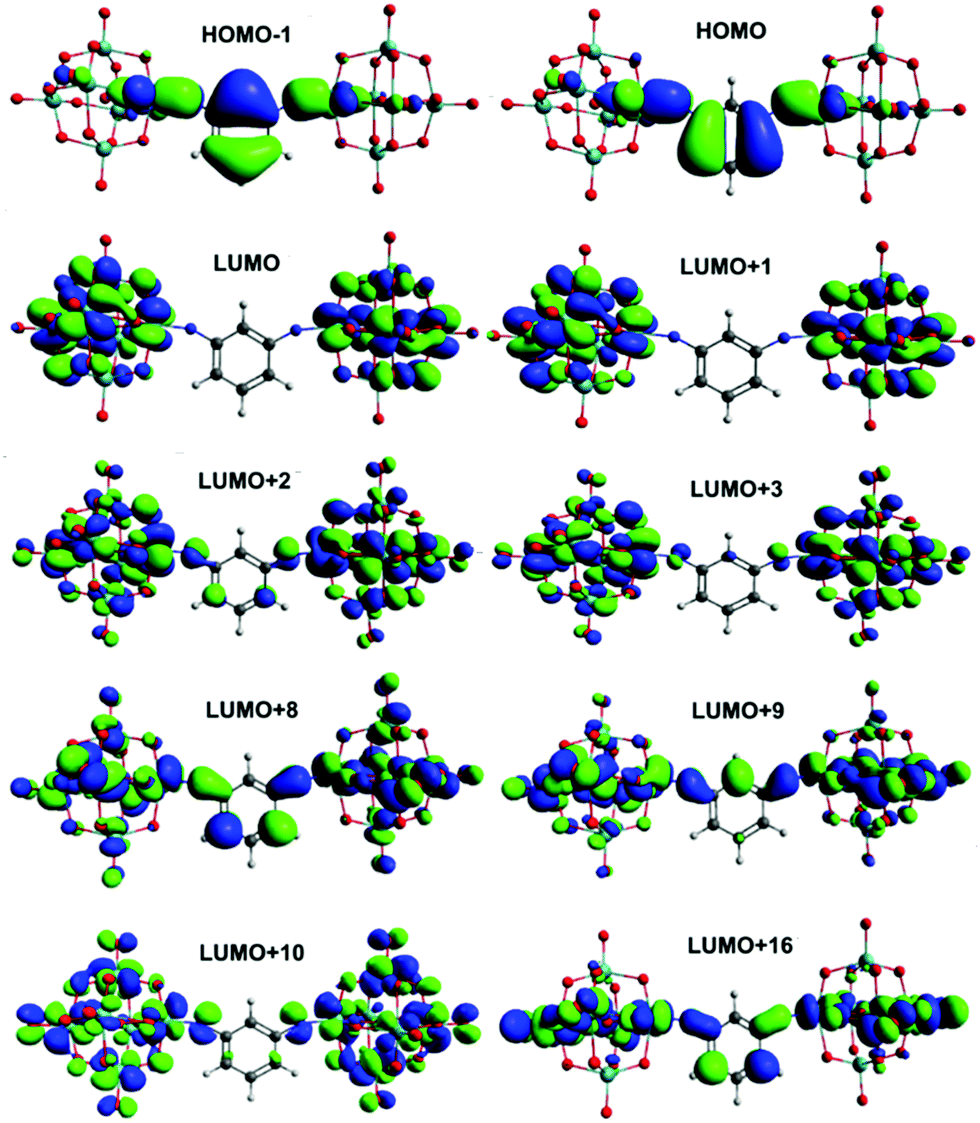 | ||
| Fig. 5 Solution phase frontier orbitals involved in significant UV-vis transitions of 6. | ||
There is very little change between the two centrosymmetric dumbbells 4 and 5 in the character of the HOMO and LUMO, and the various LUMO+x orbitals implicated in the transitions. In both cases the LUMO involves the π* orbitals of the bridge, the imido group and also d-orbitals in the equatorial plane as projected (Fig. 4, Fig. S14, ESI†), and all of the LUMO+x orbitals are entirely POM based. However, there is a switch in the main contributor to the strongest transition (3.11 eV in 4, 3.03 eV in 5), which is HOMO → LUMO in 4 and HOMO → LUMO+2 in 5. Effectively, this means that the amount of POM character increases in the transitions of 5 and there is a higher degree of charge separation for this compound. This may be a result of electron donation from the methyl groups, which raises the energy of bridge-based orbitals more than POM-based orbitals, and C–H⋯O hydrogen bonds suggested by the short C⋯O distances and near 180° C–H⋯O angles calculated for this compound. These may help enforce the observed more linear MoN–C bond and shorter POM–POM distance, while also having a slight stabilising effect on POM based orbitals – helping favour transitions to the POM.
In the C2v compounds the LUMO involves only POM orbitals, consistent with the more polar nature of these systems (Fig. 5, Fig. S15, ESI†). However, the HOMO → LUMO transition only makes a relatively small contribution to the light absorption properties which for both compounds are dominated by transitions from the HOMO and HOMO-1 to various LUMO+x. These all involve the aryl bridge but more strongly in the non-methylated compound 6, consistent with the result for the linear systems. The underlying reasons are likely similar: although potential short C–H⋯O interactions are present in both 6 and 7, 7 features more dramatic shortening of POM⋯POM distances than seen in the 4/5 pair, and methylation will have a similar effect on the energies of aryl orbitals as in the 1,4-systems. The reduced participation of the bridge in the acceptor orbitals of the transitions seen for the methylated systems is potentially significant for application of either geometry in charge separation, or of the C2v compounds as a basis for 2nd order non- linear optical chromophores, but does not seem to influence the third order NLO properties – the pattern in both 4vs.5 and 6vs.7 is for the TPA cross section to simply track the corresponding one photon cross section, which the red shift from 4 to 5 decreases, but the red shift from 6 to 7 increases.
Calculation of third order non-linear optical properties has previously only been carried out for a handful of polyoxometalate derivatives, using model potentials (LB94, SAOP, GRAC)40 or the B3LYP hybrid41 to obtain γ by finite differentiation from the DFT energy calculation, and no experimental data has been presented for comparison. Here, we have tested TD-DFT as means to compute the TPA properties of 4 to 7. Obtaining accurate calculations of TPA can be a challenge by TD-DFT,28,42 but reasonable success has been demonstrated for some organic chromophores39c,43 and it is far less computationally expensive than higher accuracy CC methods.42 As ωB97XD is not implemented for TD-DFT in GAMESS, we first calculated the linear absorption properties of compound 7 using CAM-B3LYP/6-311+G(d,p)/LanL2DZ, finding comparable results to those from Gaussian/ωB97X-D (Table S5, ESI†). TPA tensor elements, energies, probabilities and cross sections were then calculated using CAM-B3LYP and the MINI basis set with one added polarization function (Table 6). This basis set represents a very strong approximation, but larger basis sets proved too computationally heavy to be practical.
| 〈δTPA〉 | Exp. | Calculatedb | |||||
|---|---|---|---|---|---|---|---|
| δ 806/GM | S0 → S1 | S0 → S2 | |||||
| Energy/eVc | 〈δTPA〉d/a.u. | σ TPA/GM | Energyc/eV | 〈δTPA〉d/a.u. | σ TPA/GM | ||
| a Use of solvation and addition of the polarization function brought the calculated values significantly closer to experiment. b S0 → S3 was also calculated but cross sections were found to be vanishingly small. c Corresponds to 2ω (i.e. 2× the energy of photons absorbed in the TPA transition). d Rotationally averaged TPA strength. | |||||||
| 4 | 82 | 3.032 | 27.309 | 15 | 3.034 | 112.398 | 61.6 |
| 5 | 58 | 2.859 | 0.831 | 1.1 | 2.861 | 636.342 | 310 |
| 6 | 20 | 3.147 | 12.18 | 7.1 | 3.149 | 789 | 466 |
| 7 | 26 | 2.921 | 67.98 | 35 | 2.922 | 34.5 | 17.5 |
The computed σTPA values obtained for 4 and 7 are a reasonable match for experiment in terms of magnitude, but overall the computational results deviate widely from experimental trends. Notably, even for purely organic chromophores benchmarking studies of successful TD-DFT approaches have found substantial differences from experimental values as well as non-systematic deviations from experimental trends.43b In this study, the small basis set forced by the large systems, and comparison of cross-sections obtained from single-wavelength measurements with calculated values corresponding to TPA maxima can only increase the chance of such deviations. Moreover, geometry is a factor as DFT optimised geometries may not perfectly match solution averages, influencing the accessibility of the S0 → S1 and S0 → S2 transitions due to two photon selection rules (S0 → S2 is forbidden for centrosymmetric systems). Specifically, the methyl groups in 5 and 7 force much truer centrosymmetric and V-shaped geometries than seen for 4 and 5, and this is reflected in the computed S0 → S1 and S0 → S2σTPA values. Thus, considering its accessibility, TD-DFT appears to be viable as a means of calculating 3rd order NLO properties in POM derivatives. However, it is clear the approach needs refinement and optimisation, and a lot of computational power.
Conclusions
Three new, arylimido bridged bis-polyoxometalate species with centrosymmetric and C2v geometries have been synthesised and their electrochemical, linear absorption and two-photon absorption properties studied by experiment and computation in the context of a larger family of dipolar and centrosymmetric arylimido POMs. The results show communication between the POM units in all bis-POM species, resulting in formation of class II mixed valence species upon reduction of one POM. The polyoxometalate acceptor groups are able to produce significant two-photon absorption cross sections from small π-systems, and structure–activity relationships in the POM-derivatised materials are similar to those of purely organic TPA chromophores. Positioning methyl groups ortho- to the imido-linkages has a strong influence on POM–POM distances in the C2v systems, resulting in increased electrochemically observed through-space communication while through-bond communication seems to decrease. Methylation also strongly stabilises the imido-linkages against hydrolysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the UK EPSRC National Mass Spectrometry Facility (Swansea) for mass spectra. AAY thanks the Iraqi Government for financial support via the Ministry of Higher Education and Scientific Research (MOHER) – University of Kerbala for a postdoctoral fellowship. This work has been financially supported by the EPSRC through grants EP/R042675/1 and EP/M00452X/1, and the University of East Anglia. In addition to the supplementary information and deposited cif file, data can be accessed by contacting the corresponding author.Notes and references
- (a) Polyoxometalates: from Platonic Solids to Anti-Retroviral Activity, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1994 Search PubMed; (b) Polyoxometalate cluster science (special issue), ed. L. Cronin and A. Müller, Chem. Soc. Rev., 2012, vol. 41, issue 22 Search PubMed; (c) J. Fielden and L. Cronin, Coordination Clusters, in Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood and J. W. Steed, Taylor & Francis, London, 2007 Search PubMed.
- (a) Y. Ji, L. Huang, J. Hu, C. Streb and Y.-F. Song, Energy Environ. Sci., 2015, 8, 776 RSC; (b) D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615 RSC; (c) J. J. Walsh, A. M. Bond, R. Forster and T. E. Keys, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS; (d) D.-L. Long and L. Cronin, Chem. – Eur. J., 2006, 12, 3698 CrossRef CAS PubMed; (e) B. Fabre, Chem. Rev., 2016, 116, 4808 CrossRef PubMed.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed.
- (a) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed; (b) M. Sun, J. Zhang, P. Putaj, V. Caps, F. Lefebvre, J. Pelletier and J. M. Basset, Chem. Rev., 2014, 114, 981 CrossRef CAS PubMed.
- (a) C. L. Hill and D. A. Bouchard, J. Am. Chem. Soc., 1985, 107, 5148 CrossRef CAS; (b) E. Papaconstantinou, J. Chem. Soc., Chem. Commun., 1982, 12 RSC; (c) G. Marcì, E. I. García-López and L. Palmisano, Eur. J. Inorg. Chem., 2014, 21 CrossRef; (d) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232 CrossRef CAS PubMed.
- (a) A. Al-Yasari and J. Fielden, Rev. Adv. Sci. Eng, 2014, 3, 304 CrossRef; (b) C. Streb, Dalton Trans., 2012, 41, 1651 RSC; (c) J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120 RSC.
- (a) L. Yan, G. Yang, W. Guan, Z. Su and R. Wang, J. Phys. Chem. B, 2005, 109, 22332 CrossRef CAS PubMed; (b) K. J. Elliott, A. Harriman, L. Le Pleux, Y. Pellegrin, E. Blart, C. R. Mayer and F. Odobel, Phys. Chem. Chem. Phys., 2009, 11, 8767 RSC; (c) F. A. Black, A. Jacquart, G. Toupala, S. Alves, A. Proust, I. P. Clark, E. A. Gibson and G. Izzet, Chem. Sci., 2018, 9, 5578 RSC; (d) E. N. Glass, J. Fielden, Z. Huang, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Inorg. Chem., 2016, 55, 4309 CrossRef PubMed; (e) B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Enivron. Sci., 2013, 6, 1504 RSC.
- (a) Nonlinear Optics of Organic Molecules and Polymers, ed. H. S. Nalwa and S. Miyata, CRC Press, Boca Raton, FL, 1997 Search PubMed; (b) S. R. Marder, Chem. Commun., 2006, 131 RSC; (c) M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC; (d) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- (a) A. Al-Yasari, N. Van Steerteghem, H. El Moll, K. Clays and J. Fielden, Dalton Trans., 2016, 45, 2818 RSC; (b) A. Al-Yasari, N. Van Steerteghem, H. Kearns, H. El Moll, K. Faulds, J. A. Wright, B. S. Brunschwig, K. Clays and J. Fielden, Inorg. Chem., 2017, 17, 10181 CrossRef PubMed; (c) A. Al-Yasari, P. Spence, H. El Moll, N. Van Steerteghem, P. N. Horton, B. S. Brunschwig, K. Clays and J. Fielden, Dalton Trans., 2018, 47, 10415 RSC.
- (a) L. Xu, E. Wang, Z. Li, D. G. Kurth, X. Du, H. Zhang and C. Qin, New J. Chem., 2002, 26, 782 RSC; (b) F. K. Shehzad, N. Qu, Y. Zhou, L. Zhang, H. Ji, Z. Shi, J. Li and S. ul Hassan, Dalton Trans., 2016, 45, 17948 RSC; (c) G. Zhi, Y. Long, H. Ren, Y. Zhou, L. Zhang, Z. Shi, F. K. Shehzad and H. M. Asif, J. Phys. Chem. C, 2016, 120, 22549 CrossRef; (d) Z. Shi, Y. Zhou, L. Zhang, D. Yang, C. Mu, H. Ren, F. K. Shehzad and J. Li, Dalton Trans., 2015, 44, 4102 RSC.
- (a) H. Miao, H.-X. Wan, M. Liu, Y. Zhang, X. Xu, W.-W. Ju, D. Zhu and Y. Xu, J. Mater. Chem. C, 2014, 2, 6554 RSC; (b) Y. Dong, X. Xu, G. Zhou, H. Miao, G. Hu and Y. Xu, Dalton Trans., 2015, 44, 18347 RSC; (c) W. Jiang, L. Chen, W. Cheng, Y. Dong, J. Gong, H. Shen and Y. Xu, Polyhedron, 2018, 151, 185 CrossRef CAS; (d) X.-M. Luo, L. Chen, Y.-Y. Dong, J. Li, C.-H. Cui, J.-P. Cao and Y. Xu, Dalton Trans., 2018, 47, 9504 RSC; (e) H. Miao, Y. Dong, Z. Chen, X. He, G. Hu and Y. Xu, Dalton Trans., 2016, 45, 12717 RSC; (f) G. Hu, H. Miao, H. Mei, S. Zhou and Y. Xu, Dalton Trans., 2016, 45, 7947 RSC.
- (a) S. ul Hassan, H. M. Asif, Y. Zhou, L. Zhang, N. Qu, J. Li and Z. Shi, J. Phys. Chem. C, 2016, 122, 27587 CrossRef; (b) S. ul Hassan, F. Nawaz, Z. Ul Haq Khan, A. Firdous, M. A. Farid and M. S. Nazir, Opt. Mater., 2018, 86, 106 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Elsevier/Butterworth-Heinemann, Amsterdam, Boston, 6th edn, 2009 Search PubMed.
- W. G. Klemperer, Inorg. Synth., 1990, 27, 71 CAS.
- I. Bar-Nahum, K. V. Narasimhulu, L. Weiner and R. Neumann, Inorg. Chem., 2005, 44, 4900 CrossRef CAS PubMed.
- (a) M. Sheik-Bahae, A. A. Said, T. H. Wei, D. J. Hagan and E. W. V. Stryland, IEEE J. Quant. Electron., 1990, 26, 760 CrossRef CAS; (b) M. Sheik-Bahae, A. A. Said and E. W. V. Stryland, Opt. Lett., 1989, 14, 955 CrossRef CAS PubMed.
- P. Sengupta, J. Balaji, S. Banerjee, R. Philip, G. R. Kumar and S. Maiti, J. Chem. Phys., 2000, 112, 9201 CrossRef CAS.
- CrysAlisPro (Version 1.171.36.21), Agilent Technologies, Inc., Santa Clara, CA, United States, 2012.
- G. M. Sheldrick, SHELXS-2014, Programs for Crystal Structure Analysis (Release 2014-7), University of Göttingen, Göttingen, Germany, 2014.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- G. M. Sheldrick, SHELXL-2014, Programs for Crystal Structure Analysis (Release 2014-7), University of Göttingen, Göttingen, Germany, 2014.
- Mercury 4.1.0 (Build 235316), CCDC, Cambridge, United Kingdom, 2016.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16, rev. C.01, Gaussian, Inc: Wallingford, CT, 2016.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina and E. Guidez, et al. , J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- E. Rtibi, M. Abderrabba, S. Ayadi and B. Champagne, Inorg. Chem., 2019, 58, 11210 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- N. A. Murugan, J. Kongsted and H. Ågren, J. Chem. Theory Comput., 2013, 9, 3660 CrossRef CAS PubMed.
- M. T. P. Beerepoot, D. H. Friese, N. H. List, J. Kongsted and K. Ruud, Phys. Chem. Chem. Phys., 2015, 17, 19306 RSC.
- M. A. Salem, I. Twelves and A. Brown, Phys. Chem. Chem. Phys., 2016, 18, 24408 RSC.
- J. Zhang, F. Xiao, J. Hao and Y. Wei, Dalton Trans., 2012, 41, 3599 RSC.
- Y. Zhu, L. Wang, J. Hao, Z. Xiao, Y. Wei and Y. Wang, Cryst. Growth Des., 2009, 9, 3509 CrossRef CAS.
- (a) Y. Wei, B. Xu, C. L. Barnes and Z. Peng, J. Am. Chem. Soc., 2001, 123, 4083 CrossRef CAS PubMed; (b) B. Xu, Y. Wei, C. L. Barnes and Z. Peng, Angew. Chem., Int. Ed., 2001, 40, 2290 CrossRef CAS; (c) J. B. Strong, G. P. A. Yap, R. Ostrander, L. M. Liable-Sands, A. L. Rheingold, R. Thouvenot, P. Gouzerh and E. A. Maatta, J. Am. Chem. Soc., 2000, 122, 639 CrossRef CAS.
- (a) N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391 CAS; (b) N. S. Hush, Coord. Chem. Rev., 1985, 64, 135 CrossRef CAS; (c) D. M. D’Allesandro and F. R. Keene, Dalton Trans., 2004, 3590 Search PubMed; (d) D. E. Richardson and H. Taube, Inorg. Chem., 1981, 20, 1278 CrossRef CAS; (e) D. M. D’Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424 Search PubMed; (f) M. D. Ward, Chem. Soc. Rev., 1995, 121 RSC.
- (a) D. E. Richardson and H. Taube, Coord. Chem. Rev. 1984, 60, 107 Search PubMed; (b) D. E. Richardson and H. Taube, J. Am. Chem. Soc., 1983, 105, 40 CrossRef CAS.
- (a) J. Wu, G. Hu, P. Wang, F. Hao, H. Zhou, A. Zhou, Y. Tian and B. Jin, Dyes Pigm., 2011, 88, 174 CrossRef CAS; (b) Z. Shi, Y. Zhou, L. Zhang, S. ul Hassan and N. Qu, J. Phys. Chem. C, 2014, 118, 6413 CrossRef CAS; (c) F. K. Shehzahd, Y. Zhou, L. Zhang, Y. Long, I. Maitlo, A. Iqbal and D. Yang, J. Phys. Chem. C, 2018, 122, 1280 CrossRef.
- (a) F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem. – Eur. J., 2009, 15, 3130 CrossRef CAS PubMed; (b) A. Harriman, K. J. Elliott, M. A. H. Alamiry, L. Le Pleux, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer and F. Odobel, J. Phys. Chem. C, 2009, 113, 5834 CrossRef CAS.
- E. W. V. Stryland and M. Sheik-Bahae, in Characterization Techniques and Tabulations for Organic Nonlinear Materials, ed. M. G. Kuzyk and C. W. Dirk, Marcel Dekker Inc., 1998, pp. 655–692 Search PubMed.
- (a) M. G. Kuzyk, J. Chem. Phys., 2003, 119, 8327 CrossRef CAS; (b) J. Pérez Moreno and M. G. Kuzyk, J. Chem. Phys., 2005, 123, 194101 CrossRef PubMed.
- (a) N. Durand, R. Mhanna, P. Savel, H. Akdas-Kiliç, J.-P. Malval, O. Soppera and J.-L. Fillaut, Chem. Commun., 2020, 56, 12801 RSC; (b) N. Hobeika, H. Chaumeil, R. Mhanna, M. Jin, X. Wu, A. Spangenberg, D.-L. Versace, F. Morlet-Savary and J.-P. Malval, ChemPhysChem, 2020, 21, 2301 CrossRef CAS PubMed; (c) F. Kournoutas, A. Fihey, J.-P. Malval, A. Spangenberg, M. Fecková, P. le Poul, C. Katan, F. Robin-le Guen, F. Bureš, S. Achelle and M. Kakis, Phys. Chem. Chem. Phys., 2020, 22, 4165 RSC.
- G. Yang, W. Guan, L. Yan, Z. Su, K. Xu and E.-B. Wang, J. Phys. Chem. B, 2006, 110, 23092 CrossRef CAS PubMed.
- (a) F. Li, X. Hu, R. Sa and L. Niu, Struct. Chem., 2014, 539 CrossRef; (b) F. Li, X. Hu and R. Sa, Mol. Phys., 2013, 3081 CrossRef CAS.
- (a) L. Yuan, W. Lin, H. Chen, S. Zhu and L. He, Angew. Chem., Int. Ed., 2013, 52, 10018 CrossRef CAS PubMed; (b) I. H. Nayyar, A. E. Masunov and S. Tretiak, J. Phys. Chem. C, 2013, 117, 18170 CrossRef CAS.
- (a) S. Tretiak and V. Chernyak, J. Chem. Phys., 2003, 119, 8809 CrossRef CAS; (b) A. Masunov and S. Tretiak, J. Phys. Chem. B, 2003, 108, 899 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and table, synthetic procedures, CIF file. CCDC 2051212. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cp06610a |
| ‡ Present address: Department of Chemistry, Faculty of Science, University of Hail, Hail, Kingdom of Saudi Arabia. |
| This journal is © the Owner Societies 2021 |