
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion effects on Li ion transference number and dynamic ion correlations in glyme–Li salt equimolar mixtures†
Keisuke
Shigenobu
a,
Masayuki
Shibata
b,
Kaoru
Dokko
 ac,
Masayoshi
Watanabe
c,
Kenta
Fujii
*b and
Kazuhide
Ueno
*ac
ac,
Masayoshi
Watanabe
c,
Kenta
Fujii
*b and
Kazuhide
Ueno
*ac
aDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: ueno-kazuhide-rc@ynu.ac.jp; Fax: +81-45-339-3951; Tel: +81-45-339-3951
bGraduate School of Sciences and Technology for Innovation, Yamaguchi University, 2-16-1 Tokiwadai, Ube, Yamaguchi 755-8611, Japan. E-mail: k-fujii@yamaguchi-u.ac.jp; Fax: +81-836-85-9212; Tel: +81-836-85-9212
cAdvanced Chemical Energy Research Centre (ACERC), Institute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
First published on 15th January 2021
Abstract
To achieve single-ion conducting liquid electrolytes for the rapid charge and discharge of Li secondary batteries, improvement in the Li+ transference number of the electrolytes is integral. Few studies have established a feasible design for achieving Li+ transference numbers approaching unity in liquid electrolytes consisting of low-molecular-weight salts and solvents. Previously, we studied the effects of Li+–solvent interactions on the Li+ transference number in glyme- and sulfolane-based molten Li salt solvates and clarified the relationship between this transference number and correlated ion motions. In this study, to deepen our insight into the design principles of single-ion conducting liquid electrolytes, we focused on the effects of Li+–anion interactions on Li ion transport in glyme–Li salt equimolar mixtures with different counter anions. Interestingly, the equimolar triglyme (G3)–lithium trifluoroacetate (Li[TFA]) mixture ([Li(G3)][TFA]) demonstrated a high Li+ transference number, estimated via the potentiostatic polarization method (tPPLi = 0.90). Dynamic ion correlation studies suggested that the high tPPLi could be mainly ascribed to the strongly coupled Li+–anion motions in the electrolytes. Furthermore, high-energy X-ray total scattering measurements combined with all-atom molecular dynamics simulations showed that Li+ ions and [TFA] anions aggregated into ionic clusters with a relatively long-range ion-ordered structure. Therefore, the collective motions of the Li ions and anions in the form of highly aggregated ion clusters, which likely diminish rather than enhance ionic conductivity, play a significant role in achieving high tPPLi in liquid electrolytes. Based on the dynamic ion correlations, a potential design approach is discussed to accomplish single-ion conducting liquid electrolytes with high ionic conductivity.
1. Introduction
The feasibility of achieving rapid charge and discharge capabilities in Li secondary batteries has spurred significant interest in enhancing Li+ ion transport properties, including the ionic conductivity and Li+ transference number of liquid electrolytes.1–3 In general, the ionic conductivity of the electrolyte is believed to be the most important parameter for improving the rapid charge/discharge ability, and many studies have focused on enhancing the ionic conductivity of electrolyte materials. Liquid electrolytes with a salt concentration of ∼1 mol dm−3 are usually employed for battery applications because of their maximum conductivity in this concentration range. Over the past decade, however, highly concentrated electrolytes including molten Li salt solvates have captured significant attention owing to their unique electrochemical, physicochemical, and thermal properties, despite their low ionic conductivity. Li secondary batteries using concentrated liquid electrolytes have demonstrated improved rate capability, long-term cycling, and high coulombic efficiency.4–7 The unique cation transport decoupled from viscosity and the relatively high cationic transference number of the concentrated electrolytes have been considered as possible reasons for the improved rate capability.8–10In the design of liquid electrolytes with high Li+ transport properties, the enhancement of the Li+ transference number remains challenging. Indeed, few studies have undertaken a profound analysis of the Li+ transference number, although this parameter has often been used to discuss the rapid charge and discharge capability.11 The literature values of Li+ transference numbers have been summarized and discussed for carbonate-based organic electrolyte solutions,12,13 and the anion dependence of the Li+ transference number has been studied for organic14,15 and polymer electrolytes.16–19 To deepen our fundamental understanding of Li ion conduction along with the Li+ transference number, we previously investigated solvent effects on the Li+ transference number for molten Li salt solvates of lithium bis(trifluoromethanesulfonyl)amide (Li[TFSA]) with glymes (CH3O(CH2CH2O)nCH, Gn) and sulfolane (SL) and their relationship with ion cross-correlations (cation–cation, anion–anion, and cation–anion) based on the Onsager reciprocal relations.20 We found that the extremely low Li+ transference number (0.028) of [Li(G4)][TFSA], estimated via the potentiostatic polarization, was predominantly caused by the anti-correlated cation–cation motions of crown-ether-like, robust [Li(G4)]+ complex cations, while the relatively high Li+ transference number (∼0.7) of SL-based molten Li salt solvates was ascribed to weaker anti-correlations of cation–cation and cation–anion motions in a unique SL-bridged Li+ ion coordination structure. Thus, these different ion–solvent interactions and ion correlations in the electrolytes are crucial determinants for the Li+ transference number.
The interaction between Li+ ions and counter anions is also a key factor governing the Li+ ion coordination structure and Li+ transport properties. In a previous study, the effects of Li+–anion interactions on the stability of the complex cation and ionic transport properties of the glyme–Li salt equimolar mixtures with different Li salts ([Li(glyme)]X) were systematically studied.21 Robust [Li(glyme)]+ complex cations were formed only with weakly coordinating, bulky anions (such as [TFSA]), while the glyme molecules remained uncoordinated to some extent in [Li(glyme)]X with strongly Lewis basic anions. [Li(glyme)]X with dissociative perfluorinated sulfonylamide anions (such as [TFSA]) showed higher ionic conductivities and apparent dissociation degrees (or ionicities) than [Li(glyme)]X with more Lewis basic anions.
Herein, we report the effects of Li+–anion interactions on the Li+ transference number of [Li(glyme)]X, for X ranging from weakly coordinating anions (such as [TFSA]) to moderately basic anions (such as trifluoromethanesulfonate ([TfO]), BF4, and ClO4) to strongly Lewis basic anions (such as NO3 and trifluoroacetate [TFA]). The anion dependence of the Li+ ion transport mechanism is further discussed in the context of dynamic ion correlations based on Roling and Bedrov's concentrated solution theory.22 Finally, we conducted high-energy X-ray total scattering (HEXTS) experiments combined with all-atom molecular dynamics (MD) simulations to clarify the relationship between the dynamic ion correlations and solution structures in the equimolar glyme–Li salt mixtures.
2. Experimental
2.1 Materials
Purified triglyme (G3) and tetraglyme (G4) were kindly supplied by Nippon Nyukazai Co. (Japan). Lithium bis(trifluoromethanesulfonyl)amide (Li[TFSA]) was kindly provided by Solvay Japan. Battery-grade Li salts–lithium bis(fluorosulfonyl)amide (Li[FSA]), lithium bis(pentafluoroethanesulfonyl)amide (Li[BETA]), lithium trifluoromethanesulfonate (Li[TfO]), LiBF4, and LiClO4—were purchased from Kishida Chemical Co. (Japan). The other Li salts–lithium bis(nonafluorobutanesulfonyl)amide (Li[NFSA]), lithium trifluoroacetate (Li[TFA]) and LiNO3—were obtained from Mitsubishi Materials Electronic Chemicals Co. (Japan), Alfa Aesar and FUJIFILM Wako Pure Chemical Co. (Japan), respectively. All glyme–Li salt mixtures were prepared in an inert argon-filled glove box ([H2O] < 1 ppm; [O2] < 1 ppm).2.2 Measurements
The experiments to determine two different Li+ transference numbers (tNMRLi and tPPLi), the salt diffusion coefficients, and electrode potentials of Li/Li+ conducted in this study were performed according to our previous report.20 In brief, for the Li+ transference number determined by the potentiostatic polarization method (tPPLi), experiments were performed using a Li symmetric cell. A porous glass filter paper (Advantec, GA55, 17 mm in diameter) or two pieces of microporous propylene membrane separator (Celgard, Celgard3501, 17 mm in diameter) soaked in the liquid electrolyte were inserted into two Li foil electrodes (Honjo Metal, 16 mm in diameter) in a 2032 coin-type cell. Impedance spectra were measured in the frequency range of 100 mHz–1 MHz with a voltage amplitude of 10 mV using a potentiostat (Solartron Analytical, ModuLab XM ECS). The salt diffusion coefficients (Dsalt) were determined using the same Li symmetric coin cell according to the literature.23 The cell was polarized at 10 mV in potentiostatic mode and the relaxation of the cell potential was recorded after the applied potential was removed. Dsalt was determined by fitting the potential relaxation with a single exponential decay function. The electrode potentials of Li/Li+ (Δφ) were measured via a reference electrode (Li/Li+ in 1 mol dm−3 Li[TFSA]/G3) using a multi-compartment concentration cell (Fig. S1, ESI†).24 Vycor glass was used for the junction between the reference electrolyte and the sample electrolytes. The potential was recorded using a potentiostat. A slope dΔφ/d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(c) at a given concentration was obtained from the slope of the concentration dependence of the concentration cell potential.22 All the above measurements were performed at 30 °C unless otherwise noted.
ln(c) at a given concentration was obtained from the slope of the concentration dependence of the concentration cell potential.22 All the above measurements were performed at 30 °C unless otherwise noted.
High-energy X-ray total scattering measurements for the electrolytes were conducted at ambient temperature using an X-ray diffraction apparatus25 (BL04B2 beamline at SPring-8, JASRI, Japan). Monochrome 61.6 keV X-rays were obtained using a Si(220) monochromator. The observed X-ray scattering intensities were corrected for adsorption, polarization, and incoherent scattering to obtain coherent scattering intensities, Icoh(q).26–28
The X-ray structure factor SHEXTS(q) per stoichiometric volume was expressed as follows:
 | (1) |
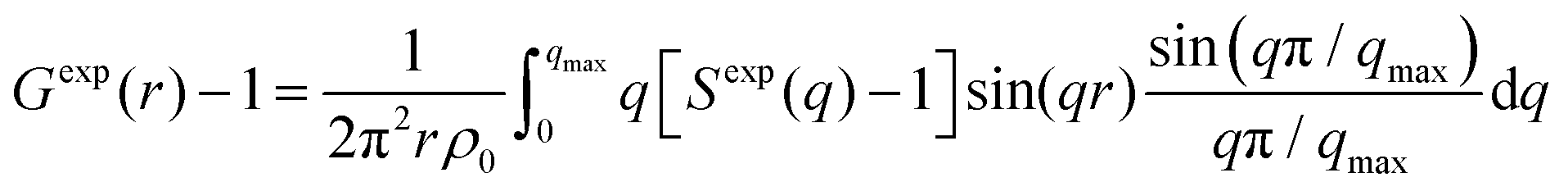 | (2) |
2.3 MD simulations
The MD simulations were performed with the GROMACS 2016.5 program under the NTP ensemble condition controlled by the Nosé–Hoover thermostat29,30 (at 298 K and 1 atm). To ensure the random starting configuration, firstly, we mixed Li+ [TFA]− and G3 molecules at high temperature and pressure (1000 K and 1000 atm, respectively) for 1 ns. The composition (i.e., the number of Li[TFSA] and Li[TFA] ion pairs, respectively, and G3 molecules) in a cubic cell was as follows: Li[TFSA]/G3 = 790/790 and Li[TFA]/G3 = 790/790. The resulting box size and density at the equilibrium state are listed in Table S1 (ESI†). The total simulation time was set as 15.0 ns for both systems. The X-ray weighted structure factors SMD(q) and radial distribution functions GMD(q) were obtained by analyzing the data collected at 0.1 ps intervals during the last 500 ps. CLaP and OPLS-AA force fields, including intermolecular Lennard-Jones (LJ) and coulombic interactions and intramolecular interactions with (1) bond stretching, (2) angle bending, and (3) torsion of dihedral angles, were used for [TFSA],31 [TFA]32–34 and G3.32,33,35 The LJ parameter for Li ions (see the Li salt/carbonate solvent system36) was used. The partial charges (q+ and q−) for TFA molecules (Fig. S2, ESI†) were obtained from quantum chemical calculations (ChelpG method37); the resulting values are listed in Table S2 (ESI†). Those for TFSA and G3 were similar to the values reported in a previous work.38The SMD(q) functions were calculated using the trajectories from the MD simulations as follows:
 | (3) |
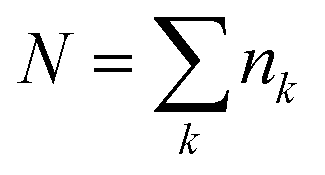 , and gMDij(r) is the atom–atom pair correlation function between atoms i and j. The GMD(q) functions were calculated from the corresponding SMD(q)s, conducting a similar procedure to that of HEXTS.
, and gMDij(r) is the atom–atom pair correlation function between atoms i and j. The GMD(q) functions were calculated from the corresponding SMD(q)s, conducting a similar procedure to that of HEXTS.
3. Results and discussion
3.1 Li+ transference number under anion-blocking condition
In our previous work, Li+ transference numbers derived from the pulsed filed gradient (PFG)-NMR method (tNMRLi) and potentiostatic polarization method (tPPLi) were determined for Li-ion conducting electrolytes.21,39,40 The PFG-NMR method is based on the Nernst–Einstein equation: tNMRLi = DLi/(DLi + Danion), where DLi and Danion are the self-diffusion coefficients of Li+ ions and anions, respectively, and the ions are assumed to be completely dissociated and move independently as ideal dilute electrolyte solutions. Hence, for highly concentrated electrolytes, tNMRLi would not reflect a realistic transference number because of their dynamic ion correlations.The Li+ transference number based on the potentiostatic polarization combined with electrochemical impedance spectroscopy41,42 (tPPLi) is defined as follows:
 | (4) |
Fig. 1(a) shows the tPPLi of the glyme–Li salt equimolar mixtures plotted as a function of ionicity. Ionicity, defined as the molar conductivity ratio (Λ/ΛNE), expresses the extent to which the self-diffusion of ionic species contributes to the actual ionic conduction and is relevant to the apparent degree of dissociation or dynamic ion correlations in electrolyte solutions.39,44–48Λ is the experimental molar conductivity and ΛNE is calculated from self-diffusion coefficients using the Nernst–Einstein equation, ΛNE = F2(DLi + Danion)/RT, where F is the Faraday constant, R is the gas constant, and T is the absolute temperature. As noted in previous papers,21,49 the strength order of Lewis basicity for the selected ions is [TFSA] < ClO4 < BF4 < [TfO] < NO3 < [TFA], whereas their order of ionicity in [Li(glyme)]X displays the exact opposite trend.
 | ||
| Fig. 1 Relationships between (a) tPPLi and ionicity, and (b) tPPLi and DG/DLi for [Li(glyme)]X. Ionicity and DG/DLi values were obtained from ref. 21. | ||
Interestingly, a good linear relationship is observed between tPPLi and ionicity (Fig. 1(a)). [Li(glyme)]X with weakly coordinating anions such as the bis(perfluorosulfonyl)amides ([FSA], [TFSA], [BETA], [NFSA]) and ClO4 show significantly low tPPLi values of less than 0.1. The value of tPPLi increases with decreasing ionicity (i.e., increasing Lewis basicity of the anions). The highest value of tPPLi is 0.93 for [Li(G4)][TFA], with the lowest ionicity of 0.04, whereas the lowest tPPLi value of 0.016 is observed for [Li(G3)][FSA], with the highest ionicity of 0.71. In Fig. 1(b), tPPLi values are plotted against the ratios of the self-diffusion coefficients of the glyme and Li+ ions (DG/DLi). Here, DG/DLi reflects the stability of [Li(glyme)]+ complex ions in the equimolar glyme–Li salt mixtures.21DG/DLi ≈ 1 (for [Li(glyme)]X with weakly coordinating anions, such as [Li(G3)][TFSA]) indicates the coupled diffusion of the glyme and Li+ ions in the form of [Li(glyme)]+ complex ions. In contrast, if DG/DLi > 1 (for [Li(glyme)]X with strongly Lewis basic anions, such as [Li(G3)][TFA]), the complex ions are considered to be short-lived or unstable, and the glymes remain uncoordinated to some extent in the equimolar glyme–Li salt mixtures.
The anion-dependent stability of [Li(glyme)]+ complex ions was also evaluated in light of the Li+–glyme residence time using MD simulations.50 The residence time of the Li+–glyme complex was much shorter than that of the Li+–anion pair for the glyme–Li salt mixtures with strongly Lewis basic anions (such as [TFA]). As a consequence of strong Li+–anion interactions, uncoordinated glymes and Li+–anion pairs mainly comprise the glyme–Li salt mixtures with Lewis basic anions. Therefore, the presence of uncoordinated (or highly exchangeable) glymes and/or Li+–anion pairs is suggested to be responsible for the high tPPLi values of [Li(glyme)]X with strongly Lewis basic anions. MD simulations by Bedrov et al. demonstrated that the enhancement of the Li+ transference number under anion-blocking conditions is attributable to the reduction of Li+–solvent residence time by shortening the chain length of the glymes.22 In this regard, our results shown in Fig. 1(b) are in good agreement with their notion: the presence of free (or highly exchangeable) glyme, which is accompanied by strong Li+–anion interactions in the present system, results in high tPPLi values.
The relationship between tPPLi and the ionic conductivity of [Li(glyme)]X is illustrated in Fig. 2. The lowest ionic conductivity of 0.083 mS cm−1 is observed for [Li(G4)][TFA], with the highest tPPLi value of 0.93, whereas the highest ionic conductivity of 1.7 mS cm−1 is achieved for [Li(G3)][FSA], with the lowest tPPLi value of 0.016. The figure clearly indicates that more Lewis basic anions contribute to the enhancement of tPPLi, while decreasing the ionic conductivity. Balsara et al. confirmed a similar “trade-off” between the Li+ transference number and ionic conductivity for Li+ ion-conducting solid polymer electrolytes.43 The Li+ ionic conductivity characterized by the product of tPPLi and the conductivity is larger for [Li(glyme)]X with NO3, [TfO] and BF4 anions than [Li(glyme)]X with strongly Lewis basic [TFA] and weakly coordinating anions such as [FSA] (see Fig. S3, ESI†). Thus, higher Li+ transport properties can be achieved with the moderately basic anions. In the following sections, we investigate the causes underlying the enhancement of tPPLi for strongly Lewis basic anions and the inverse relationship between tPPLi and ionic conductivity from the viewpoint of dynamic ion correlations and liquid structures.
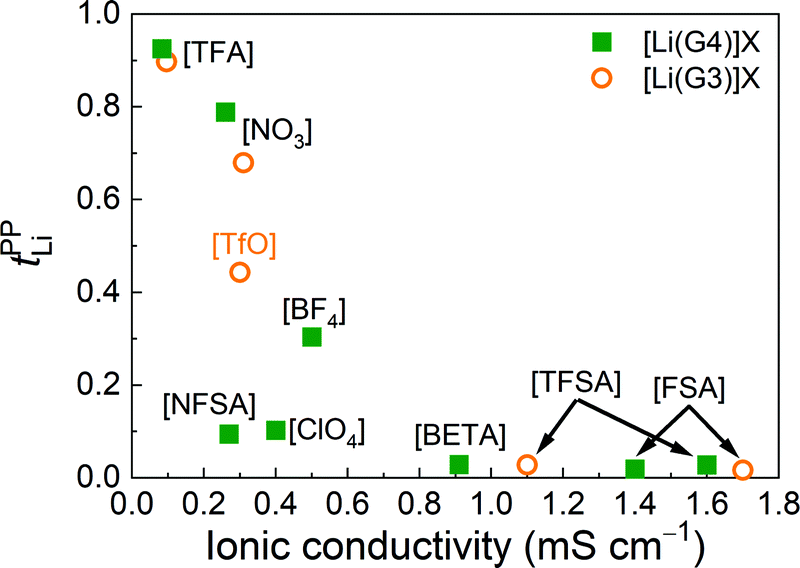 | ||
| Fig. 2 Plots of tPPLiversus ionic conductivity for [Li(glyme)]X. | ||
3.2 Dynamic ion correlations
For aqueous electrolyte solutions, dynamic ion correlations were systematically evaluated based on the velocity cross-correlation functions by Woolf and Harris.51,52 For solid polymer electrolytes, recently, Balsara and Newman investigated the correlations by introducing the Stefan–Maxwell diffusion coefficients.43,53,54 Regarding non-aqueous liquid electrolyte solutions, dynamic ion correlations were derived from the Onsager transport coefficients and were first applied for the glyme molten Li salt solvates of Li[TFSA] by Roling and Bedrov et al.22,55,56 In our previous work, we discussed transport behavior in glyme- and SL-based molten Li salt solvates by introducing experimentally determined transport coefficients. The Onsager transport coefficients can describe the ionic conductivity σion as follows:| σion = σ++ + σ−− − 2σ+− | (5) |
− consist of self-terms and distinct terms, respectively, and the ionic conductivity σion is expressed as| σion = σself+ + σdistinct++ + σself− + σdistinct−− − 2σ+−. | (6) |
All the Onsager transport coefficients (σ++, σ−−, and σ+−) can be calculated from four experimental quantities—σion, tPPLi, Dsalt, and dΔφ/dln(c); these data are shown in Table S3 (ESI†). The detailed procedure for estimating the transport coefficients was described in a previous paper.20 The dynamic cross-correlations of the ions (cation–cation, anion–anion, cation–anion) are indicated by the signs of σdistinct++, σdistinct−−, and σ+−, respectively. The sign of the cross-correlation is positive in the case of correlated ion motions, while the sign is negative for anti-correlated motions. In addition, a value of zero for these correlations indicates non-correlated interionic dynamics, as predicted in ideal electrolyte solutions.
To clarify the effects of the interionic dynamics on the anionic dependency of tPPLi and the trade-off between tPPLi and ionic conductivity, the ion dynamics for the extraordinary case of [Li(G3)][TFA] was compared with those of another extreme case, [Li(G3)][TFSA], reported in our prior study.20 As discussed in the previous section, both glyme–Li salt mixtures show exactly opposite properties: [Li(G3)][TFA] shows high tPPLi but poor ionic conductivity, and [Li(G3)][TFSA] displays high ionic conductivity but very low tPPLi.
The normalized Onsager transport coefficients (σself+, σself−, σdistinct++, σdistinct−−, and σ+− divided by σion) of [Li(G3)][TFA] are shown in Fig. 3(a), and the numerical data are listed in Table S4 (ESI†). Interestingly, all five coefficients (σself+/σion, σself−/σion, σdistinct++/σion, σdistinct−−/σion, and σ+−/σion) are positive for [Li(G3)][TFA]. This is distinctly different from the results for the previously studied [Li(G3)][TFSA], for which σdistinct++/σion, σdistinct−−/σion, and σ+−/σion were all negative.20Fig. 3(b) depicts the σ+−/σion values for [Li(G3)][TFSA], [Li(G4)][BF4], and [Li(G3)][TfO]. σ+−/σion shifts from a negative value for [Li(G3)][TFSA] to moderately positive values for [Li(G4)][BF4] and [Li(G3)][TfO], and a largely positive value for [Li(G3)][TFA] (Fig. 3(a)), confirming that the σ+−/σion value is reflective of the Li+–anion interactions in [Li(glyme)]X.
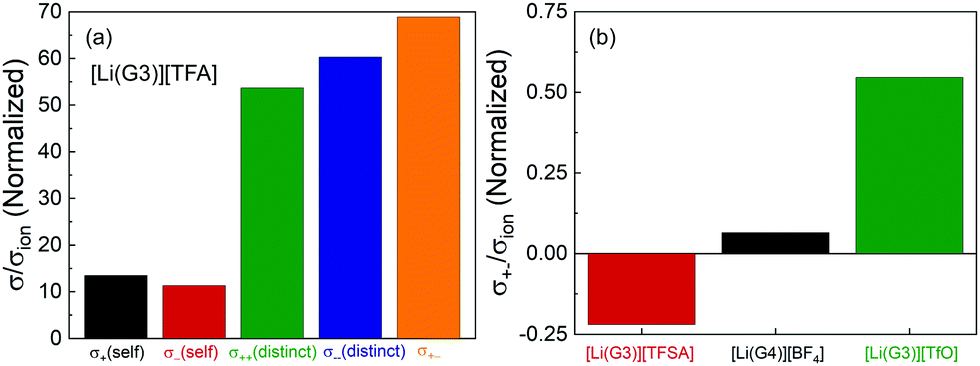 | ||
| Fig. 3 (a) All Onsager transport coefficients (σself+, σself−, σdistinct++, σdistinct−− and σ+−: left to right) normalized by σion of [Li(G3)][TFA], and (b) differences in σ+− (cross-correlation of cation–anion) of several G3- or G4-based glyme–Li salt equimolar mixtures. | ||
The correlated motions of the ion species with the same sign contribute to the enhancement of the ionic conductivity; however, the correlated motions of the cation–anion negatively affect the ionic conductivity. Thus, the correlated cation–anion motions almost offset the positive contribution from σself+, σself−, σdistinct++, and σdistinct−− to the ionic conductivity; therefore, [Li(G3)][TFA] exhibits very low ionic conductivity among the glyme–Li salt mixtures studied here.
In addition, the normalized values of the five Onsager transport coefficients for [Li(G3)][TFA] (Fig. 3(a)) are nearly hundreds of times larger than those of [Li(G3)][TFSA] simply because of its low ionic conductivity. The calculation of the five transport coefficients is affected by the self-diffusion coefficients determined for Li+ and the anions. The larger values of the normalized coefficients suggest that the measured self-diffusion coefficients of the ions in [Li(G3)][TFA] contribute little to the actual ionic conductivity (σion). The largely positive values of σdistinct++/σion, σdistinct−−/σion, and σ+−/σion for [Li(G3)][TFA] (Fig. 3(a)) suggest that all of the ions tend to move preferentially in the same direction. These results raise the possibility that Li+ ions and [TFA] anions form ionic aggregates or ion clusters of [Lix+([TFA]−)y](x−y) as well as neutral ion pairs of Li–[TFA].
3.3 Liquid structures
The solution structures of [Li(G3)][TFA] and [Li(G3)][TFSA] were investigated HEXTS and all-atom MD simulations. Fig. S4 (ESI†) shows the X-ray structure factors, S(q)s, obtained from HEXTS and MD simulations within the q range of 0–20 Å. In addition, Fig. S5 (ESI†) illustrates the corresponding X-ray radial distribution functions, r2{G(r) − 1}, in the r range of 0–25 Å. In both cases, the S(q)s and corresponding [r2{G(r) − 1}]s obtained from the MD simulations reproduce the HEXTS profiles accurately. Therefore, we can further consider the coordination structures and intramolecular/intermolecular interactions through analysis of MD simulations. It is possible to transform the [r2{G(r) − 1}]s into atom–atom pair correlation functions for each component. The correlation functions of the Li+–Li+ and Li+–anions are depicted in Fig. 4. | ||
| Fig. 4 Atom–atom pair correlation functions [gMDatom–atom(r)]s between (a) Li-ions and (b) for the O atoms of anions around Li ions and integrated profiles (cumulative coordination number: N(r)) for [Li(G3)][TFSA] and [Li(G3)][TFA]. | ||
Fig. 4(a) shows the atom–atom pair correlation function (g) between the Li ions of [Li(G3)][TFSA] and [Li(G3)][TFA]. Regarding [Li(G3)][TFSA], two sharp peaks appear at 3.0 and 9.2 Å, corresponding to the [gMDLi–Li(r)] for Li+–glyme complexes. The first peak can be assigned to the Li+–Li+ correlation, which is attributable to the two Li ions coordinated with same G3 molecule, suggesting the formation of dimers such as [Li2(G3)2]2+ in this electrolyte (Fig. S6, ESI†); however, because the cumulative coordination number (N(r)) is lower than 0.15 (see Fig. S7, ESI†), the existence probability of this structure is low. The second peak (9.2 Å) can be attributed to a correlation between [Li(G3)]+ complexes. This result is strongly supported by the previous study for [Li(G4)][TFSA].35 The potential mean force (PMF) for the Li+–Li+ pair correlation (PMF = −RTlngMDLi–Li(r)) of [Li(G4)][TFSA] has a minimum value at 9.5 Å, suggesting that stable [Li(G4)]+ complex cations predominantly exist by maintaining the distance between the other complex cations.35
In contrast, multiple sharp peaks appear in the correlation function for [Li(G3)][TFA]. These peaks start to emerge at 3.8 Å and continue until 12.8 Å with nearly regular intervals between peaks. This strongly suggests that the Li+–Li+ correlation in [Li(G3)][TFA] can be ascribed not only to Li+–solvent (G3) interactions but also to Li+–anion interactions, and a continuous Li+-ordered structure is formed as a polynuclear complex over a relatively long distance. Similar long-range ion-ordering was also observed in a highly concentrated electrolyte of Li[TFSA] in N,N-dimethylformamide.57Fig. 4(b) shows the Li+–anion pair correlations and the integrated profile (cumulative coordination number: N(r)) for each electrolyte, where the distance for the correlations of Li+–[TFA] anions is shorter than that of Li+–[TFSA]. In addition, the intensity of the peak for the Li+–[TFA] anion correlation is four times larger and the shape of the peak is much sharper. Moreover, the cumulative number of Li+–[TFA] anions is much higher than that of Li+–[TFSA]. These findings are consistent with the solution structure presumed from the dynamic ion correlations of [Li(G3)][TFA] investigated in the previous section: [TFA] anions strongly interact with Li+, and the aggregating structure is dominant in [Li(G3)][TFA].
Snapshots of the coordination structures of [Li(G3)][TFSA] and [Li(G3)][TFA] are presented in Fig. 5. As shown in Fig. 5(a), the four O atoms of G3 coordinate to one Li ion to form a stable [Li(G3)]+ complex cation along with one [TFSA] anion to compensate for the positive charge in [Li(G3)][TFSA]. This structure is in good agreement with those reported in previous experimental and computational works.50,58,59
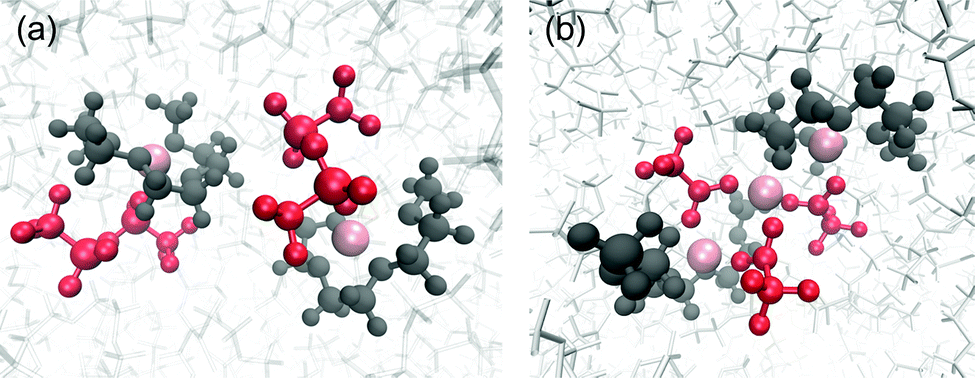 | ||
| Fig. 5 Snapshots of the coordination structures found in (a) [Li(G3)][TFSA] and (b) [Li(G3)][TFA] obtained from MD simulations. Pink: Li+, red: anions and dark grey: G3. | ||
In contrast, based on the results of the correlation function (g(r)), a continuously ordered Li+ structure is formed in [Li(G3)][TFA] by coordinating both G3 and [TFA] anions (Fig. 5(b)). A similar coordination structure was confirmed in the solid crystal structure of a Li[TFA] and G4 mixture (Fig. S8, ESI†).60 The peak positions of the Li+–Li+ correlation in [Li(G3)][TFA] (Fig. 4(a)) have a good agreement with the Li+–Li+ distances obtained from the crystal structure (see Table S5, ESI†). It is feasible that Li+–[TFA] anion aggregation and the continuously ordered Li+ structure observed in the crystal are maintained to some extent even in the liquid state.
3.4 Relationship between dynamic ion correlations and Li+ transference number
Here, we investigate the relationship between the dynamic ion correlations and the Li+ transference number. tPPLi can be defined using the Onsager transport coefficients as follows,55,56 | (7) |
 | (8) |
If the ion motions are non-correlated (β = 0 for cation–anion correlations), as postulated in ideal solutions, tPPLi is equivalent to the parameters α and tNMRLi. In most cases of liquid electrolytes used in battery systems, however, ion correlations cannot be ignored. Thus, tPPLi is significantly varied by the interplay between the cation–cation and cation–anion cross-correlations.
In Fig. 6, tPPLi is simulated versus β for different α values. As predicted by eqn (7) (visualized in Fig. 6), the parameter α must be not less than 0.5 (α ≥ 0.5) in order to achieve tPPLi = 1. The parameter β is well explained by the strength of the cation–anion correlations represented by σ+−. If the ion motions of the cation–anion are strongly anti-correlated, the value of β approaches −1.0. However, if strong correlations exist between cations and anions in the electrolytes, the β value approaches 1.0. Eqn (7) suggests that a tPPLi value of unity is attainable in the range of 0 ≤ β ≤ 1. Consequently, the concentrated solution theory predicts the possibility of a single Li+ ion conducting liquid electrolyte, and there are several approaches within α ≥ 0.5 and 0 ≤ β ≤ 1.
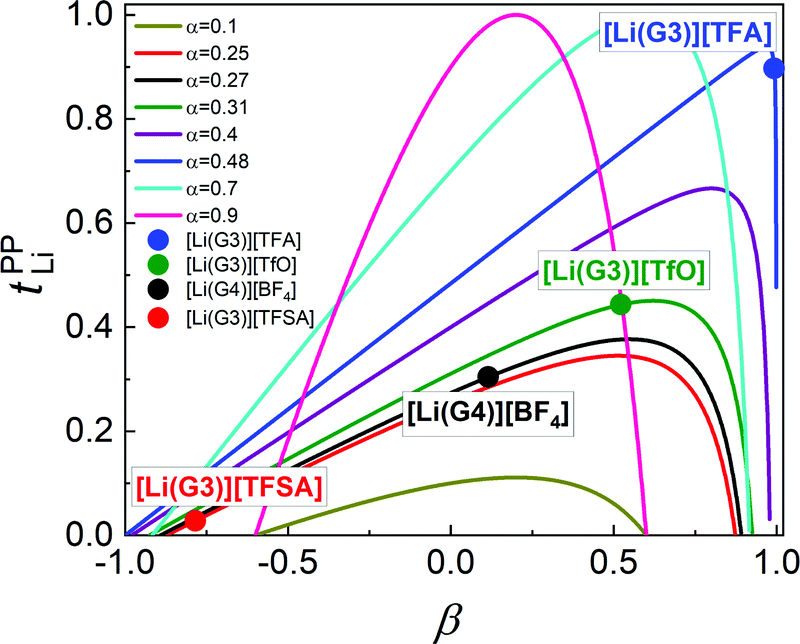 | ||
| Fig. 6 Possible range of values for tPPLi as a function of β for different α values. The experimental data for [Li(G3)][TFSA], [Li(G4)][BF4], [Li(G3)][TfO], and [Li(G3)][TFA] are plotted in the figure. | ||
The α and β parameters for [Li(G3)][TFSA], [Li(G4)][BF4], [Li(G3)][TfO], and [Li(G3)][TFA] are listed in Table 1. Furthermore, we illustrate the possible range of tPPLi as a function of β for different α values in Fig. 6, where the experimental values for [Li(glyme)]X are plotted. The Lewis basicity of the anions has an impact on both parameters α and β. According to our previous results,20 the low value of α (0.25) for [Li(G3)][TFSA] chiefly stems from the strong anti-correlation of cation–cation motions. However, the value of α is close to 0.5 for [Li(G3)][TFA] and higher than that of [Li(G3)][TFSA]. Almost the same values of σ−− and σ++, which are attributed to the coupled ion motions in the form of ion clusters, would result in α → 0.5 for [Li(G3)][TFA]. The parameter β predominantly depends on the Lewis basicity of the anion (eqn (8)). For [Li(G3)][TFSA], the anti-correlated ion motion of the cation–anion (σ+− < 0) results in a largely negative value of β (−0.78). However, the β value gradually increases as the ion motion of the cation–anion shifts from anti-correlated (σ+− < 0 for [Li(G3)][TFSA]) to nearly non-correlated (σ+− ≈ 0 for [Li(G4)][BF4]) to correlated (σ+− > 0 for [Li(G3)][TfO] and [Li(G3)][TFA]) with the increasing Lewis basicity of the anions. Therefore, the high tPPLi of 0.90 is found to be a consequence of the correlated ion motions of the cation–cation (α ≈ 0.5) and cation–anion (β ≈ 1) in [Li(G3)][TFA].
| Sample | α | β |
|---|---|---|
| [Li(G3)][TFSA]20 | 0.25 | −0.78 |
| [Li(G3)][TfO] | 0.31 | 0.52 |
| [Li(G3)][TFA] | 0.48 | 0.99 |
| [Li(G4)][BF4] | 0.27 | 0.11 |
For [Li(G3)][TFA], in which the high tPPLi is derived from strongly correlated ion motions (α ≈ 0.5, β ≈ 1), the ionic conductivity becomes very low (0.096 mS cm−1) because the large positive value of σ+− negatively affects the ionic conductivity. Therefore, highly associating systems such as [Li(G3)][TFA] may not be adequate for simultaneously achieving high ionic conductivity and high tPPLi. In contrast, the previously studied SL-based molten Li salt solvates ([Li(SL)2][TFSA] and [Li(SL)3][TFSA]) showed a higher α value of ∼0.9 and a β parameter close to zero (β = −0.18 in the case of [Li(SL)3][TFSA]), and exhibited a relatively high tPPLi value of ∼0.7 and ionic conductivity of 1.0 mS cm−1.20 Therefore, liquid electrolyte systems with non-correlated cation–anion ion motions (β ≈ 0) and strongly correlated cation–cation motions (α ≈ 1) would be more appropriate for solving the trade-off problem between tPPLi and the ionic conductivity, as shown in Fig. 2.
4. Conclusions
The tPPLi behavior of [Li(glyme)]X was investigated as a function of ionicity and was enhanced with as the ionicity decreased (i.e., as the Lewis basicity of the counter anions increased). However, a trade-off between the tPPLi and ionic conductivity was observed, which is similar to that of solid polymer electrolyte systems. The highest tPPLi of 0.93 was achieved for [Li(G4)][TFA], which had the lowest ionic conductivity of 0.083 mS cm−1, whereas the lowest tPPLi value of 0.016 was found for [Li(G3)][FSA], with the highest ionic conductivity of 1.7 mS cm−1. To clarify this trade-off, we studied the dynamic ion correlations for each electrolyte based on Roling and Bedrov's concentrated solution theory. The largely positive value of σ+−/σion was found to be the primary cause of the high tPPLi of [Li(G3)][TFA], and conversely, led to its low ionic conductivity.We further explored the liquid structures of [Li(G3)][TFSA] and [Li(G3)][TFA] using a combination of HEXTS measurements and MD simulations. From the results of the Li+–Li+ and Li+–anion pair correlation functions, it can be stated that Li+ ions preferentially interact with G3 and form [Li(G3)]+ complex cations in [Li(G3)][TFSA]. In contrast, Li+ ions form an ordered structure through interaction with not only G3 molecules but also [TFA] anions in [Li(G3)][TFA]. These results strongly support the existence of ionic aggregates or ion clusters of [Lix+([TFA]−)y](x−y) as well as neutral ion pairs of Li–[TFA], as expected from the dynamic ion correlations.
We elucidated the Li+ transference number as a function of the parameters α and β. [Li(glyme)]X with strongly associating anions, such as [Li(G3)][TFA], exhibited a high tPPLi with low ionic conductivity due to strongly correlated ion motions (α ≈ 0.5, β ≈ 1). However, our analysis also suggested a possible approach to solving the trade-off using liquid electrolytes with non-correlated cation–anion ion motions (β ≈ 0) and strongly correlated cation–cation motions (α ≈ 1). To develop an elaborate design principle for single-ion conducting liquid electrolytes with high ionic conductivity, future studies will focus on the achievement of α ≈ 1 and β ≈ 0 values in low-viscosity liquid electrolyte systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant No. 20H02837 to K. U., 20H02823 to K. F. and 18H03926 to K. D.) and by JST ALCA-SPRING (Grant Number JPMJAL1301), Japan.References
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, J. Electrochem. Soc., 2018, 165, A2654–A2666 CrossRef CAS.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef CAS.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938 CrossRef CAS.
- J. Alvarado, M. A. Schroeder, M. Zhang, O. Borodin, E. Gobrogge, M. Olguin, M. S. Ding, M. Gobet, S. Greenbaum, Y. S. Meng and K. Xu, Mater. Today, 2018, 21, 341–353 CrossRef CAS.
- M. Forsyth, H. Yoon, F. Chen, H. Zhu, D. R. MacFarlane, M. Armand and P. C. Howlett, J. Phys. Chem. C, 2016, 120, 4276–4286 CrossRef CAS.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122, 10736–10745 CrossRef CAS.
- X. Gao, F. Wu, A. Mariani and S. Passerini, ChemSusChem, 2019, 12, 4185–4193 CrossRef CAS.
- E. R. Logan and J. R. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS.
- L. O. Valóen and J. N. Reimers, J. Electrochem. Soc., 2005, 152, A882 CrossRef.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- D. B. Shah, K. R. Olson, A. Karny, S. J. Mecham, J. M. DeSimone and N. P. Balsara, J. Electrochem. Soc., 2017, 164, A3511–A3517 CrossRef CAS.
- J. Popovic, D. Höfler, J. P. Melchior, A. Münchinger, B. List and J. Maier, J. Phys. Chem. Lett., 2018, 9, 5116–5120 CrossRef CAS.
- P. Ravn Sørensen and T. Jacobsen, Electrochim. Acta, 1982, 27, 1671–1675 CrossRef.
- M. Armand, Solid State Ionics, 1983, 9–10, 745–754 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- Y. Tominaga, K. Yamazaki and V. Nanthana, J. Electrochem. Soc., 2015, 162, A3133–A3136 CrossRef CAS.
- K. Shigenobu, K. Dokko, M. Watanabe and K. Ueno, Phys. Chem. Chem. Phys., 2020, 22, 15214–15221 RSC.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323–11331 CrossRef CAS.
- D. Dong, F. Sälzer, B. Roling and D. Bedrov, Phys. Chem. Chem. Phys., 2018, 20, 29174–29183 RSC.
- D. B. Shah, H. Q. Nguyen, L. S. Grundy, K. R. Olson, S. J. Mecham, J. M. DeSimone and N. P. Balsara, Phys. Chem. Chem. Phys., 2019, 21, 7857–7866 RSC.
- H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 20246–20256 CrossRef CAS.
- S. Kohara, K. Suzuya, Y. Kashihara, N. Matsumoto, N. Umesaki and I. Sakai, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 1030–1033 CrossRef CAS.
- S. Sakai, National Laboratory for High Energy Physics, Tsukuba, Japan, 1990 Search PubMed.
- D. T. Cromer, J. Chem. Phys., 1969, 50, 4857–4859 CrossRef CAS.
- J. H. Hubbell, W. J. Veigele, E. A. Briggs, R. T. Brown, D. T. Cromer and R. J. Howerton, J. Phys. Chem. Ref. Data, 1975, 4, 471–538 CrossRef CAS.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef.
- J. N. Canongia Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- E. K. Watkins and W. L. Jorgensen, J. Phys. Chem. A, 2001, 105, 4118–4125 CrossRef CAS.
- S. Saito, H. Watanabe, K. Ueno, T. Mandai, S. Seki, S. Tsuzuki, Y. Kameda, K. Dokko, M. Watanabe and Y. Umebayashi, J. Phys. Chem. B, 2016, 120, 3378–3387 CrossRef CAS.
- J.-C. Soetens, C. Millot and B. Maigret, J. Phys. Chem. A, 1998, 102, 1055–1061 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- Y. Kamiyama, M. Shibata, R. Kanzaki and K. Fujii, Phys. Chem. Chem. Phys., 2020, 22, 5561–5567 RSC.
- K. Hayamizu, Y. Aihara, S. Arai and C. G. Martinez, J. Phys. Chem. B, 1999, 103, 519–524 CrossRef CAS.
- Y. Ugata, R. Tatara, K. Ueno, K. Dokko and M. Watanabe, J. Chem. Phys., 2020, 152, 104502 CrossRef CAS.
- P. G. Bruce, J. Evans and C. A. Vincent, Solid State Ionics, 1988, 28–30, 918–922 CrossRef.
- M. Watanabe, S. Nagano, K. Sanui and N. Ogata, Solid State Ionics, 1988, 28–30, 911–917 CrossRef.
- M. D. Galluzzo, J. A. Maslyn, D. B. Shah and N. P. Balsara, J. Chem. Phys., 2019, 151, 020901 CrossRef.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962–4967 RSC.
- K. R. Harris, J. Phys. Chem. B, 2010, 114, 9572–9577 CrossRef CAS.
- K. Ueno, H. Tokuda and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 1649–1658 RSC.
- H. K. Kashyap, H. V. R. Annapureddy, F. O. Raineri and C. J. Margulis, J. Phys. Chem. B, 2011, 115, 13212–13221 CrossRef CAS.
- C. Austen Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS.
- W. Shinoda, Y. Hatanaka, M. Hirakawa, S. Okazaki, S. Tsuzuki, K. Ueno and M. Watanabe, J. Chem. Phys., 2018, 148, 193809 CrossRef.
- L. A. Woolf, J. Phys. Chem., 1978, 82, 959–962 CrossRef CAS.
- L. A. Woolf and K. R. Harris, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 933–947 RSC.
- D. M. Pesko, K. Timachova, R. Bhattacharya, M. C. Smith, I. Villaluenga, J. Newman and N. P. Balsara, J. Electrochem. Soc., 2017, 164, E3569–E3575 CrossRef CAS.
- I. Villaluenga, D. M. Pesko, K. Timachova, Z. Feng, J. Newman, V. Srinivasan and N. P. Balsara, J. Electrochem. Soc., 2018, 165, A2766–A2773 CrossRef CAS.
- F. Wohde, M. Balabajew and B. Roling, J. Electrochem. Soc., 2016, 163, A714–A721 CrossRef CAS.
- N. M. Vargas-Barbosa and B. Roling, ChemElectroChem, 2020, 7, 367–385 CrossRef CAS.
- K. Fujii, M. Matsugami, K. Ueno, K. Ohara, M. Sogawa, T. Utsunomiya and M. Morita, J. Phys. Chem. C, 2017, 121, 22720–22726 CrossRef CAS.
- S. Tsuzuki, W. Shinoda, M. Matsugami, Y. Umebayashi, K. Ueno, T. Mandai, S. Seki, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 126–129 RSC.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2015, 17, 8248–8257 RSC.
- W. A. Henderson, N. R. Brooks and V. G. Young, Chem. Mater., 2003, 15, 4685–4690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of the Li/Li+ electrode potential against the natural logarithm of the Li salt concentration in the mixture of LiX in glyme (G3 or G4); the product of tPPLi and σionversus ionicity; X-ray structure factors S(q) and corresponding radial distribution functions in the form of [r2[G(r) − 1]]s; snapshot of the coordination structure of [Li2(G3)2]2+; Li+–Li+ pair correlation function [gMDLi–Li(r) for [Li(G3)][TFSA]; numerical data for the composition of the systems and density values; force field parameters; the six experimentally obtained parameters for the calculation of the Onsager and normalized transport coefficients; Li–Li distances obtained from the crystal structure of Li[TFA] and G4 mixture. See DOI: 10.1039/d0cp06381a |
| This journal is © the Owner Societies 2021 |