
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A quinone based single-molecule switch as building block for molecular electronics†
Herbert
Früchtl
 and
Tanja
van Mourik
*
and
Tanja
van Mourik
*
EaStCHEM School of Chemistry, University of St Andrews, St Andrews, UK. E-mail: tanja.vanmourik@st-andrews.ac.uk
First published on 4th January 2021
Abstract
Using a model molecule, we show that it is possible to create molecules that show the required properties for use as elements in a molecular circuit or computer: two conformations with similar energy but different electric conductivity, and the possibility to switch between those by applying an external electric field.
While the miniaturisation of semiconductor-based circuits is still continuing, it is no longer following Moore's law, which predicted a doubling of transistors per surface area every 18 months. This miniaturisation must reach its physical limits in the foreseeable future. One possible avenue of overcoming this barrier is the use of molecular electronics, where individual molecules would act as the building blocks of electronic devices, such as transistors or memory elements. A recent review article by Zhang1 demonstrates an active research area.
Schaub et al.2,3 reported a controllable switch consisting of an azophenine molecule deposited on a Cu-(110) surface. If a voltage greater than 0.3 V is applied, one of two symmetry-related tautomers can be produced, depending on the position of a Scanning Tunnelling Microscope (STM) tip. A smaller voltage allows the current tautomeric state of the molecule to be determined without changing it. Translated into the language of computing, this constitutes a memory element that can be written and read.
Unfortunately, the need for an STM tip to be moved to the correct position above the molecule precludes the operation at a frequency that might be competitive with current microelectronics. An additional problem is that changes in conductance are only relevant in the direction perpendicular to the surface, because the supporting metal would short-circuit any voltage parallel to the surface.
In order to create a molecule that can be used in electronic devices, three prerequisites have to be in place: bistability, i.e. the existence of two states of the same or similar energy, a means of forcing the molecule to switch between these states, and a significant difference in conductance between those conformers. We will demonstrate that for a simple model system based on the same quinone core (Fig. 1), with a coordinated iron atom above the centre of the ring, an analogous amino–imino tautomerisation mechanism to that reported in the azophenine switch can be predicted, which addresses all three of these requirements.
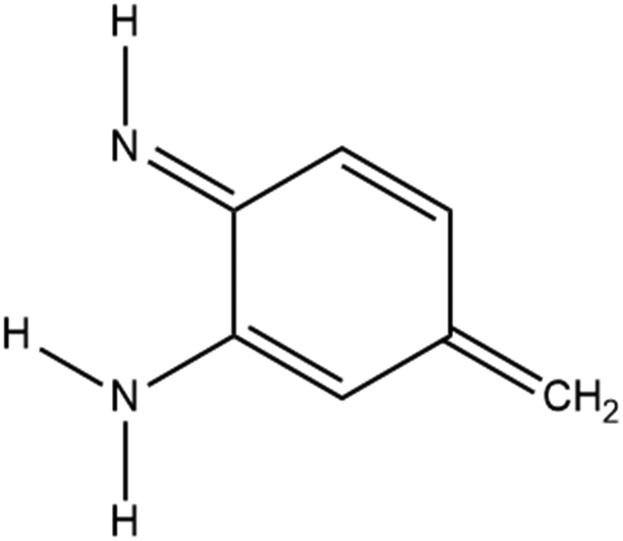 | ||
| Fig. 1 Model system considered in this work. | ||
If such a switch could be produced, it would be possible to create networks of switches, connected by molecular wires, in two or three dimensions. Fig. 2 shows a possible example, using acetylene as a molecular wire, and for the sake of simplicity a quinone molecule with only one imino–amino switch. Assuming that the imino double bond is conducting and the amino single bond insulating, after switching the circled hydrogen atom as indicated by the arrow, the current will flow from the left bottom ring to the left upper ring, instead of from the left lower ring to the right upper ring.
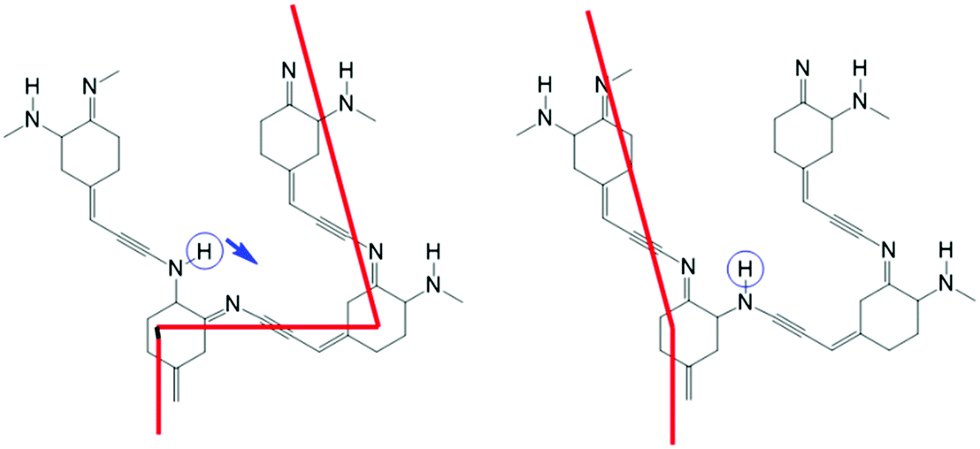 | ||
| Fig. 2 An example network of switches showing the conduction path through conjugated double bonds before (left) and after (right) the tautomeric switch of a hydrogen. | ||
The switching mechanism investigated in this study was an external electric field in the direction of the moving hydrogen (see below). We chose a value of ±1 V Å−1, which is an experimentally feasible strength, for example in an STM. External electric fields have been used on, for example, fullerene-based switches.4,5
The molecular geometry and energy profile were determined using the PBE6 density functional with Grimme D3 dispersion correction and Becke–Johnson damping.7,8 The def2-TZVP basis set9 was used for all calculations. As the aim of this study is a proof-of-principle on a model system, this level of theory was considered sufficient. All calculations were carried out using Gaussian.10
The transition path was determined using relaxed potential energy scans, varying the distance of the switching hydrogen to either of the two nitrogen atoms. The coordinate of this hydrogen atom was then projected onto the C–C bond at the opposite side of the ring to the two nitrogen containing groups, as this creates a profile that is not biased to either minimum. The molecule with a coordinated Fe atom was treated as a neutral open-shell triplet, as this resulted in the lowest energy. Without a metal present, the molecule was treated as closed shell. Where an electric field was applied, this was oriented along the C–C bond described above. This will be generally parallel to the switching direction, but the geometry of the carbon atoms defining the direction of the current will be less prone to geometric changes during the transition than atoms closer by.
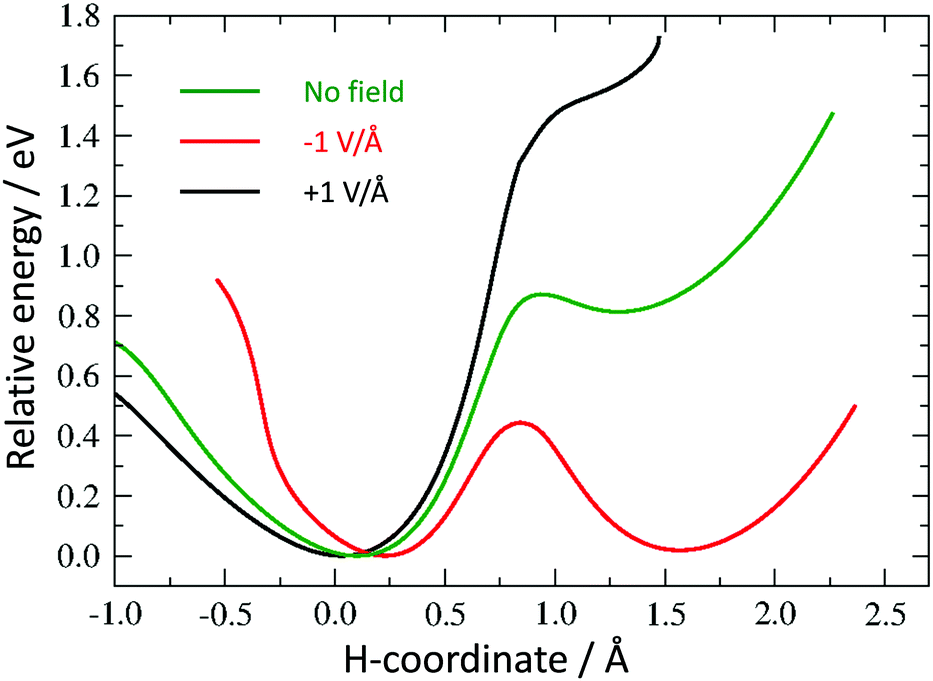
As discussed above, we need to demonstrate the existence of two conformations of similar energy. This can be achieved by coordinating an iron atom above the quinone ring. The optimised structure of the iron-doped quinone is shown in Fig. 3 and the Cartesian coordinates are included in the ESI.† The iron atom is located 1.7 Å above the plane of the quinone ring. Fig. 4 and 5 show the tautomerisation barrier without and with this coordinated atom, respectively.
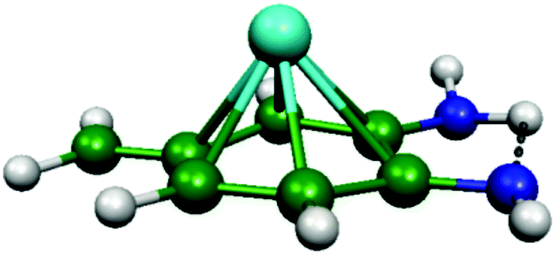 | ||
| Fig. 3 The PBE-D3/def2-TZVP optimised structure of the Fe-coordinated quinone molecule. Colour scheme: green = carbon; blue = nitrogen; white = hydrogen; and cyan = iron. | ||
 | ||
| Fig. 4 Potential energy curves for hydrogen tautomerisation in the model system without coordinated Fe atom. | ||
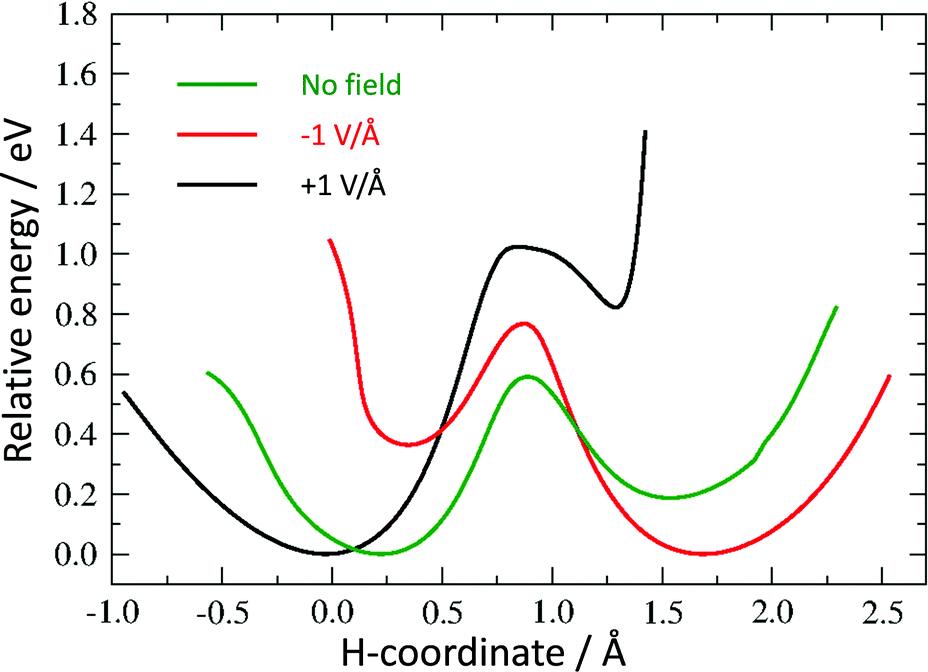 | ||
| Fig. 5 Potential energy curves for hydrogen tautomerisation in the model system with coordinated Fe atom. | ||
Without a metal atom, the quinone-like structure is, as expected, much more stable than the “switched” alternative (see “no field” profile in Fig. 4). The latter is metastable, but with a barrier of 0.87 eV to reach, and a much smaller barrier (0.06 eV) for the backward transition. The corresponding profile with a coordinated Fe atom (Fig. 5) shows a much more symmetric double well, with an energy difference of only 0.19 eV between the minima, which is much smaller than the barrier height (0.59 eV). We believe that this difference in energy between the two minima may be further reduced by changes in the molecule or its surroundings. Thus, the first requirement for a molecular switch is fulfilled. Whereas it is not strictly necessary that the two energy minima are of the same value, a (near-)symmetric double well potential would make it easier to include a reversible switch in an actual device if, for example, the electric field is pulsed by a reversible capacitor. It would also guarantee that the kinetic barrier is the same from both directions. Since, in addition to the barriers having to be large enough for the two states to be kinetically stable, they also have to be low enough to be overcome by an electric field, an asymmetric profile would narrow the range of acceptable barrier heights.
The second criterion is the existence of a considerable difference in conductivity between the two tautomers along the chosen bonds. This can be assumed to be the case, since the imino bond has double bond character and can function as the starting point of a molecular wire consisting of conjugated double or triple bonds, such as, e.g., [–CH–]n or [C2]n, which are known to be conducting, whereas the amino nitrogen is connected to both of its neighbours via single bonds, which will break the conduction path.
The final requirement for a useable molecular switch is a means to trigger it. We tested the effect of an external electric field on the energy barrier. This would open up the opportunity to “clock” an electronic device through, for example, an oscillating external capacitor. We repeated our calculation of the energy barrier in the presence of a field of 1 V Å−1 in the positive and negative direction along the switching path as described above (see Fig. 4 and 5).
All curves are calibrated to show their lowest point at zero. While not removing the barriers completely, an electric field introduces considerable asymmetry in the direction of the field. For the Fe-containing model system, we have a difference of 0.36 eV and 0.82 eV between the minima in the negative and positive direction, respectively, a large barrier of 0.77 eV and 1.02 eV for a negative and positive field, respectively, in the unfavoured direction and a much smaller barrier (0.40 eV and 0.20 eV in the negative and positive direction, respectively) towards the lower-energy minimum.
While this asymmetry may not be sufficient to trigger an immediate transfer of hydrogen in case of a reversal of the electric field for our example molecule, it indicates that such a scenario should be possible, either by using slightly larger fields or lowering the barrier through a change in the chemical environment, such as electron donating or withdrawing side groups.
In conclusion, the current work shows that a non-surface-based molecular switch satisfying our three criteria for good switching behaviour is in principle feasible.
It should be pointed out that this is a proof-of-principle study on a model system. Whereas the (counterpoise-corrected) gas-phase interaction energy of the Fe atom with the quinone molecule (−1.4 eV) is favourable, in a realistic experiment there will be competing interactions, and a means would have to be found to keep the coordinated metal atom in place, such as caging it in surrounding organic groups, or sandwiching it between two rings. A better understanding of the electronic structure features enabling the switch with a coordinated metal atom may facilitate the design of other molecules with similar properties but without the need for metal atoms. We also note that factors other than bonds being single or double may affect the current flow and that the presence of a metal atom may change the nature of those bonds. A more quantitative investigation of the conductivity of different connections to a switch molecule as described here would require the study of its transport properties with methods such as the non-equilibrium Green's function (NEGF) method. However, a chain of conjugated bonds can be expected to be electrically conducting, as demonstrated for various conducting polymers as well as insulated molecular wires.11
If a network of switches was to be built, it would initially need to be constructed in two dimensions on a non-conducting surface or built into the third dimension as an organic or metal–organic framework. Besides an electric field, as explored in the current Communication, other switching mechanisms may include electronic excitations through photons or the current through the molecule. In the latter case, the network would be self-modifying and it would be possible to program various types of logic into it.
Author contributions
Both authors contributed equally to the research and manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EaStCHEM for computer resources supplied by the EaStCHEM Research Computing Facility. The research data underpinning this publication can be accessed at https://doi.org/10.17630/335fed3b-81ef-4f7b-a0ea-09a65b56220a.Notes and references
- J. L. Zhang, J. Q. Zhong, J. D. Lin, W. P. Hu, K. Wu, G. Q. Xu, A. T. S. Wee and W. Chen, Chem. Soc. Rev., 2015, 44, 2998 RSC.
- J. A. Garrido Torres, G. J. Simpson, C. J. Adams, H. A. Früchtl and R. Schaub, Nano Lett., 2018, 18, 2950 CrossRef CAS.
- G. J. Simpson, S. W. L. Hogan, M. Caffio, C. J. Adams, H. Früchtl, T. van Mourik and R. Schaub, Nano Lett., 2014, 14, 634 CrossRef CAS.
- A. Jaroš, E. F. Bonab, M. Straka and C. Foroutan-Nejad, J. Am. Chem. Soc., 2019, 141, 19644 CrossRef.
- K. Zhang, C. Wang, M. Zhang, Z. Bai, F.-F. Xie, Y.-Z. Tan, Y. Guo, K.-J. Hu, L. Cao, S. Zhang, X. Tu, D. Pan, L. Kang, J. Chen, P. Wu, X. Wang, J. Wang, J. Liu, Y. Song, G. Wang, F. Song, W. Ji, S.-Y. Xie, S.-F. Shi, M. A. Reed and B. Wang, Nat. Nanotechnol., 2020, 15, 1019 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Terao, Y. Tanaka, S. Tsuda, N. Kambe, M. Taniguchi, T. Kawai, A. Saeki and S. Seki, J. Am. Chem. Soc., 2009, 131, 18046 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the PBE-D3/def2-TZVP optimised structures of the undoped and doped quinone molecules. See DOI: 10.1039/d0cp06250b |
| This journal is © the Owner Societies 2021 |