
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular features toward high photo-CIDNP hyperpolariztion explored through the oxidocyclization of tryptophan†
Felix
Torres
 a,
Alexander
Sobol‡
a,
Jason
Greenwald
a,
Alois
Renn
a,
Olga
Morozova
b,
Alexandra
Yurkovskaya
b and
Roland
Riek
*a
a,
Alexander
Sobol‡
a,
Jason
Greenwald
a,
Alois
Renn
a,
Olga
Morozova
b,
Alexandra
Yurkovskaya
b and
Roland
Riek
*a
aLaboratory of Physical Chemistry, Swiss Federal Institute of Technology, ETH-Hönggerberg, CH-8093 Zürich, Switzerland. E-mail: roland.riek@phys.chem.ethz.ch
bInternational Tomography Center, Siberian Branch of the Russian Academy of Science, Novosibirsk 630090, Russia and Novosibirsk State University, Novosibirsk 630090, Russia
First published on 12th February 2021
Abstract
Photo-chemically induced dynamic nuclear polarization (photo-CIDNP) is a promising solution to the inherent lack of sensitivity in NMR spectroscopy. It is particularly interesting in biological systems since it operates in water, at room temperature, and it can be repeated if the bleaching of the system can be controlled. However, the photo-CIDNP signal enhancement is well below those of other hyperpolarization techniques. While DNP, PHIP, and SABRE reach polarization enhancements of 103 to 104-fold, photo-CIDNP enhancement is typically only one order of magnitude for 1H and two orders of magnitude for 13C in the amino-acids tryptophan and tyrosine. Here we report on a photo-oxidation product of tryptophan that is strongly photo-CIDNP active under continuous wave light irradiation. In conjunction with the dye Atto Thio 12, a 1H signal enhancement of 120-fold was observed on a 600 MHz spectrometer, while at 200 MHz the enhancement was 380-fold. These enhancements in signal to noise correspond to a reduction in measurement time of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400-fold and 144400-fold, respectively. The enhancement for 13C is estimated to be over 1200-fold at 600 MHz which corresponds to an impressive measurement time reduction of 1440000-fold. This photo-CIDNP active oxidation product of tryptophan has been identified to be 3α-hydroxypyrroloindole. The reasons for its improved signal enhancement compared to tryptophan have been further investigated.
400-fold and 144400-fold, respectively. The enhancement for 13C is estimated to be over 1200-fold at 600 MHz which corresponds to an impressive measurement time reduction of 1440000-fold. This photo-CIDNP active oxidation product of tryptophan has been identified to be 3α-hydroxypyrroloindole. The reasons for its improved signal enhancement compared to tryptophan have been further investigated.
Introduction
Nuclear magnetic resonance (NMR) is extensively used in biology to obtain structural information at atomic resolution. However, despite impressive technological innovations including the increasing strength of magnetic fields, the introduction of the cryo-probe and improvements in the pulse-sequences, the issue of sensitivity remains the major barrier to applying NMR to a broader set of problems in biology. NMR is inherently insensitive because of the low Boltzmann population difference between up and down nuclear spins at room temperature. To overcome this hurdle, several hyperpolarization techniques have been proposed during the last 60 years such as dynamic nuclear polarization (DNP),1,2para-hydrogen induced polarization (PHIP),3 signal amplification by reversible exchange (SABRE),4,5 and chemically induced dynamic nuclear polarization (CIDNP), the last of which was independently discovered by Bargon et al.6 and Ward et al.7 in 1967. The photo-CIDNP setup is straightforward since it only requires irradiation of the sample, which is here performed thanks to the combination of a light source and an optic guide. The CIDNP theory is well established and relies on the radical pair mechanism proposed by Closs Kaptein and Oosterhoff in 1969.8,9 The extension by Adrian,10 which uses the Noyes11 diffusion theory constitutes an approximate theoretical framework including the present study.This study was put in motion by a serendipitous observation, while measuring the photo-CIDNP activity of fluorescein12 and Atto Thio 12 (AT12)13 on the amino acid tryptophan (Fig. 1, see also below) under continuous wave (CW) irradiation. After a series of photo-CIDNP measurements on the same tryptophan/AT12 sample, a number of new peaks appeared in the light irradiated spectra that remained undetectable in the dark spectra, evidence of an unexpected polarization (Fig. 1). Interestingly, traces of this photo-CIDNP-active product can also be found in the literature, notably in Okuno et al.,14 where a photo-CIDNP active impurity is visible for the spectrum of irradiated tryptophan in the presence of fluorescein and 3-iodo-1-propanol. The highly polarized impurity was obviously suspected to be a photoinduced degradation product of tryptophan. Indeed, we identified it as an oxidocyclization product of tryptophan that results from the generation of 1O2 by the triplet state of AT12. Subsequently, the resulting 3α-hydroxypyrroloindole was successfully synthesized, characterized and its photo-CIDNP activity measured. Moreover, the combination of CW photo-CIDNP, time-resolved (TR) photo-CIDNP experiments and density functional theory (DFT) simulations provided valuable information about the reasons for such an improvement of the signal enhancement in the 3α-hydroxypyrroloindole as compared to that of tryptophan. The relevant parameters for CIDNP such as the Δg and the hyperfine coupling constants (HFCCs) were related to the observed anomalous lines in TR-photo-CIDNP experiments. Furthermore, insights into the reaction kinetics of the photo-CIDNP reaction emphasized the possible importance of conformation, and the molecular geometry for photo-CIDNP activity. All these mechanistic studies were done in order to evaluate the potential of using CW photo-CIDNP as a signal enhancement tool in biologically and chemically focused NMR.
 | ||
| Fig. 1 Unexpected polarisation of a trace degradation product. Photo-CIDNP of a 1 day old 1 mM tryptophan sample measured by 1H 1D NMR at 14.1 T (i.e. 600 MHz 1H NMR frequency), irradiated in the presence of 25 μM AT12 with a 1 W 532 nm laser. The dark spectrum is in black and the irradiated spectrum is in green. The red dashed boxes emphasize the highly polarized signals from an unknown molecule. | ||
Theoretical background for photo-CIDNP
The anomalous lines observed in the CIDNP experiment are explained by the radical pair mechanism.9 The following section provides a brief overview about the photo-CIDNP theory. Nevertheless, several good and recent reviews should be consulted for a more extensive description of this phenomenon, such as Okuno et al., and Morozova et al.15,16The radical pair can be generated by a light-excited dye that undergoes intersystem crossing into a triplet state.17 As a consequence of its electronic structure, the triplet state dye is long-lived and highly redox-active. Hence, when the TD collides with an aromatic molecule, it can generate a radical pair (Fig. 2, step a). The newly formed radical pair is in a triplet state, where the Pauli principle applies and forbids the electron back transfer. The recombination of the radical pair is only possible when the pair has evolved into a singlet state. When the two radicals diffuse away from each other, the electron-exchange coupling vanishes (Fig. 2, step b) and the triplet–singlet mixing happens, according to eqn (1):
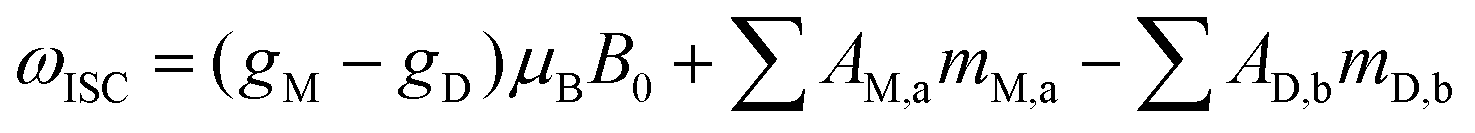 | (1) |
| |〈S|ψ(τ)〉|2 = sin2(ωISCτ) | (2) |
| PG ∼ |ωISC,α|1/2 − |ωISC,β|1/2 | (3) |
 | ||
| Fig. 2 Schematic representation of the photo-CIDNP mechanism. The different steps are described in more detail in the text. D is the symbol of the photosensitizer and M isthe symbol for any aromatic molecule that can form a radical pair with D. The nuclear spins before and after radical pair mechanism are represented to emphasize the polarization (P), which is the difference between the nuclear spin populations, Nα and Nβ. | ||
The radical pairs that recollide in a T spin state cannot recombine and therefore diffuse apart in the bulk solution (Fig. 2, step d). These free radicals can collide with a new partner and form a radical pair (F-pair), in singlet or triplet states. The singlet state pairs recombine directly and have no effect on the final polarization. The T radical pair can generate polarization again by the nuclear spin-dependent triplet–singlet electron spin mixing. The polarization arising from the repeated cycles of free M and D radical collisions is called F-pair polarization19–21 (PF), and is added to the PG to yield the final polarization (P).
In order to reach the maximal possible polarization, the study was conducted under continuous wave laser irradiation on the order of seconds. Hence, the hyperpolarization present after CW irradiation in the second timescale is a mix resulting from the PG and the PF. However, only the use of time-resolved (tr) photo-CIDNP enables us to observe the pure geminate polarization16,22,23 necessary to obtain the fine information about the key physical properties governing the singlet–triplet mixing of the radicals such as the g-factors and the HFCCs. As described in this section the intensity of photo-CIDNP spectra is driven by the efficiency of the quenching of the triplet state dye by the molecule of interest and the magnetic parameters such as g-factor and the HFCCs.
Materials and methods
The NMR measurements were performed at 298 K either on a Bruker Avance III 600 MHz spectrometer equipped with a cryoprobe or on a Bruker Avance 200 MHz spectrometer equipped with a room temperature probe. The irradiation of AT12 samples was performed with a Coherent Verdi V10 diode-pumped solid state laser emitting at a wavelength of 532 nm. The laser used for the fluorescein samples was a Thorlabs L450P1600MM, with a diode laser emitting at 450 nm. The laser light was coupled (using appropriate coupling optics) to an optical fiber (Thorlabs, FG950UEC) of length 10 m and a diameter of 0.95 mm. The end of the fiber was inserted into the sample solution in a 3 mm NMR tube to a depth of about 5 mm above the NMR coil region. The TR-photo-CIDNP experiments were performed on a Bruker DPX 200 MHz NMR spectrometer, and irradiation was performed with a COMPEX Lambda Physik XeCl excimer laser (wavelength 308 nm, and pulse energy up to 150 mJ).The HPLC analysis and initial purification was performed on an Agilent Technologies® 1200 LC system using a semi-preparative Kinetex® C18 column. The runs were performed with an acetonitrile (0.1% trifluoroacetic acid, TFA) gradient going from 3 to 30% in water (1% TFA) in 15 min at a flow rate of 3 mL min−1, at 40 °C. The UV spectra were recorded with a Jasco® V-650 UV-vis spectrometer at a concentration of 0.01 mM. Tyrosine and tryptophan (Sigma) were prepared as stock solutions of 0.2 mg ml−1 and 0.18 mg ml−1, respectively, in a 0.1 M sodium/potassium phosphate buffer (pH 7.1) with 5% D2O. The stock solution of Atto Thio 12 (AT12) was 1 mg ml−1 in H2O. To prevent dye quenching the enzyme cocktail of glucose oxidase (Go, 120 kDa), catalase (Cat, 240 kDa) and D-glucose (G, 180 Da) was used at a concentration of 14 nM for each enzyme and 2.5 mM of glucose, as described elsewhere.12,24 The stock solutions were 0.25 μM for Go and 0.16 μM for Cat, respectively. The glucose stock solution was 500 mM in D2O with 0.02% NaN3. All the samples were prepared in 100 mM of a KPO4 buffer at pH = 7.1 with either 20 μM of AT12 or 25 μM of fluorescein, and 100 μM of target molecule (i.e. tyrosine, tryptophan or HOPI), unless otherwise specified. The sample for TR-photo-CIDNP was prepared using 4 mM of HOPI and 4 mM of TCBP dye at pH = 7.5.
Results and discussion
HOPI characterization
In order to identify the molecule that showed the large polarization enhancement depicted in Fig. 1, the irradiated samples in which the highly polarized impurities could be observed were pooled together and purified by reversed-phase HPLC. Several peaks from the chromatogram were collected separately and screened by NMR with the photo-CIDNP experiment. More than one HPLC peak had significant photo-CIDNP signal enhancement and after identifying the peak with the highest photo-CIDNP signal enhancement, state-of-the-art NMR experiments were used to characterize its chemical structure (Fig. S1–S3, ESI†). In addition, the product was characterized by ESI-TOF mass spectrometry yielding a positive m/z of 221. Dyes can relax from an excited singlet state towards a triplet state towards intersystem crossing. This later state relaxes very slowly to the ground state and is therefore prone to react with oxygen to form the very oxidative singlet oxygen (Fig. 3). As the literature provided many characterized tryptophan oxidation products as potential candidates for the unknown molecules,25–27 these species were investigated further. The m/z ratio suggests the addition of an oxygen atom, and the 1D 1H-NMR spectrum shows that all the hydrogens on the benzo group remain, eliminating all the hydroxy-tryptophans as candidates. The 3α-hydroxypyrroloindole (HOPI) depicted in Fig. 3 is consistent with the NMR data and the m/z ratio from MS. The reaction mechanism leading to HOPI involves an epoxy intermediate that can form on both sides of the double bond of the pyrrole moiety of tryptophan. The direct consequence of this is the presence of two diastereoisomers when the nitrogen of the amine group opens the epoxide following an SN2 mechanism. This last step leads to cyclization. The two diastereoisomers have been previously characterized.28 The trans isomer is more polar than the cis, based on the C18 HPLC retention times (Fig. S4, ESI†). Moreover, the chemical shifts associated with the different diastereoisomers were consistent with the ones previously described in the literature;28 in particular for the Hβ and the Hα. Next, the HOPI diastereoisomer pair was synthesized according to the formerly described procedure.29 The full experimental protocol for the HOPI synthesis is detailed in the dedicated ESI.† The synthesized products exhibited the same NMR spectra and polarization properties during photo-CIDNP experiments as observed for the degradation products of tryptophan (compare Fig. 1 with Fig. 4A and B). | ||
| Fig. 3 Schematic representation of the photoinduced oxidocyclization of tryptophan with singlet oxygen yielding the 3α-hydroxypyrroloindole diastereoisomers. The reaction is caused by the relaxation of the triplet state dye to its ground state by the excitation of oxygen into its highly oxidative excited singlet state. The rates are from Chmyrov et al.30 | ||
Photo-CIDNP findings for HOPI
The oxidocyclization of tryptophan into HOPI greatly affects its CW-photo-CIDNP performance. Of particular interest is the strong AT12-based polarization found for 1H with a factor of 73 for trans-HOPI and 68 for cis-HOPI at 100 μM HOPI concentration in a 14.1 T magnetic field (i.e. 600 MHz 1H NMR frequency). Similarly, polarization in the presence of fluorescein yields a factor of −70 for trans-HOPI and −47 for cis-HOPI (Fig. 4A, B and Table 1). Interestingly, both HOPIs show a strong concentration-dependence of the signal enhancement but display contrary behaviours for the two diastereoisomers. At the lower concentration of 20 μM, trans-HOPI shows an enhancement of 120 for the H5 resonance, whereas for cis-HOPI it is only ca. 40 (Fig. 4C), while at a concentration of ca. 0.5 mM both show a similar enhancement of ca. 60. At 4.7 T (200 MHz) the trans-HOPI is enhanced by 380-fold in the presence of AT12, and 160-fold in the presence of fluorescein (Table 1). The signal enhancement for the trans-HOPI at 4.7 T is illustrated in Fig. 4C. In contrast to the single-scan photo-CIDNP 1D 1H NMR experiment the reference dark spectrum with a factor of about three-fold less signal to noise ratio took 16 h to acquire with 11 520 scans, a time difference of five orders of magnitude. | ||
| Fig. 4 Photo-CIDNP 1H 1D NMR spectra of HOPI, 100 μM, in the presence of AT12, 20 μM, (green lines) or fluorescein, 25 μM (blue lines). In black are the dark 1H 1D NMR spectra. The irradiation was performed for 4 s at 1 W power level, and wavelengths of 450 nm for fluorescein and 532 nm for AT12. (A) trans-HOPI at 600 MHz and (B) cis-HOPI at 600 MHz. Note that trans-HOPI displays two H2 and H7 signals due to different conformations. (C) trans-HOPI at 200 MHz. The dark spectrum was accumulated over 11520 scans. For representation purposes the spectra were normalized to the noise level, hence the dark spectra are scaled by a factor of 0.0093, which correspond to 1/√11520. (D) Photo-CIDNP of 13C detected on the attached 1H of naturally abundant trans-HOPI at 600 MHz. The dark spectrum (no light) was accumulated over 2560 scans. For representation purposes the spectra were normalized to the noise level; hence, the dark spectrum is scaled by 0.02, which corresponds to 1/√2560. The (very weak) signal at 3.8 ppm of the dark spectrum is due to the sugars, which are at a concentration of 2.5 mM (ESI†), and were used for the cross-calculation of the signal enhancement. | ||
| Molecule | AT12 at 14.1 T (600 MHz) | Fluorescein at 14.1 T (600 MHz) | AT12 at 4.7 T (200 MHz) | Fluorescein at 4.7 T (200 MHz) |
|---|---|---|---|---|
| The signal-to-noise enhancement factors correspond to the strongest aromatic resonance of HOPI (H5) and tryptophan (H4), both at 100 μM. | ||||
| trans-HOPI | 73 | −70 | 380 | −160 |
| cis-HOPI | 68 | −42 | N.D. | N.D. |
| TRP | 18 | 53 | 90 | 225 |
While tryptophan is typically better polarized with fluorescein than AT12, the trans-HOPI and to a lesser extend the cis-HOPI are equally hyperpolarized after irradiation in the presence of AT12 or fluorescein dyes (Table 1). Furthermore, the polarization sign for HOPI is inverted with fluorescein compared to that of AT12 (Fig. 4A and B). Moreover, the pattern of the signal enhancement is perturbed. The photo-CIDNP spectrum of tryptophan with AT12 shows only absorptive lines for the aromatic protons and emissive lines for the Hβ (Fig. S5, ESI†), while the HOPI spectrum presents both absorptive and emissive lines for the aromatics and is emissive for the newly aliphatic H2 (Fig. 4A and B).
The finding that dyes react preferentially with some molecules over others, yielding to preferential polarization, has already been reported. This is in particular the case for fluorescein and tryptophan, which are a better pair than fluorescein and tyrosine12 and conversely for tyrosine, which is more efficiently hyperpolarized with AT12 than tryptophan.13 In the high-field limit for fast tumbling molecules this difference is attributed to the offset in the Zeeman splitting (Δg) of the two single electrons of the respective dye and molecule (eqn (1)), while the changes in sign and relative intensities are explained by the changes in the HFCC values upon chemical structure modification.
Photo-CIDNP of 13C resonances of HOPI
It is important to note that the here-documented polarization enhancements are for 1H, while it is common that the polarization enhancements are reported for 13C or other nuclei, which have either a lower gyromagnetic ratio or larger HFCC when compared with 1H. The anisotropy of the 13C HFCC needs to be considered for a proper analysis since their anisotropic character is much more pronounced than for hydrogen. For the carbons C5, C7, and C2 we could calculate HFCC values for the z-axis (in the molecular frame) of 25.1, 22.3, and 27.9 Gauss, respectively. Thus, for the following 3 reasons it is expected that HOPI will display an even stronger enhancement for 13C: (1) its gyromagnetic ratio is 3.98-fold lower than that of 1H, (2) the 13C HFCCs are at least a factor of 2 higher than that for the protons (Tables S2 and S3, ESI†) the longitudinal relaxation of 13C is slower than that of 1H yielding less signal decay during the CW irradiation. Hence, at least a factor of 8 more is expected. Indeed, a 13C signal enhancement of 1217-fold is estimated in a 1D single scan photo-CIDNP experiment on trans-HOPI and AT12 performed on a 600 MHz spectrometer starting with 13C (steady state) magnetization that is detected on its covalently bounded 1H via a refocusing INEPT31 (Fig. S14B ESI† and Fig. 4D). In this case, only an estimation was possible since the trans-HOPI sample contained naturally abundant 13C, yielding a dark reference spectrum of extremely low signal intensity. It is noted that the estimated signal enhancement equals a 106-fold reduction in measurement time. The irradiated spectrum measured in 1 scan (5 seconds) would have required 1 440 000 scans for the dark spectrum, which corresponds to 2000 hours of measurement or approximately 83 days.g-Factor differences (Δg)
In the following, the Zeeman splitting, the HFCC values and reaction kinetics of both HOPI diastereoisomers are investigated in some detail starting with Δg. As shown above in the photo-CIDNP theory section, in the high-field limit for fast tumbling molecules (i.e. <1 ns), the singlet–triplet state mixing is mainly driven by the Δg. This parameter results from the spin-orbital coupling of the unpaired electron with the SOMO. The oxidocyclization of the HOPI transforms the aromatic system from an indole ring to an aniline-like aromatic ring after the conversion of the pyrrole part of the indole group into a pyrrolidine. This modification of the shape and expansion of the SOMO is suggested to be the reason for the different g-factor values of tryptophan and HOPI. An experimental support of the g-factor modification through the chemical reaction is the sign alteration of the HOPI photo-CIDNP when the dye is changed from fluorescein to AT12 (Fig. 3A and B). According to the Kaptein rules,32 this polarization sign alteration is explained by the dye-dependent sign alteration of the radical pair Δg. Indeed, no other conditions are changed between the two experiments except the dye. Furthermore, the observed magnetic field-dependence of the enhancements support the important role of the Δg in the photo-CIDNP signal enhancement. At 4.7 T (i.e. 200 MHz), the H5 signal of trans-HOPI is enhanced 380-fold in the presence of excited AT12 (triplet at 6.98 ppm), and −160-fold in the presence of excited fluorescein (Table 1). The tryptophan signal is enhanced by 225 and 90-fold in the presence of fluorescein and AT12, respectively. The field-dependent normalized photo-CIDNP polarizations have been calculated according to eqn (3), and plotted into Fig. 5. The experimental signal enhancement values were normalized by the relative sensitivity of the 600 MHz (14.1 T) and 200 MHz (4.7 T) NMR spectrometers to obtain the normalized polarizations, and were fitted to the calculated curves. The photo-CIDNP polarizations monitored with AT12 dye (Fig. 5A) had to be calculated for the different possible g-values estimated from the photo-CIDNP anomalous line sign alteration of HOPI, and the g-values of tyrosine and tryptophan.13 The fitting of the experimental polarization to the calculated curves provided satisfactory results for all the possible g-values of AT12 except 2.0036, thus refining the range of g-values for HOPI from 2.0037 to 2.0040. Moreover, the optimal field for HOPI is strongly shifted to high field as compared to that of tryptophan, which is explained by the cooperativity of the smaller Δg and higher HFCCs as compared to that of tryptophan. The slightly higher field-dependence of the AT12-monitored photo-CIDNP of HOPI as compared to that of tryptophan can be explained by this shift. The fluorescein-monitored photo-CIDNP field-dependence polarization of HOPI is shifted towards higher field values, due to the particularly low difference in g-values (Fig. 5B). Therefore, the optimal field for the HOPI-fluorescein photo-CIDNP polarization is 9–10 T, which would correspond to a 400 MHz spectrometer. This surprising finding is corroborated by the relatively lower polarization of HOPI at 200 MHz as compared to that at 600 MHz. In comparison, the tryptophan-fluorescein photo-CIDNP couple has a higher Δg and presents a slight increase of the polarization at 200 MHz as compared to that at 600 MHz. All these findings together show a stronger field-dependence of the AT12-based enhancement compared to that of fluorescein. This dye-specific field-dependence is in large part explained by the Δg, the difference of the g-factor of the dye to the g-factor of the radicals. In principle, at lower fields, the HFCCs have a greater influence and the polarization resulting from each spin sorting step is higher and reaches a maximum when the condition μBΔgB0 = HFCC/2 is fulfilled. However, as it appears with the HOPI fluorescein couple, this greater influence of the HFCC can be observed at higher field if the Δg is small enough. Fig. S11 (ESI†) shows the ranking of the molecules according to the g-factor of their radicals. Fluorescein has a smaller Δg with tryptophan than that of AT12 as demonstrated elsewhere.12 Furthermore, based on our calculation, the Δg of the fluorescein-HOPI pair is expected to be very low and on the order of 0.0001 and thus its effect on the polarization is not very magnetic field-sensitive, while the Δg of the AT12-HOPI pair is several times larger yielding a stronger magnetic field-dependence of the polarization. Whether the higher quantum yield of AT12 compared to that of fluorescein is also an important factor in the magnetic field-dependence of polarization is open for discussion.12,30 Furthermore, the calculated g-factors for the HOPI radicals yielded values in the vicinity of 2.0035 (see the ESI†). Considering the sign of the anomalous lines, the g-factor of the HOPI radicals have been estimated above fluorescein's g-factor (2.0034) and below AT12's (between 2.0034 and 2.0041). However, the sign of the polarization when the experiments are conducted with 3,3′,4,4′-tetracarboxy-benzophenone (TCBP) dye with a g-factor = 2.0035 (Fig. S7 and S8, ESI†) enables one to place the g-factor value of HOPIs in between 2.0034 and 2.0035. Furthermore, the polarization of HOPI at 200 MHz in the presence of TCBP is 3-fold lower as compared to that of tryptophan, which is expected since the Δg of HOPI with TCBP is estimated to be smaller than the Δg of HOPI with fluorescein, and shifts the optimal magnetic field to higher values. | ||
| Fig. 5 Field dependent photo-CIDNP polarization calculated from the HFCC of HOPI (red) and tryptophan (green) in the presence of AT12 (A), or fluorescein (B). The g-value of AT12 is unknown and the curves were simulated for different potential values: 2.0036 (solid), 2.0037 (dashed), 2.0038 (dot-dashed), 2.0039 (dotted), and 2.0040 (loosely dotted). | ||
Finally, since the Δg between HOPI and fluorescein is lower than the Δg between HOPI and AT12, one would expect the HOPI fluorescein couple to yield higher signal enhancements, while considering only the magnetic parameters. However, it is the AT12, which provides the best performance as a photo-CIDNP sensitizer for HOPI, suggesting that other parameters than magnetics are to be considered to address the polarization quantitation.
Hyperfine couplings
Despite the fact that triplet–singlet state mixing of the radical pair electron spins are mainly driven by the Δg in fields above 1 Tesla (high-field), the spin sorting, and thus the hyperpolarization is the result of the HFCCs. These coupling constants are not field-dependent and are specific for each nucleus within the radical. The HFCC values depend on the Fermi contact interaction between the nucleus and the unpaired electron, and on the spin density.33 As a consequence of the differences in the conjugated systems of tryptophan and HOPI, their spin densities are different. Unlike the absorptive signals of the aromatic protons of tryptophan, the aromatic anomalous signals are emissive and absorptive for HOPI. Moreover, the H2 proton, which was aromatic in the tryptophan is now in the α-position of the conjugated system and has an intense emissive signal after irradiation in the presence of AT12. To get further insights into the HFCC constants of HOPI with an emphasis on understanding which of the chemical features are important to generate a highly polarizable molecule, HFCCs were computed from DFT simulations run on Gaussian (Table S2, ESI†).In order to determine experimentally the HFCC and mechanistic insights (see below) time-resolved (tr) photo-CIDNP is necessary because in a CW-photo-CIDNP experiment the predominance of F-pair over geminate polarization (slightly) deviates from the total polarization and only the geminate polarization is purely related to the HFCC.34 For TR-photo-CIDNP the experimental conditions were changed (see the ESI for details) using TCBP as the dye because the hyperfine couplings of its radicals have been determined from EPR35 and CIDNP23,36 and because its radicals show clear CIDNP patterns that are distinctly different for electron transfer (ET) and proton coupled electron transfer (PCET) reactions. The experiments were performed at 4 mM concentration of HOPI and at a pH of 7.5 (Fig. S7 and S8, ESI†)
The experimental HFCCs determined by the TR-photo-CIDNP anomalous line intensities (Fig. S7 and S8, ESI†) were then correlated with the calculated ones shown in Fig. 6A and B showing determination coefficients of 0.97 and 0.98 for trans-HOPI and cis-HOPI, respectively (Fig. 6A and B). It is thereby interesting to note that the H2 HFCC was dramatically increased (i.e. by a factor of 5–6) by the aromaticity loss of the five membered-ring. The high HFCC of beta protons in tyrosine and tryptophan have already been predicted. However, because of the angular dependence of the HFCC,37 the rotation around the σ molecular bond was averaging out the expected hyperpolarization effect. From this observation, we conclude that in general, aliphatic protons in the α position to an aromatic system with hindered rotational mobility (i.e. that in a cyclic aliphatic ring) yield high HFCC.
 | ||
| Fig. 6 Correlation between the 1H CIDNP intensities of the geminate products detected in the reversible photoreaction of TCBP and the calculated 1H HFCCs of the cis-HOPI (A) and trans-HOPI (B) radicals (circles) and the combination of TCBP radicals I, III, and IV (squares). Solid line: best fit by the function P1i = −CA1i (fitting to circles), P2j = −CA2j (fitting to squares). Concentration-dependent H5 Photo-CIDNP enhancement for trans- and cis-HOPI. (C) Concentration-dependence of the enhancement of trans-HOPI shown as red circles and cis-HOPI data shown in grey circles. (D) Concentration dependence of the polarized spin concentration. For the calculation it is assumed that the polarization at 14.1 T was 100 ppm. The data points shown in red are: trans-HOPI (350, 175, 88, 44, 22, 11, and 5.5 μM) and in grey: cis-HOPI (440, 220, 110, 55, 27.5, 13.8, and 6.9 μM). | ||
Electron transfer
Since the HFCC of the TCBP dye depend on its protonation state, the electron transfer mode can be addressed by analyzing the TR-photo-CIDNP anomalous line intensities.23 In the case of tryptophan or N-acetyl tryptophan, the TCBP anomalous line intensities match with the unprotonated TCBP's HFCCs and thus a simple electron transfer (ET). However, the time-resolved CIDNP spectra for cis/trans-HOPI yield a mix of protonated TCBP radicals as it can be observed from the correlation plot between the anomalous line intensities and the HFCCs of TCBP/H+ mix (Fig. 6A, B and Fig. S9, S10, Table S2, ESI†). This last observation suggests a PCET reaction mechanism for the HOPI radicals. The degenerate electron exchange takes place when the radical and the diamagnetic molecule differ only by one electron. It is worth noting that PCET is also the formation mechanism of tyrosyl radicals,23 which also shows improved signal enhancement when photo-CIDNP experiments are performed in the presence of AT12.13Diastereomer-dependent reaction kinetics
Under the given experimental conditions, the photo-CIDNP enhancement of the H5 signal is similar for trans-HOPI and cis-HOPI (73 versus 68, Table 1). However, the signal enhancement of the H2 proton is different for the two diastereoisomers, and each exhibits two different chemical shifts that correspond to two slowly interconverting conformers. The larger enhancement factor for trans-HOPI H2 is -82-fold for its upfield conformer and for cis-HOPI H2 the larger enhancement is -47-fold for its downfield conformer. Moreover, the HOPI concentration-dependence of the signal enhancement displays completely different behaviours for the two diastereoisomers. At the lower concentration of 20 μM, trans-HOPI shows an enhancement of 120 for the H5 resonance, whereas for cis-HOPI it is only ca. 40 (Fig. 6C). Towards an understanding of this difference it is noted that the radical pair formation obeys bimolecular steady state kinetics, which is outlined in the ESI† “Reaction kinetics”. It is demonstrated that the slope of the concentration-dependent photo-CIDNP spin polarization shown in Fig. 4D provides the time-independent rate constant ratio k1/k−1 under the assumption that both the HOPIs are in surplus, where k1 is the radical pair formation rate constant and k−1 the overall radical pair recombination rate constant. Based on the fits of the experimental data from Fig. 4D, this ratio is higher for trans-HOPI (54 M−1) than cis-HOPI (39 M−1).Similar mechanistic insights are obtained from competition experiments with photo-CIDNP of tryptophan with either cis-HOPI or trans-HOPI with the latter showing a stronger interference on the photo-CIDNP of tryptophan (Fig. S6, ESI†). In principle, the conformational dependence of the g-factors and the HFCCs could modulate the recombination rate k-1. However, neither the HFCC nor the g-factor calculations showed a significant difference between the two diastereoisomers (Tables S2 and S3, ESI†) suggesting that the reason for different polarization properties is solely resulting from the reaction yield of the radical pair formation. This suggests other modulation parameters such as the conformational flexibility, redox potential,38 hydrodynamic radius or the dye–HOPI interaction potential to be the key for the different polarizations observed. We propose here a straightforward explanation referring to the Marcus theory for electron transfer, or in this case proton coupled electron transfer.39,40 Shortly, according to the Franck–Condon principle, the electron transfer timescale being orders of magnitude faster than the nuclei motions, the radicals are generated in the wrong solvation environment, and in the wrong vibrational state. Marcus revised the well-known Franck–Condon principle to satisfy energy conservation. Hence, the vibrational states of the diamagnetic reactant must match the vibrational states of its radical product to enable the electron transfer. The quantum mechanics simulations predict that the cis-HOPI has an internal H-bond between the hydroxy and the carboxy groups, while the trans-HOPI does not. However, T1 relaxation experiments revealed no decrease of the relaxation time T1 for cis-HOPI as compared to that of trans-HOPI with T1 values of 3.88 s and 3.26 s, respectively. This last experiment invalidates the H-bond hypothesis. Nevertheless, the HPLC retention times suggest that the trans diastereoisomer may be less hydrophobic than the cis. This difference in hydrophobicity is very likely related with a different solvation profile and may be the result of the different conformations. This particular conformation of the cis-HOPI interacts differently with its environment and is responsible for a higher energy penalty, while jumping to the higher vibrational states as compared to the trans-HOPI. The result of this is a higher free energy barrier for the generation of the radical in the case of the cis-HOPI and then a slower reaction rate according to eqn (3):
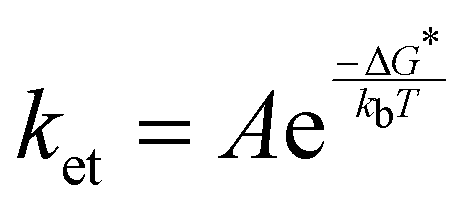 | (4) |
On the AT12-HOPI pair – an outlook
The present study starting from a serendipitous observation identified a powerful CIDNP dye-ligand pair – AT12 and HOPI – yielding at relatively low concentrations signal enhancement in the order of two to three orders of magnitude, explained by the symbiotic contributions of several factors such as hyperfine couplings, g-factor difference between dye and ligand and reaction kinetics. This pair opens on one hand an avenue towards high throughput screening or NMR experiments at very low concentrations of ligand protein interaction studies relevant in biological sciences and drug research, if the HOPI can be covalently linked as a CIDNP-NMR probe to the molecules of interest as usually required in fluorescence-based experiments/assays. The HOPI is thereby a rather small molecule when compared to the usual fluorescent dyes. Moreover, HOPI photo-CIDNP can be monitored with AT12, which is excited at 532 nm, an important redshift when compared to fluorescein (450 nm), FMN (450 nm), bipyridyl (308 nm), or TCBP (308 nm). On the other hand, the HOPI studies indicate that many small molecules of chemical and biological interest are potentially CIDNP-active as they comprise more often than rare tryptophan-like or HOPI-like entities showing that there is a significant chemical space of CIDNP-active molecules to be investigated and potentially used.Conclusions
Understanding the chemical features that make photo-CIDNP highly efficient is of great value in designing highly polarizable radical pairs. The previously reported maximum 1H signal enhancement was for tyrosine,13 which could be enhanced up to 60-fold with AT12, while trans-HOPI could be polarized up to 120-fold at a magnetic field of 14.1 T. At a magnetic field of 4.7 T the 1H signal enhancement observed is up to 380-fold and the 13C enhancement at 14.1 T is estimated to be on the order of 1000-fold. The optimal field for the fluorescein-HOPI pair was estimated to be 400 MHz and improved photo-CIDNP performances are expected at this field. In addition, the optimal field for the HOPI-TCBP is potentially higher than 400 MHz. The optimization of photo-CIDNP pairs for high field could yield to unprecedented signal to noise performance. The reasons for such enhancements are the larger HFCCs, reduced Δg and improved yields of the reactive species. It was already observed that aliphatic protons in the alpha position relative to an aromatic cycle presented the highest HFCC. Unfortunately, the dihedral-dependence of the HFCC combined with the rotation of the aliphatic bonds was averaging these HFCCs to lower values.33,41–43 The constraining of the aliphatic proton H2 in a 5-membered ring impedes the rotation and provides a high HFCC that is responsible for a high photo-CIDNP signal enhancement. Moreover, smaller conjugated systems generate higher spin density and higher HFCCs, as well as modify the SOMO shape and the resulting HFCCs and g-factors. Furthermore, the difference in signal enhancement for the two diastereoisomers underlines the importance of the reactivity of the molecule in relation to the dye. It is an elegant illustration of the Marcus theory and suggests that diastereoisomers should be investigated separately. Furthermore, given the intrinsic complexity of the CIDNP phenomenon, the selection of a dye-molecule couple for CW-photo-CIDNP application cannot be simply derived from the quenching rate and magnetic parameters obtained with TR-photo-CIDNP. The latter is necessary to obtain the information about the electron transfer reaction and the HFCC. It gives also a clear idea of the quenching rates in play. Therefore, it is concluded that the selection of an optimal dye-polarizable aromatic molecule pair may yield unprecedented signal enhancement on the order of several orders of magnitude.Author contributions
F. T. performed most of the experiments and wrote the manuscript. A. S. made the first observation of the HOPI lines. J. G. helped to identify the oxidation product of tryptophan. A. R. helped with the experiments and wrote the manuscript. O. M. performed and analysed the TR-photo-CIDNP experiments. A. Y. helped with analysing TR-photo-CIDNP results. R. R. helped with the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Daniel Zindel for the synthesis of the 3α-hydroxypyrroloindole diastereoisomer. Y. A. and O. M. acknowledge support by Russian Foundation for Basic Research (projects 20-03-00234 and 19-29-10028).References
- A. Abragam, The principles of nuclear magnetism, Clarendon Press, Oxford, 1961 Search PubMed.
- J. H. Ardenkjaer-Larsen, K. Golman, A. Gram, M. H. Lerche, R. Servin, M. Thaning and J. Wolber, Discovery Med., 2003, 3, 37–39 Search PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. Green, J. Chem. Phys., 2009, 131, 194505 CrossRef PubMed.
- J. Bargon, H. Fischer and U. Johnsen, Z. Naturforsch. A., 1967, A 22, 1551–1555 Search PubMed.
- H. R. Ward and R. G. Lawler, J. Am. Chem. Soc., 1967, 89, 5518–5519 CrossRef CAS.
- G. L. Closs, J. Am. Chem. Soc., 1969, 91, 4552–4554 CrossRef CAS.
- R. Kaptein and J. L. Oosterhoff, Chem. Phys. Lett., 1969, 4, 195–197 CrossRef CAS.
- F. J. Adrian, J. Chem. Phys., 1971, 54, 3912–3917 CrossRef CAS.
- R. M. Noyes, J. Chem. Phys., 1954, 22, 1349–1359 CrossRef CAS.
- Y. Okuno and S. Cavagnero, J. Phys. Chem. B, 2016, 120, 715–723 CrossRef CAS PubMed.
- A. Sobol, F. Torres, A. Aicher, A. Renn and R. Riek, J. Chem. Phys., 2019, 151, 234201 CrossRef PubMed.
- Y. Okuno and S. Cavagnero, J. Magn. Reson., 2018, 286, 172–187 CrossRef CAS PubMed.
- Y. Okuno and S. Cavagnero, Emagres, 2017, 6, 283–313 CAS.
- O. B. Morozova and K. L. Ivanov, ChemPhysChem, 2019, 20, 197–215 CrossRef CAS PubMed.
- M. Cocivera, J. Am. Chem. Soc., 1968, 90, 3261–3263 CrossRef CAS.
- G. L. Closs, in Advances in Magnetic and Optical Resonance, ed. J. S. Waugh, Academic Press, San Diego, 1974, vol. 7, pp. 157–229 Search PubMed.
- J. K. Vollenweider, H. Fischer, J. Hennig and R. Leuschner, Chem. Phys., 1985, 97, 217–234 CrossRef CAS.
- J. K. Vollenweider and H. Fischer, Chem. Phys., 1986, 108, 365–372 CrossRef CAS.
- J. K. Vollenweider and H. Fischer, Chem. Phys., 1988, 124, 333–345 CrossRef CAS.
- A. S. Kiryutin, K. L. Ivanov, O. B. Morozova, A. V. Yurkovskaya, H. M. Vieth, Y. A. Pirogov and R. Z. Sagdeev, Dokl. Phys. Chem., 2009, 428, 183–188 CrossRef CAS.
- O. B. Morozova, K. L. Ivanov, A. S. Kiryutin, R. Z. Sagdeev, T. Kochling, H. M. Vieth and A. V. Yurkovskaya, Phys. Chem. Chem. Phys., 2011, 13, 6619–6627 RSC.
- J. H. Lee and S. Cavagnero, J. Phys. Chem. B, 2013, 117, 6069–6081 CrossRef CAS PubMed.
- G. E. Ronsein, M. C. de Oliveira, M. H. de Medeiros and P. Di Mascio, J. Am. Soc. Mass Spectrom., 2009, 20, 188–197 CrossRef CAS PubMed.
- M. Gracanin, C. L. Hawkins, D. I. Pattison and M. J. Davies, Free Radical Biol. Med., 2009, 47, 92–102 CrossRef CAS PubMed.
- W. E. Savige, Aust. J. Chem., 1975, 28, 2275–2287 CrossRef CAS.
- G. E. Ronsein, M. C. B. Oliveira, S. Miyamoto, M. H. G. Medeiros and P. Di Mascio, Chem. Res. Toxicol., 2008, 21, 1271–1283 Search PubMed.
- M. Nakagawa, S. Kato, S. Kataoka, S. Kodato, H. Watanabe, H. Okajima, T. Hino and B. Witkop, Chem. Pharm. Bull., 1981, 29, 1013–1026 CrossRef CAS.
- A. Chmyrov, J. Arden-Jacob, A. Zilles, K. H. Drexhage and J. Widengren, Photochem. Photobiol. Sci., 2008, 7, 1378–1385 CrossRef CAS PubMed.
- J. H. Lee, A. Sekhar and S. Cavagnero, J. Am. Chem. Soc., 2011, 133, 8062–8065 CrossRef CAS PubMed.
- R. Kaptein, J. Chem. Soc., Chem. Commun., 1971, 732–733 RSC.
- R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231–1253 CrossRef CAS PubMed.
- A. S. Kiryutin, O. B. Morozova, L. T. Kuhn, A. V. Yurkovskaya and P. J. Hore, J. Phys. Chem. B, 2007, 111, 11221–11227 CrossRef CAS PubMed.
- O. Säuberlich, J. Brede and D. Beckert, J. Phys. Chem., 1996, 100, 18101–18107 CrossRef.
- O. B. Morozova, M. S. Panov, N. N. Fishman and A. V. Yurkovskaya, Phys. Chem. Chem. Phys., 2018, 20, 21127–21135 RSC.
- A. Torolabbe and J. Maruani, J. Magn. Reson., 1985, 61, 254–261 CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 979–989 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- I. Kuprov, T. D. Craggs, S. E. Jackson and P. J. Hore, J. Am. Chem. Soc., 2007, 129, 9004–9013 CrossRef CAS PubMed.
- F. Khan, I. Kuprov, T. D. Craggs, P. J. Hore and S. E. Jackson, J. Am. Chem. Soc., 2006, 128, 10729–10737 CrossRef CAS PubMed.
- I. Kuprov and P. J. Hore, J. Magn. Reson., 2004, 168, 1–7 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp06068b |
| ‡ Deceased. |
| This journal is © the Owner Societies 2021 |