
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntersystem crossing processes in the 2CzPN emitter: a DFT/MRCI study including vibrational spin–orbit interactions†
Angela
Rodriguez-Serrano
 ,
Fabian
Dinkelbach
and
Christel M.
Marian
*
,
Fabian
Dinkelbach
and
Christel M.
Marian
*
Institut für Theoretische Chemie und Computerchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Christel.Marian@hhu.de; Tel: +49 211 8113209
First published on 12th January 2021
Abstract
Multireference quantum chemical calculations were performed in order to investigate the (reverse) intersystem crossing ((R)ISC) mechanisms of 4,5-di(9H-carbazol-9-yl)-phthalonitrile (2CzPN). A combination of density funcional theory (DFT) and multireference configuration interaction methods (MRCI) was used. The excellent agreement of the computed absorption spectrum with available experimental absorption spectra lends confidence to the chosen computational protocol. Vertically, two triplet excited states (T1 and T2) are found below the S1 state. At the excited state minima, the calculated adiabatic energies locate only the T1 state below the S1 state. The enhanced charge transfer (CT) character of the geometrically relaxed excited states causes their mutual (direct) spin–orbit coupling (SOC) interaction to be low. Contributions of vibronic SOC to the (R)ISC probability, evaluated by a Herzberg–Teller-like procedure for a temperature of 300 K, are small but not negligible. For ISC, the S1 → T1 channel is the fastest (8 × 106 s−1), while the S1 → T2 channel is found to be thermally activated (9 × 104 s−1) and less efficient when proceeding from the adiabatic S1 state. Our calculations also reveal, however, a barrierless S1 → T2 ISC pathway near the Franck–Condon region. RISC is found to essentially proceed via the T1 → S1 channel, with a rate constant of (3 × 104 s−1) if our adiabatic singlet–triplet energy gap in vacuum (ΔEST = 0.12 eV) is employed. Shifting the potentials to match two experimentally reported singlet–triplet energy gaps in toluene (ΔEST = 0.21 and 0.31 eV, respectively) leads to a drastic reduction of the computed rate constant by up to 4 orders of magnitude. The T2 state is not expected to play a major role in mediating triplet–singlet transitions in 2CzPN unless it is directly populated by hot excitons. No indication for a strong vibronic coupling of the T2 and T1 potentials is found, which could help overcome the negative exponential dependence of the RISC rate constant on the magnitude of the energy gap.
1. Introduction
The carbazolyl dicyanobenzene (CDCB) family of compounds has been widely investigated as thermally-activated delayed fluorescence (TADF) emitters in organic light emitting diodes (OLEDs).1 The devices, built using these prototype compounds have shown high luminescence efficiencies and excellent operational stability.2–8As first proposed by Uoyama et al.,9 CDCB emitters were constituted by carbazole (Cz) units as donors (D) and the dicyanobenzene (DCB) moiety acting as an acceptor (A). These systems showed strong intramolecular charge-transfer (ICT) emission with the luminescence colour varying (from sky blue to orange) with the number and relative position of the Cz and cyano groups in the DCB unit. Of this series, the sky blue emitter 2CzPN (4,5-di(9H-carbazol-9-yl)-phthalonitrile, Fig. 1) had the smallest external quantum efficiency (EQE) of 8.0% in toluene and 2,8-bis(diphenylphosphoryl)-dibenzo[b,d]thiophene (PPT) film, while the green emitter 4CzIPN showed the highest external quantum efficiency (EQE) in toluene and 4,4′-N,N′-dicarbazole-biphenyl (CBP) film (19.3%).9 The lower efficiency of 2CzPN has been attributed to a large roll-off with current increase, which is dominated by strong contributions of singlet–triplet annihilation (STA) and triplet–triplet annihilation (TTA) as shown by Masui et al. by using an exciton-quenching model.10 Later, its efficiency was further improved by including a mixed co-host system, which included electron/hole transporting materials in the device.11 This architecture improved its efficiency to an EQE of 21.8% that is stated to be one of the highest for blue TADF OLEDs.
 | ||
| Fig. 1 Chemical structure of 4,5-di(9H-carbazol-9-yl)-phthalonitrile (2CzPN). | ||
In TADF, the chromophore exhibits delayed fluorescence (DF) from the lowest-lying (S1) singlet state, which is thermally populated from an energetically close-by triplet excited state (Tn) by reverse intersystem crossing (RISC). Therefore, with RISC being the rate limiting process in TADF, a small singlet–triplet energy gap (ΔEST) in the range of the thermal energy in conjunction with an effective spin–orbit coupling (SOC) between target states is crucial for facilitating the up-conversion of the triplet population into emissive singlets.12 This is particularly critical for 2CzPN and its derivatives,5,6 where the reported ΔEST values are large compared to those of other TADF emitters.1 For 2CzPN, ΔEST values between 0.21 eV13 and 0.31 eV7 have been determined in toluene solution yielding RISC rate constants (kRISC) of 6 × 103 s−1 and 1.7 × 105 s−1, respectively. The latter value is remarkably high in view of the sizable singlet–triplet splitting. An exceptionally small ΔEST value of 0.09 eV was determined in a 1,3-bis(N-carbazolyl)benzene (mCP) OLED, yielding a kRISC value of 5.6 × 103 s−1.10 These observations bring to light interesting optical properties of these emitters that are influencing their kRISC and, consequently, the TADF efficiencies.
The optical and electronic properties of the sky blue 2CzPN emitter have been extensively studied in solution5,11,13 and in a variety of OLED ensembles.8,10,11,14–17 In toluene, hexane and dichloromethane (DCM) solutions, 2CzPN shows similar ultraviolet absorption spectra, which are constituted by three bands: two bands (a maximum and a shoulder) in the range of 300 and 350 nm that were assigned to LE on the Cz moiety and a wide CT band peaking at ca. 375 nm.11,13,16 The absorption spectrum in tetrahydrofuran (THF) solution is found to be blue-shifted by a few nanometers.5 The emission of 2CzPN shows up at ca. 475 nm in toluene and is blue-shifted to 447 nm in hexane,16 while this peak is red-shifted to 501 nm in diluted THF solution.13 Bathochromic shifts of the emission maxima with increasing solvent polarity are typical for CT transitions in liquid solution. For all solvents, the reported rate constant for the radiative decay is similar (∼107 s−1).
Experimental works supported by theoretical calculations have played a significant role in rationalizing mechanistic details about the (R)ISC of TADF emitters. A D–A type of architecture in multi-chromophoric systems leads to a large spatial separation between the frontier molecular orbitals (HOMO and LUMO) and thus promoting a small ΔEST. Consequently, their lowest-lying excited states are typically of charge transfer (CT) character. According to the empirical El-Sayed's rules for SOC, the coupling between singlet and triplet states of the same character is forbidden, e.g. between (ππ*) CT states. Therefore, vibrationally induced SOC becomes of great importance in mediating efficient (R)ISC for TADF emitters, which in fact has been elegantly demonstrated in experiments and via theoretical calculations.18–24 Vibronic coupling can boost the coupling between two (ππ*) CT states by borrowing its intensity from a close-lying state with a local excitation (LE) character. This state can typically bridge the energy gap between the lowest singlet and triplet states as shown by Gibson et al.20 and Etherington et al.22 for D–A and D–A–D TADF active compounds between phenothiazine as a D and dibenzo thiophene-S-S-dioxide as an A. The influence of multiple excited states and vibrational effects in SOC and their role in TADF has been recently discussed by Marian and Penfold et al.25,26
For 2CzPN, previous studies have demonstrated that conformational freedom modulates the degree of admixture of the LE character in the S1 and T1 (CT ππ*) states. These include experimental transient absorption spectroscopy (TAS)13 and transient electron-spin resonance27 as well as several theoretical TDDFT-based approaches28–33. Such studies focus on substitution effects on the electronic and excited state properties of several CDCBs performed mainly on ground state geometries. Other computational works calculate first-order SOC values in order to obtain (R)ISC rate constants via semiclassical Marcus theory using the adiabatic T1 state34,35 or involving the singlet–triplet crossing seam36.
In this study, we aim at understanding the underlying excited state decay mechanisms that drive the (R)ISC process in 2CzPN. To this end, we carried out extensive hybrid density functional theory/multireference interaction (DFT/MRCI) calculations. The DFT/MRCI method37,38 has been largely used in our laboratory for getting mechanistic insights into (R)ISC of organic39 and inorganic40,41 compounds. Furthermore, we give a detailed overview on the (R)ISC efficiencies and their implications for TADF emission.
2. Computational details
As it is well known, substantial errors are obtained when calculating excitation energies of CT states with time-dependent density functional theory (TDDFT) and standard exchange–correlation (xc) functionals. Several computational studies have evaluated the performance of TDDFT for a wide range of TADF emitters (including 2CzPN) correlating (vertical) singlet and triplet transition energies with experimental absorption energies. This, with the purpose of finding an optimal percentage of Hartree–Fock (HF) exchange contribution to the xc functional that allows the evaluation of transition energies for CT states and singlet–triplet energy gaps in a more precise manner.42–44 This empirical calibration seems to work very well for states with strong CT character but has limited applicability for mixed CT–LE states and in cases where the LE contributions to a desired state vary with geometry distortions. A more promising ansatz toward a balanced description of CT and LE states in donor–acceptor compounds in TDDFT calculations is optimal tuning where the ω parameter of a range-separated functional is adjusted such that the negative HOMO energy equals the molecular vertical ionisation.45,46In this context, we performed an evaluation of the performance of different density functionals such as B3LYP, PBE0, CAM-B3LYP and BHLYP and of ab initio methods such as RI-CC2 and ADC2 on the ground and excited state geometries (Tables S1 and S3 and Fig. S2 of the ESI†).47 For all the DFT calculations, Grimme's dispersion corrections (D3) together with Becke and Johnson (BJ) damping were accounted for.48,49
We found the best agreement with the experimental absorption spectra in toluene and DCM11,13 for a combination of PBE050,51-D3/def-SV(P)52 geometries and DFT/MRCI excitation energies and oscillator strengths. Therefore, all further calculations were performed following this computational protocol. The conductor-like screening model (COSMO) was used to mimic the solvent environment (ε = 2.380 for toluene and ε = 8.930 for DCM).53,54 The geometries of the electronic ground and excited states of 2CzPN were all optimized without symmetry constraints. The optimized minima of the low-lying singlet excited states were obtained by using TDDFT.55 For the triplet excited states, the Tamm–Dancoff approximation (TDA)56 to TDDFT was employed. The TURBOMOLE program was used for all the geometry optimizations.57 The harmonic vibrational frequency calculations were performed employing the AOFORCE module58,59 of TURBOMOLE for the ground state, and the SNF program60 for the excited states.
Vertical and adiabatic excitation energies and optical electronic properties were computed using the DFT/MRCI method.37 The tight parametrization of the Hamiltonian, reported in ref. 38, was employed (DFT/MRCI-R2016), which has been specially designed for multichromophoric systems. Up to 10 roots were calculated for each singlet and triplet manifold. For a better understanding of the relaxation of the system, linearly interpolated potential energy profiles (LIPs) between target singlet and triplet state minima were computed.
A wavefunction analysis of the singlet and triplet states was performed by using a Löwdin orthogonalization61 of the one-electron transition density matrix (1TDM) as implemented in the TheoDORE program of F. Plasser.62 This ansatz was used to characterize the nature of the electronically excited states in the singlet and triplet manifold. In this context, the triplet wavefunction analysis requires singlet–triplet transition density matrices. These densities were not available from the current DFT/MRCI code. Therefore, we extended the DFT/MRCI program to supply singlet–triplet transition density matrices and TheoDORE was modified to read these new density matrices enabling the triplet wavefunction analysis. Here, the system was decomposed into three fragments for calculating the charge transfer numbers (ΩAB), charge transfer character (ωCT), and natural transition orbitals (NTOs) of the Ω matrices for the individual states.63,64 The Cz rings of 2CzPN constituted each one fragment (separately) and the phthalonitrile (PN) moiety the third fragment.
The spin–orbit matrix elements (SOMEs) coupling target singlet and triplet states were calculated with the spin–orbit coupling kit (SPOCK) developed in our group.65–67 Herein, the spin–orbit coupling (SOC) is described by the Breit–Pauli Hamiltonian and the spin–orbit-mean-field-approximation (SOMF) is employed.68,69 Rate constants for ISC and RISC between singlet and triplet states were determined in the framework of the Fermi golden-rule approximation and a time-dependent approach to ISC mediated by SOC within the Condon and Herzberg–Teller approximations as implemented in the VIBES program.70,71 The derivatives of the SOMEs (∂SOMEs) with respect to mass-weighted normal coordinates were obtained as the average of two-point finite differences using a displacement of ±0.1 along the dimensionless coordinates.72 For fixing the phases of the DFT/MRCI wave functions, the MO phases at each displaced geometry were set to match the ones of the equilibrium geometry. The overlap matrix between them was calculated, where the off-diagonal elements give an indication of a change of phase or ordering of the MOs. Then, the phases of the wave functions were adjusted in such a way that the largest coefficient of each wave function is positive.73 Temperature effects on the rate constants were accounted for by assuming a Boltzmann distribution of the harmonic vibrational state populations in the initial electronic state.74 Zero-point vibrational energy (ZPVE) corrections are automatically included in the VIBES calculations. For further details on the theory and the methods used for calculating ISC rate constants check the previously published review article.25
3. Results and discussion
In the following we present the results obtained by our quantum chemical calculations. First, the ground state geometrical and optical properties are discussed considering also other experimental and theoretical reports. The energetics is analysed in terms of the DFT/MRCI vertical and adiabatic transition energies and interpolated pathways between target states. The characters of the low-lying excited states are analysed and the kinetics of different (R)ISC decay pathways are discussed considering direct and vibronic SOC. The Cartesian coordinates of all the computed minima, the frontier molecular orbitals (MOs) (Fig. S1 and S4–S9, ESI†) and electronic spectra (Tables S5–S8, ESI†) calculated at each of these geometries together with additional electronic structural information are provided in the ESI.†3.1. Geometries and electronic structure at the S0 state
The optimized ground state minima of 2CzPN at the PBE0/def-SV(P) level of theory are displayed in Fig. 2a and b. Two conformers were found (S0 and S0′), where the difference between them is the relative orientation of the Cz groups w.r.t. the PN moiety. As expected by steric effects, a rather distorted orientation of the two bulky Cz groups is found, thus preventing the possibility of finding stable minima of higher symmetry in this emitter. The two optimized minima of 2CzPN are not the optical reflection of one another. For the S0′, the opening angle of the Cz rings is wider. In fact, their ground state energies differ by 0.074 eV (1.7 kcal mol−1), with the S0 geometry in Fig. 2a being more stable. | ||
| Fig. 2 Selected geometrical parameters (dihedral angles) of the ground state minima of 2CzPN optimized at the PBE0-D3(BJ)/def-SV(P) level of theory. | ||
The calculated geometries agree well with the experimental X-ray structures reported by M. Wong et al.75 A comparison between these geometries and other S0 minima obtained at other theoretical levels can be found in the ESI† (Table S1). We also expect enantiomers for S0 and S0′ to exist.
The calculated DFT/MRCI-R2016 absorption spectra of both conformers of the isolated 2CzPN are presented in Fig. 3a. The first absorption band is composed of two CT transitions. While the wavelengths of the intensive S2 absorptions and hence the positions of the peak maxima are nearly identical in both cases, the widths of the first absorption band varies. Due to its red-shifted S1 absorption, interaction of the S0 conformer (Fig. 2a) with visible light yields a broader first absorption band. Also the relative intensities and energetic positions of the second absorption bands of the two conformers differ. For comparison with experimental results, the calculations were repeated in toluene and DCM environments. The DFT/MRCI-R2016 solution spectra of the S0 conformer are shown in Fig. 3b without any empirical tuning, together with two experimental absorption spectra measured in toluene13 and DCM.11 Solvent effects on the absorption spectra appear to be very small. Excellent agreement between the computed DFT/MRCI-R2016 spectra of the most stable S0 conformer in Fig. 2a and the experimental data is observed. In the following, we therefore focus the analysis on this conformer.
 | ||
| Fig. 3 DFT/MRCI-R2016 absorption spectra of 2CzPN calculated at the S0 minimum optimized at the PBE0-D3(BJ)/def-SV(P) level of theory: (a) in vacuum and (b) in toluene and dichloromethane (DCM) using the COSMO model. The experimental absorption spectra in toluene13 and DCM11 are also presented. | ||
The DFT/MRCI-R2016 vertical energies of the low-lying singlet and triplet states calculated at the S0 state geometry of 2CzPN are listed in Table 1. These are compared to other theoretical and experimental reports which are also displayed in this table. Experimental excitation energies and oscillator strengths (in toluene and DCM) agree well with the calculated values of the 2CzPN in vacuum. All these excited states can be associated with ππ* electronic transitions, and the corresponding frontier MOs relevant to these states are presented in Fig. 4a. From this figure, it can be observed that the H and H−2 MOs are mostly localized on the Cz units but present also some amplitudes on the PN unit. While the H−2 is mostly located on the Cz units, the H−6 contains strong localization on the PN unit and lower density in the Cz units. Regarding the MOs unoccupied in the electronic ground state, the L and L+1 MOs present amplitudes on the PN unit and at the nitrogen atom of the Cz units, while the L+2 and L+3 are mostly localized on the Cz units.
| State | ΔEv | Transition | % weight | f(L) | Theorya | Exp.b |
|---|---|---|---|---|---|---|
| a Other theoretical reports. b Experimental reports. c TD-PBE0(D3)/SV(P). d TD-ADC(2)/SV(P). e Ref. 42, in cyclohexane. f Ref. 35, TD-LC-w*PBE/6-31+G* in toluene (PCM). g Ref. 42, TD-MPW1B95/6-31G*. h Ref. 42, TD-CAMB3LYP/6-31G*. i Ref. 45, TDA-B2(GP)-PLYP/def2-TZVP//PBE0-D3(BJ)/def2-TZVP in toluene (COSMO). j Ref. 13, toluene solution. k Ref. 16, hexane solution. l Ref. 46, TDA-LC-w*PBE/6-31+G* in toluene (PCM). m Ref. 42, TD-B3LYP/6-31G*. | ||||||
| S0 | 0.00 | |||||
| S1 | 3.19 | H → L | 78.0 | 0.126 | 2.98,c 3.27,d 3.22,f 3.13,g 3.68,h 3.15i | 3.19,e 3.30,j 3.32k |
| S2 | 3.36 | H−2 → L | 80.3 | 0.202 | 3.17,c 3.55d | |
| S3 | 3.57 | H−1 → L | 79.0 | 0.011 | ||
| S4 | 3.72 | H → L+1 | 69.4 | 0.182 | ||
| S5 | 3.83 | H−3 → L | 81.7 | 0.006 | ||
| T1 | 2.79 | H → L | 59.1 | 2.68,c 3.02,d 2.70,g 2.81,l 2.50m | ||
| H−6 → L | 15.1 | |||||
| H−1 → L | 11.7 | |||||
| T2 | 3.13 | H−2 → L | 72.4 | 3.04f | ||
| T3 | 3.24 | H−1 → L+3 | 25.9 | |||
| H−3 → L+2 | 24.7 | |||||
| H−1 → L | 14.3 | |||||
| T4 | 3.27 | H−1 → L+2 | 31.5 | |||
| H−3 → L+3 | 28.2 | |||||
| H → L + 2 | 12.5 | |||||
| T5 | 3.50 | H → L+1 | 28.8 | |||
| H → L+2 | 21.2 | |||||
| H−2 → L+3 | 16.8 | |||||
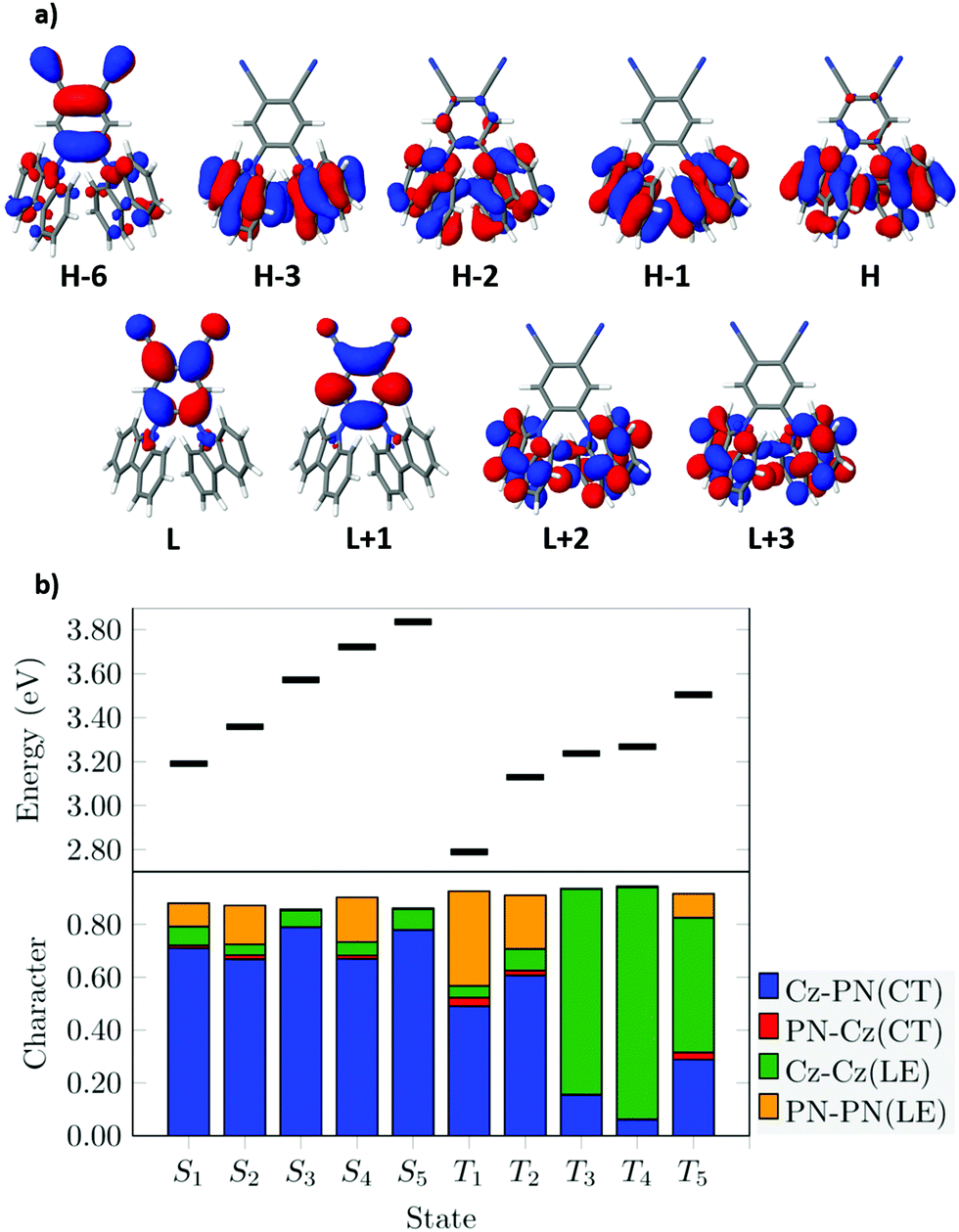 | ||
| Fig. 4 (a) BH-LYP frontier molecular orbitals of 2CzPN calculated at the S0 geometry optimized at the PBE0-D3(BJ)/def-SV(P) level of theory. (b) Fragment-based analysis for the vertical singlet and triplet state DFT/MRCI-R2016 wavefunctions. | ||
The fragmentation analysis is presented in Fig. 5b for the low-lying singlet and triplet states. This procedure will allow us to characterize the nature of the calculated DFT/MRCI-R2016 excited states of 2CzPN in a simple manner. The lowest excited states present a considerably large CT character from the Cz substituents to the PN unit (in blue region of the bars) combined with some (lower) contributions of local excitations within the PN (in orange) unit and the Cz units (in green).
 | ||
| Fig. 5 Selected geometrical parameters (dihedral angles) of the ground and excited state minima of 2CzPN optimized at the PBE0-D3(BJ)/def-SV(P) level of theory. TDDFT was used for the optimization of the excited singlet states, while TDA was used for the optimization of the triplet states. | ||
Among the low-lying singlets, there are three states with considerable oscillator strength (Table 1), namely S1, S2 and S4. The wavefunction of the S1 CT state of 2CzPN is strongly dominated by an H → L electronic transition and shows up at 3.19 eV (in the gas phase), which is expected to be slightly red-shifted due to solvent–solute interactions. This value agrees well with the experiment (3.19 eV) in toluene. Here it is interesting to note that functionals like CAM-B3LYP and ωB97X-D yield excitation energies above 3.6 eV for this state.34,42 At 0.17 eV above, the S2 state is found arising from a H−2 → L transition. Like S1, the S2 excitation has predominantly CT character, mixed with LE on the PN unit. Together, they form the first absorption band with maximum around 375 nm. At variance with the interpretation of Hosokai et al.13 who assigned the band with maximum at approx. 325 nm to originate from ππ* electronic transitions within the Cz units, our fragmentation analysis reveals the S4 state to have mainly CT character. It is a multiconfigurational state with a leading term corresponding to a H → L+1 excitation. The S3 state, with much lower oscillator strength, corresponds to a H−1 → L transition and shows up 0.38 eV above the S1 state.
The electronic structure of the low-lying triplet states is more mixed in the Franck–Condon (FC) region, i.e., the weight percentage of their dominant contributions is lower than in the corresponding singlet states and have a more combined CT – LE character as shown by the fragmentation analysis of the transition densities (see Fig. 4b). Vertically, two triplet states below the S1 state can be found. The T1 state shows up at 2.79 eV (ΔEST = 0.40 eV) and arises from a linear combination of three excitations: H → L (59%), H−6 → L (15%) and H−1 → L (12%). Here, the H−6 → L transition is largely occurring between MOs localized at the PN moiety. As such, this state has a higher LE character compared to its singlet counterpart (S1 state) in this geometry. Other theoretical reports have also assigned the T1 state of 2CzPN with a strongly mixed CT-LE character, e.g. by using optimally tuned long-range corrected functionals and PCM54 and uncorrected TDA-PBE028 and TD-B3LYP34 functionals. In addition, Huang et al.42 have shown how the LE contribution of this state increases with the increase of the HF exchange in conventional functionals.
Only 0.06 eV below the S1 state is the T2 state, characterized by a large contribution from a H−2 → L (72%) CT excitation. This energy separation is smaller than when using an empirically corrected TD-LC-w*PBE functional and the 6-31+G* basis set (ca. 0.1 eV).35 The following (T3–T5) states present a substantial LE character emerging from transitions between the Cz units as shown in Fig. 4.
3.2. Minima of the low-lying excited states
The energy distribution and nature of the adiabatic minima of the low-lying singlet and triplet excited states of 2CzPN may drive the plausible energy relaxation channels, for instance, ISC and RISC. The DFT/MRCI-R2016 adiabatic excitation energies of the optimized low-lying singlet and triplet states are listed in Table 2. The corresponding geometries are presented in Fig. 5.| State | ΔE (eV) | Transition | % weight | ZPVE | Exp. | Theoryb |
|---|---|---|---|---|---|---|
| a Ref. 13, onset of the fluorescence and phosphorescence (77 K) spectra in toluene. b Other theoretical reports. c Ref. 42, TD-B3LYP/6-31G*, UDFT for the triplet states. d Ref. 42, TD-PBE0/6-31G*, UDFT for the triplet states. e Ref. 7, calculated from the highest peak of the emission spectrum in toluene at 77 K. | ||||||
| S1 | 2.91 | H → L | 89.3 | −0.09 | 2.94,a 2.96e | 2.84,c 2.99d |
| S1′ | 2.92 | H → L | 89.4 | −0.10 | ||
| S2 | 3.13 | H−2 → L | 83.1 | −0.01 | ||
| T1 | 2.79 | H → L | 84.7 | −0.10 | 2.75,a 2.65e | 2.50,c 2.53,d |
| T2 | 3.01 | H−2 → L | 83.4 | −0.07 | 2.71c | |
Minima for the S1, S2, T1 and T2 states were found. Two enantiomeric minima for each excited state related to the opposite orientation of the Cz groups in space are obtained. We have considered the two geometries for the S1 state (S1 and S1′) for the present analysis. In this manner, we are able to cover relaxation pathways arising from both directions of coordinate space. The minimum geometries of the S2′, T1′ and T2′ states are given in Fig. S3 of the ESI.†
A common feature of these optimized excited state minima is the altered orientation of the Cz rings. They arrange in a more parallel fashion with respect to one another and in a more perpendicular fashion with regard to the PN ring. This trend is the largest for the S1(S1′) minima, followed by the T1 and T2 geometries (Fig. 5). Concomitantly, the CT character of these states increases upon geometry relaxation (compare Fig. 4, 6 and Fig. S10, ESI†). The change in electronic structure is especially pronounced for the T1 state, which exhibits a considerable CT-LE mixing in the FC region but adiabatically it is mainly a CT state. This result is at variance with conclusions drawn by other authors who predict a much higher LE character for the adiabatic T1 state of 2CzPN (near 50%, LC-ω*PBE/6-31G*) on the basis of CT indices obtained by orbital composition analysis42 and natural transition orbital analysis35.
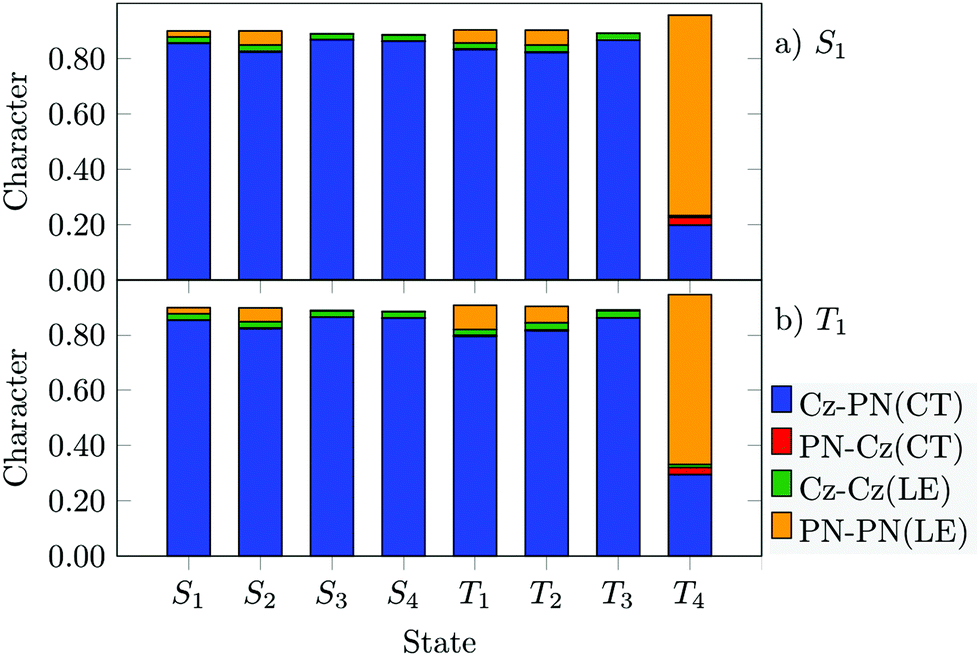 | ||
| Fig. 6 Fragment-based analysis of the DFT/MRCI-R2016 wavefunctions performed at the (a) S1 and (b) T1 state minima. | ||
In line with the large geometry change, the geometry relaxation effect leads to an energy stabilization of the S1 state by ca. 0.28 eV w.r.t. the FC region. The onsets of the experimental emission spectra in toluene7,13 (2.94–2.96 eV) match the DFT/MRCI-R2016 adiabatic energy for the S1 state of 2.91 eV very closely (Table 2). A similar stabilizing effect is found for the S2 state (by 0.23 eV). In contrast, the geometry relaxation effect on the T1 state energy is significantly lower than in the S1 state, with the consequence that the ΔEST between the S1 and T1 adiabatic states is substantially smaller than that in the FC region. Experimental estimates of ΔEST vary between 0.09 eV and 0.35 eV, depending on the molecular environment and on the underlying model.5,7,10,13,76 Our adiabatic ΔEST gap of 0.12 eV (0.13 eV after inclusion of ZPVE corrections) agrees well with the experimental value in an mCP OLED (0.09 eV), estimated from peak emission fluorescence (300 K) and phosphorescence (5 K) wavelengths.10 By considering the threshold energies of the experimental fluorescence and phosphorescence bands (77 K) in toluene solution as a reference, a ΔEST gap of 0.21 eV is derived.13 An even larger ΔEST value of 0.31 eV is obtained if the T0–01 transition energy is taken to coincide with the first phosphorescence band maximum (77 K), while S0–01 is derived from the crossing point of the absorption and fluorescence bands at 300 K.7,42 Other TDDFT and TDA reports best agreeing with this (latter) value were ΔEST values of e.g. 0.49, 0.34 and 0.46 eV, which can be obtained when using TD-ωB97X/6-31G*,30 TD-B3LYP and TD-PBE034 with the same basis set.
The T2 CT state is influenced by a small percentage of LE character (Fig. S10, ESI†). Its minimum shows up 0.10 eV above the adiabatic S1 state. This situation is different from the FC region where this state is located below the S1 state. In contrast, TD-B3LYP calculations predict this state to be located adiabatically below the S1 state (by 0.17 eV) being the only study reporting about the participation of this state in the (R)ISC decay processes on 2CzPN.34 Preliminary DFT/MRCI calculations locate two adiabatic triplet states (T1 and T2) below the S1 state in the 4CzIPN TADF emitter,12 with the T2 state being halfway from the T1 and S1 states. This strongly suggests marked differences between the TADF mechanisms of both emitters: for 4CzIPN, the presence of the intermediate T2 state helps to accelerate the (R)ISC processes, while for 2CzPN, pathways involving the adiabatic T2 state are thermally activated.
3.3. Relaxation channels
To shed light on possible energy dissipation mechanisms arising upon photoexcitation, we constructed LIPs connecting the optimized electronic states as displayed in Fig. 7. These LIPs offer a qualitative view of the overall photophysics of the excited molecule. The LIPs were calculated using the respective adiabatic minimum internal coordinates.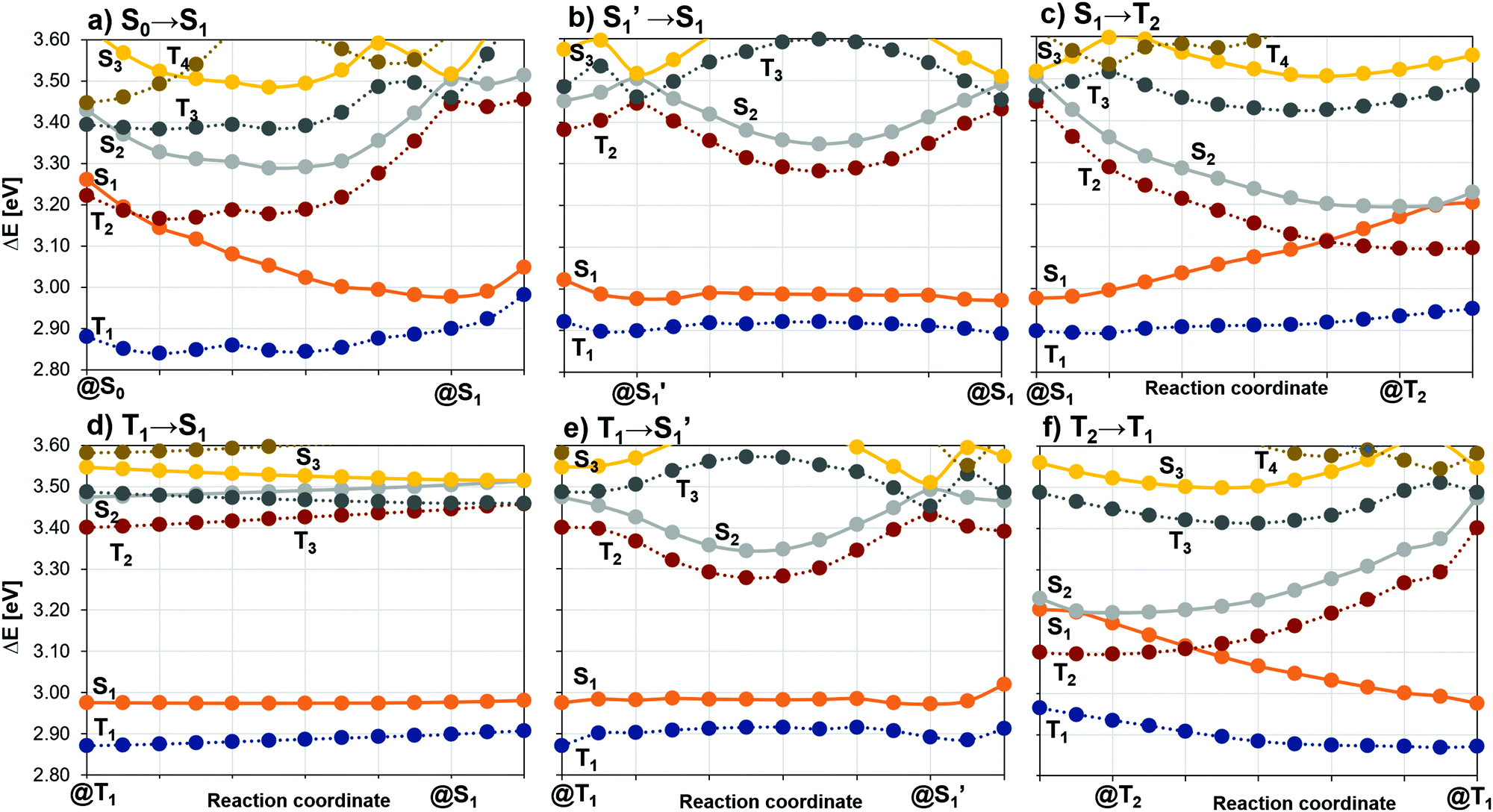 | ||
| Fig. 7 DFT/MRCI-R2016 energies calculated at linear interpolated pathways (LIPs) between target electronic state minima of 2CzPN. Dashed lines correspond to triplet state PESs, while continuous lines correspond to singlet states. Excitation energies calculated relative to the S0 state at its corresponding minimum. | ||
Fig. 7a shows the LIP between the minima of the S0 and the S1 states. Photoexcitation at the FC point eventually results in the population of the adiabatic S1 state due to Kasha's rule. Along this relaxation path that includes marked changes in the Cz torsional angles, we found a crossing between the S1 and T2 PESs in the vicinity of the FC region. A value of 0.18 cm−1 for their mutual SOC at this crossing point indicates that ISC is possible, in principle. However, while these two states are separated by only −0.06 eV in the FC region, their energy gap rises to 0.44 eV at the S1 minimum. A crossing of the S1 and T2 PESs also occurs along a pathway connecting their adiabatic minima (Fig. 7c), in this case with a small energy barrier (∼0.1 eV). In contrast, the T1 and T2 PESs do not intersect (Fig. 7f), suggesting that their vibronic coupling is small. The interconversion of the S1 and S1′ enantiomers proceeds without a substantial barrier (Fig. 7b). As the Cz units are nearly perpendicular to the PN ring, the interconversion coordinate is mainly a sheering distortion. The S1 and T1 PESs show parallel tracks during this interconversion. Note the close energetic proximity of the S1 and T1 PESs at these geometries, a consequence of the similar nuclear arrangements at the S1 and T1 minima and the drastically reduced LE character of the adiabatic T1 state in comparison to those of the FC region.
From the S1 minimum, only the T1 state is accessible in a downhill process, whereas the T2 state is lying 0.10 eV above the adiabatic S1 state. Therefore, ISC channels in 2CzPN may arise from S1 → T1 and S1 → T2 decay, where the latter is thermally activated. In electroluminescent devices, RISC is expected to proceed mainly through a T1 → S1 channel as this is the only state lying energetically close by (see Fig. 7d). However, contributions involving the T2 state cannot be excluded per se. Several factors are decisive for efficient (R)ISC between target states such as the size of SOC, their adiabatic energy difference and their mutual vibrational overlap, which in turn also displays temperature dependence. In the following, the properties driving the mentioned (R)ISC channels are discussed in detail.
The component-averaged SOMEs between these target singlet and triplet states calculated at each minimum are displayed in Table 3. As expected from El-Sayed's rule, the magnitude of these SOMEs is in general small as also commonly found for transitions between ππ* CT states of organic molecules. The coupling between the S1 and T1 CT states (∼0.2 cm−1 at their corresponding minima) arises mainly from the minor LE contributions to the triplet wavefunction (Fig. 6). The larger 〈T1|ĤSO|S1〉 value at the S0 geometry correlates with the higher percentage of LE PN configurations and the increased ΔEST gap in the FC region. Other computational studies report higher SOC values at the excited-state minima as well, e.g., SOMES1→T1 = 0.737 cm−1@T1 minimum.34 The authors mention, however, that the underlying B3LYP-TDA/6-31G* calculations significantly overestimate the S1 and T1 excitation energies.
| SOMEs | @S0 | @S1 | @S1′ | @T1 | @T2 |
|---|---|---|---|---|---|
| 〈T1|ĤSO|S1〉 | 0.479 | 0.211 | 0.217 | 0.292 | 0.499 |
| 〈T2|ĤSO|S1〉 | 0.183 | 0.223 | 0.249 | 0.240 | 0.228 |
| 〈T3|ĤSO|S1〉 | 0.033 | 0.085 | 0.086 | — |
The effect of vibronic SOC in 2CzPN has been analysed in the literature by combining molecular dynamics (MD) and TDDFT-TDA calculations.27,28 Torsional motions modulating the CT–LE mixture were found to facilitate the SOC-driven ISC in 2CzPN. The polarization of experimentally observed ESR signals also supported a spin–vibronic ISC mechanism in this emitter.27 In contrast, a study applying time-resolved infrared vibrational spectroscopy in conjunction with (TD)DFT calculations comes to the conclusion that the suppression of structural changes upon S1–T1 conversion assists the TADF process in Cz benzonitrile derivatives.15
As an alternative to non-adiabatic MD, static approaches based on Herzberg–Teller (HT)-like expansions can be used to determine spin–vibronic interactions.26 To this end, derivatives of the SOMEs (∂SOMEs) with respect to distortions along the dimensionless normal mode (vi) displacements have to be computed. The resulting ∂SOME values (Table S10, ESI†) are very small. For the S1–T1 coupling, the maximum variation of the ∂SOMEs is found in the second decimal place, using either the S1 or T1 minimum nuclear arrangements as the expansion origin, while for the S1–T2 coupling a few values between 0.2 and 0.4 cm−1 can be found. As a consequence, vibronic contributions to the S1–T1 (R)ISC rate constants are expected to be weak.
The (R)ISC rate constants (kISC/kRISC) calculated in the framework of the FC and HT approximations at 300 K for 2CzPN are collected in Table 4. In this table, the rate constants involving the S1 → T1 and T1 → S1 channels were also calculated considering the reported experimental ΔEST gaps of 0.21 eV13 and 0.31 eV7 in toluene solution. Pathways connecting different enantiomers (e.g. S1′ → T1,2 or T1 → S1′) result in very low values for the associated (R)ISC rate constants. They are therefore not displayed in Table 4. Due to the large geometrical (spatial) difference between the initial and final states, their vibrational overlaps are very small. Instead, interconversion of the enantiomers can easily proceed on the S1 or T1 PESs, as shown in Fig. 7b and e.
| ISC | ΔEST (eV) | k CISC (300 K) | k HTISC (300 K) | k ISC (exp.) |
|---|---|---|---|---|
| a Ref. 10, in mCP OLED. b Ref. 13, toluene (10−4 M) solution. c Ref. 76, co-deposited films (mCBP host matrix). d Ref. 7, toluene (10−5 M) solution. e Ref. 5, THF solutions. f Ref. 77, toluene (NR: not reported). | ||||
| S1–T1 | +0.12 | 3.2 × 106 | 7.6 × 106 | |
| +0.09a | 3.1 × 107![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
|||
| +0.21b | 8.9 × 105 | 3.7 × 106 | 2.1 × 107b |
|
| +0.29c | — | — | 2.9 × 107c |
|
| +0.31d | 5.8 × 105 | 1.6 × 106 | 3.5 × 107d |
|
| +0.35e | — | — | 3.0 × 107e |
|
| S1–T2 | −0.10 | 1.1 × 104 | 9.3 × 104 | |
Vibronic SOC enhances the (R)ISC rate constants roughly by a factor of 2 at 300 K, as may be seen when comparing the kHT(R)ISC values with the direct kCISC values in Table 4. While an exponential decrease of the nonradiative transition rate constant may be expected from the energy gap law for weak coupling cases,26 no correlation between the experimentally derived kISC and ΔEST values of 2CzPN can be made out. All experimental studies agree that kISC (S1 → T1) is in the range of 2.1–3.5 × 107 s−1. In contrast, kRISC (T1 → S1) depends substantially (varying by ca. three orders of magnitude) on the experimental conditions and the model for determining the kinetic constants from the measurements. The Adachi group reports kRISC (T1 → S1) values varying slightly in the range of 5.4–6.0 × 103 s−1, despite appreciable ΔEST shifts (0.09–0.29 eV).10,13,76 Much higher RISC rate constants (7.9 × 104–1.7 × 105 s−1) were published by two Chinese work groups although their experimentally deduced ΔEST values (0.31–0.35 eV) are even larger.5,7 Our computed S1 → T1 ISC rate constant (7.6 × 106 s−1 at 300 K for an energy gap of ΔEST = 0.12 eV) is smaller than the experimentally determined ones by a factor of about 3 (Table 4). Unlike the experimental values, the computed ISC rate constants decrease to 1.6 × 106 s−1, if the PESs are vertically shifted to match ΔEST = 0.31 eV. The impact of the energy gap on the RISC rate constant is even more pronounced, as may be expected for TADF from the Boltzmann relation. Our computed kRISC (T1 → S1) value of 2.7 × 104 s−1 (300 K) is approximately in the right ballpark given the high sensitivity of the results with respect to the singlet–triplet energy gap. With the increase of ΔEST from 0.12 to 0.31 eV, we observe a substantial reduction of the computed RISC rate constant by 4 orders of magnitude.
So far, we have not discussed the involvement of the T2 state in the ISC and RISC processes. According to our calculations, T2 is located adiabatically 0.22 eV above the T1 state and 0.10 eV above S1. Population of the T2 state thus requires thermal activation. With a computed HT rate constant of approximately 9.3 × 104 s−1, the S1 → T2 transition cannot compete with the much faster S1 → T1 process. The reverse T2 → S1 transition is a downhill process, but requires the population of the T2 levels either by ISC from the S1 state or by activated internal conversion from T1. Assuming a Boltzmann relation for the latter spin-allowed process yields a prefactor of about 10−4, showing that the T2 → S1 RISC does not essentially contribute to the TADF process unless hot excitons are available.
By using semiclassical Marcus theory to compute the Franck–Condon-weighted density of states and TDDFT-TDA potentials, Xu et al.34 computed much higher rate constants. They obtained values of 1.3–21.1 × 107 s−1 for the S1 → T1 ISC and of 0.2–2.8 × 106 s−1 for the T1 → S1 RISC processes, respectively, depending on the choice of reorganization energy. The very fast S1 → T2 transition (kISC = 2.9 × 107 s−1) reported by these authors is caused by the fact that the T2 state is placed adiabatically below the S1 state in their calculations at the B3LYP-TDA/6-31G* level of theory. Interestingly, Aizawa et al.36 have recently calculated the RISC constants for a variety of TADF emitters using a method involving the singlet–triplet crossing seam-optimized geometry (TDA-LC-BLYP/6-31+G*) and Marcus non-adiabatic theory. In agreement with our findings, the authors state that there is no crossing between the S1 and T1 states of 2CzPN but that the optimized intersection corresponds to a crossing with the T2 state. A RISC rate constant in the order of 103 s−1 together with an activation energy of ca. 0.25 eV has been reported for isolated 2CzPN using their methodology.
4. Conclusions
By means of combined PBE0/SV(P) and DFT/MRCI-R2016 quantum chemical computations, we have studied in detail excited-state decay pathways and (R)ISC mechanisms of the blue TADF emitter 2CzPN. To this end, vertical and adiabatic electronic excitation energies, vibrational frequencies and wave functions as well as (R)ISC rate constants including direct and vibronic SOC were determined.Two minima differing by 0.074 eV (1.7 kcal mol−1) in energy were located in the electronic ground state of 2CzPN. Their nuclear arrangements differ mainly in the orientation and the opening angle of the Cz molecular planes. Based on energetic considerations and on their spectral properties, we decided to focus on the more stable conformer in the following. Enantiomers of both structures exist that are separated from their respective mirror images by high barriers in the electronic ground state.
In the FC region, two triplet excited states are located below the S1 state with energy separations of ΔE(S1–T1) = 0.40 eV and ΔE(S1–T2) = 0.06 eV according to the DFT/MRCI-R2016 calculations. The excited-state transition densities, studied through fragmentation analysis of the natural transition density, revealed the nature of these states to have a strong CT component. The admixture with LE character is substantial in the triplet manifold (e.g. for the T1 state) and less pronounced in the low-lying excited singlet states. The excellent agreement of the computed absorption spectrum in vacuum with available experimental absorption spectra in toluene, THF and DCM solutions and the negligible solvent effect on the spectral properties lends confidence to the chosen computational protocol.
The calculated adiabatic DFT/MRCI-R2016 excitation energies in vacuum locate only the T1 state below the S1 state (by 0.12 eV), while the T2 state lies 0.10 eV above. The intersection of the S1 and T2 PESs occurs close to the FC point. Considering the small magnitude of the electronic SOC (component averaged 〈T2|ĤSO|S1〉 = 0.18 cm−1) renders the dynamic ISC S1 → T2 along the S1 geometry relaxation path not very likely. At the optimized equilibrium structures of the S1 and T1 states, the Cz rings are markedly more perpendicular to the PN plane than in the ground state. The electronic decoupling of their π-systems facilitates the charge separation in these donor–acceptor systems, thus enhancing their CT character. Moreover, it enables a nearly barrierless interconversion of the enantiomers in the first excited singlet and triplet states by a sheering motion of the Cz units. Because of the small overlaps of the vibrational wavefunctions of the two enantiomeric potential energy wells, we nevertheless have indications that cross-couplings, such as S1′ → T1 ISC, do not play an important role among the excited-state decay processes.
The mutual (direct) SOCs between the S1 and T1 states at their respective minimum geometries are reduced in comparison to the FC point, a direct consequence of the varying LE contributions to the wave functions. Furthermore, the calculated derivatives of the S1–T1 SOMEs with respect to dimensionless normal mode displacements are small, but not negligible indicating that the contributions of Herzberg–Teller vibronic SOC to the k(R)ISC constants are moderate. Spin–vibronic interactions are found to speed up the S1–T1 ISC and T1–S1 RISC processes by a factor of about 2. The triplet state formation is predicted to proceed predominantly via the S1 → T1 channel with a rate constant of 8 × 106 s−1 at room temperature, somewhat smaller than the experimentally derived values of 2–3 × 107 s−1,5,7,10,13,76 but in the right ballpark. In addition, a thermally activated S1 → T2 channel is available, but predicted to be less efficient (kISC = 9 × 104 s−1).
Our calculated RISC rate constant associated with the back population of the S1 state of 2CzPN in vacuum through the T1–S1 channel (kRISC = 3 × 104 s−1) certainly is one order of magnitude larger than the smallest constants (5–6 × 103 s−1) reported in solution and in OLEDs,10,13,76 but significantly smaller than the highest value (3.5 × 106 s−1)77 determined by kinetic modelling of transient triplet absorption spectra in toluene solution. Astonishingly, the experimentally derived RISC rate constants and ΔEST values do not appear to be correlated. On one hand, nearly constant kRISC values have been reported, despite appreciably different ΔEST gaps (0.09–0.29 eV).10,13,76 On the other hand, a variation of experimentally derived rate constants by ca. 2 orders of magnitude can be made out when comparing different measurements of 2CzPN RISC in toluene solutions with similar singlet–triplet energy gaps of 0.31 and 0.29 eV, respectively.7,13 Following this consideration, we repeated the (R)ISC rate constant calculations using ΔEST values of two of these experimental reports, i.e. 0.2113 and 0.31 eV7,42 in addition to our computed value of 0.12 eV to test the impact of singlet–triplet separation on the transition probabilities. As expected for a thermally activated process in the weak coupling limit, the RISC rate constant decreases exponentially (by ca. 4 orders of magnitude) when increasing ΔEST from 0.12 eV to 0.31 eV, while keeping the electronic coupling terms constant. The downhill S1 → T1 ISC process is less affected (less than one order of magnitude) by this variation of the energy gap.
Although the T2 state lies adiabatically close to S1, it is not expected to play a major role in mediating the spin-flips in 2CzPN unless the T2 state is directly populated by hot excitons from the embedded OLED layer. Starting from the equilibrated S1 state, the S1 → T2 ISC is an activated process that cannot compete with the much faster S1 → T1 ISC. Moreover, we found no indication for a strong vibronic coupling of the T2 and T1 potentials that could help overcome the negative exponential dependence of the RISC rate constant on the magnitude of the energy gap.
In comparison, DFT/MRCI calculations on the very efficient 4CzIPN TADF emitter12 show a significant CT-LE mixing in the low-lying triplet states and minor effects in the S1 state. This complements with a T2 state energetically located halfway from the S1 and T1 state, acting as an intermediate between these states, thereby accelerating the (R)ISC processes.
Our calculations show that the (R)ISC probability in 2CzPN depends mainly on the interplay between the S1 and T1 states with spin–vibronic interactions accelerating the nonradiative transitions only marginally. One possible explanation for the strange findings with regard to the RISC rate constants in relation to the S1–T1 energy gap in 2CzPN is that the models employed for deriving ΔEST values from spectroscopic data are not adequate. Could, alternatively, a larger singlet–triplet splitting, caused by higher LE contributions to the excited-state wave functions, be compensated by an increase of the electronic SOC strength? In principle, this is possible but difficult to imagine in this particular case. At the S0 geometry, where the ΔE(S1–T1) is relatively large (0.40 eV), the magnitude of 〈T1|ĤSO|S1〉 differs by a factor of ca. 2 from the values at the relaxed excited-state geometries. The increased electronic coupling would lead to an acceleration of the ISC probability by a factor of about 4 in FC approximation, as opposed to a deceleration by 4 orders of magnitude caused by the reduced vibrational overlaps of the shifted potentials. The most plausible explanation for the high RISC rates, reported in some experimental studies,5,7,77 is the participation of other processes such as triplet–triplet annihilation or hot exciton transfer in the delayed fluorescence.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (DFG) through projects MA 1051/17-1 and 396890929/GRK 2482 is gratefully acknowledged.References
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- D. R. Lee, S.-H. Hwang, S. K. Jeon, C. W. Lee and J. Y. Lee, Chem. Commun., 2015, 51, 8105–8107 RSC.
- R. Wang, Y.-L. Wang, N. Lin, R. Zhang, L. Duan and J. Qiao, Chem. Mater., 2018, 30, 8771–8781 CrossRef CAS.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Sci. Rep., 2015, 5, 7859 CrossRef CAS.
- S. Gan, S. Hu, X.-L. Li, J. Zeng, D. Zhang, T. Huang, W. Luo, Z. Zhao, L. Duan, S.-J. Su and B. Z. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 17327–17334 CrossRef CAS.
- G. H. Kim, R. Lampande, J. B. Im, J. M. Lee, J. Y. Lee and J. H. Kwon, Mater. Horiz., 2017, 4, 619–624 RSC.
- Y. Zhang, D. Zhang, T. Tsuboi, Y. Qiu and L. Duan, Sci. China: Chem., 2019, 62, 393–402 CrossRef CAS.
- Y. J. Cho, B. D. Chin, S. K. Jeon and J. Y. Lee, Adv. Funct. Mater., 2015, 25, 6786–6792 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- J. W. Sun, K.-H. Kim, C.-K. Moon, J.-H. Lee and J.-J. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 9806–9810 CrossRef CAS.
- H. Yersin, Highly efficient OLEDs: materials based on thermally activated delayed fluorescence, Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2019 Search PubMed.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef.
- S. Haseyama, A. Niwa, T. Kobayashi, T. Nagase, K. Goushi, C. Adachi and H. Naito, Nanoscale Res. Lett., 2017, 12, 268 CrossRef.
- M. Saigo, K. Miyata, S. Tanaka, H. Nakanotani, C. Adachi and K. Onda, J. Phys. Chem. Lett., 2019, 10(10), 2475–2480 CrossRef CAS.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS.
- M. Inoue, T. Serevičius, H. Nakanotani, K. Yoshida, T. Matsushima, S. Juršėnas and C. Adachi, Chem. Phys. Lett., 2016, 644, 62–67 CrossRef CAS.
- D. de Sa Pereira, C. Menelaou, A. Danos, C. Marian and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10(12), 3205–3211 CrossRef CAS.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- I. Lyskov and C. M. Marian, J. Phys. Chem. C, 2017, 121, 21145–21153 CrossRef CAS.
- C. M. Marian, J. Phys. Chem. C, 2016, 120(7), 3715–3721 CrossRef CAS.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS.
- E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend and N. C. Greenham, J. Phys. Chem. Lett., 2018, 9, 4053–4058 CrossRef CAS.
- Y. Olivier, B. Yurash, L. Muccioli, G. D’Avino, O. Mikhnenko, J. C. Sancho-García, C. Adachi, T.-Q. Nguyen and D. Beljonne, Phys. Rev. Mater., 2017, 1, 075602 CrossRef.
- D. Kim, Bull. Korean Chem. Soc., 2017, 38, 899–903 CrossRef CAS.
- K. Lee and D. Kim, J. Phys. Chem. C, 2016, 120, 28330–28336 CrossRef CAS.
- Y. Olivier, J.-C. Sancho-Garcia, L. Muccioli, G. D’Avino and D. Beljonne, J. Phys. Chem. Lett., 2018, 9, 6149–6163 CrossRef CAS.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- Y. Olivier, M. Moral, L. Muccioli and J.-C. Sancho-Garcia, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- S. Xu, Q. Yang, Y. Wan, R. Chen, S. Wang, Y. Si, B. Yang, D. Liu, C. Zheng and W. Huang, J. Mater. Chem. C, 2019, 7, 9523–9530 RSC.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS.
- N. Aizawa, Y. Harabuchi, S. Maeda and Y.-J. Pu, Nat. Commun., 2020, 11, 3909 CrossRef CAS.
- S. Grimme and M. Waletzke, J. Chem. Phys., 1999, 111, 5645–5655 CrossRef CAS.
- I. Lyskov, M. Kleinschmidt and C. M. Marian, J. Chem. Phys., 2016, 144, 034104 CrossRef.
- C. M. Marian, A. Heil and M. Kleinschmidt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1394 Search PubMed.
- J. Föller and C. M. Marian, J. Phys. Chem. Lett., 2017, 8, 5643–5647 CrossRef.
- N. Lüdtke, J. Föller and C. M. Marian, Phys. Chem. Chem. Phys., 2020, 22, 23530–23544 RSC.
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS.
- X. Tian, H. Sun, Q. Zhang and C. Adachi, Chin. Chem. Lett., 2016, 27, 1445–1452 CrossRef CAS.
- M. Alipour and N. Karimi, J. Chem. Phys., 2017, 146, 234304 CrossRef.
- R. Baer, E. Livshits and U. Salzner, Annu. Rev. Phys. Chem., 2010, 61, 85–109 CrossRef CAS.
- H. Sun, C. Zhong and J.-L. Brédas, J. Chem. Theory Comput., 2015, 11, 3851–3858 CrossRef CAS.
- M. Loebnitz, Einfluss des Torsionswinkels zwischen Donor- und Akzeptor-Einheiten von Isomeren des bis(Carbazolyl)-Dicyanobenzols auf die spektroskopischen Eigenschaften, Bachelor's thesis, Heinrich-Heine Düsseldorf Universität, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Schäfer, A. Klamt, D. Sattel, J. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- TURBOMOLE V7.0 2015, A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- P. Deglmann and F. Furche, J. Chem. Phys., 2002, 117, 9535–9538 CrossRef CAS.
- J. Neugebauer, M. Reiher, C. Kind and B. A. Hess, J. Comput. Chem., 2002, 23, 895–910 CrossRef CAS.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS.
- F. Plasser, TheoDORE 1.7.2: A Package for Theoretical Density, Orbital Relaxation, and Exciton Analysis, n.d.
- A. A. Voityuk, J. Chem. Phys., 2014, 140, 244117 CrossRef.
- S. Mai, F. Plasser, J. Dorn, M. Fumanal, C. Daniel and L. González, Coord. Chem. Rev., 2018, 361, 74–97 CrossRef CAS.
- M. Kleinschmidt, J. Tatchen and C. M. Marian, J. Comput. Chem., 2002, 23, 824–833 CrossRef CAS.
- M. Kleinschmidt and C. M. Marian, Chem. Phys., 2005, 311, 71–79 CrossRef CAS.
- M. Kleinschmidt, J. Tatchen and C. M. Marian, J. Chem. Phys., 2006, 124, 124101 CrossRef.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- B. Schimmelpfennig, AMFI, an atomic mean-field spin–orbit integral program, Stockholm University, 1996 Search PubMed.
- M. Etinski, J. Tatchen and C. M. Marian, J. Chem. Phys., 2011, 134, 154105 CrossRef.
- M. Etinski, V. Rai-Constapel and C. M. Marian, J. Chem. Phys., 2014, 140, 114104 CrossRef.
- J. Tatchen, N. Gilka and C. M. Marian, Phys. Chem. Chem. Phys., 2007, 9, 5209–5221 RSC.
- F. Dinkelbach and C. M. Marian, J. Serb. Chem. Soc., 2019, 84, 819–836 CrossRef CAS.
- M. Etinski, J. Tatchen and C. M. Marian, Phys. Chem. Chem. Phys., 2014, 16, 4740–4751 RSC.
- M. Y. Wong, S. Krotkus, G. Copley, W. Li, C. Murawski, D. Hall, G. J. Hedley, M. Jaricot, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, D. Beljonne, L. Muccioli, M. Moral, J.-C. Sancho-Garcia, M. C. Gather, I. D. W. Samuel and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2018, 10, 33360–33372 CrossRef CAS.
- T. Furukawa, H. Nakanotani, M. Inoue and C. Adachi, Sci. Rep., 2015, 5, 8429 CrossRef CAS.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. Goodson, J. Am. Chem. Soc., 2020, 142, 8074–8079 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the optimized electronic states in terms of geometries, MOs, vertical excitation energies, fragmentation analyses and Cartesian coordinates, derivatives of the SOMEs and vibrational coupling modes. See DOI: 10.1039/d0cp06011a |
| This journal is © the Owner Societies 2021 |