
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs the H4 histone tail intrinsically disordered or intrinsically multifunctional?†
Konstantin
Röder

Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: kr366@cam.ac.uk
First published on 17th February 2021
Abstract
The structural versatility of histone tails is one of the key elements in the organisation of chromatin, which allows for the compact storage of genomic information. However, this structural diversity also complicates experimental and computational studies. Here, the potential and free energy landscape for the isolated and bound H4 histone tail are explored. The landscapes exhibit a set of distinct structural ensembles separated by high energy barriers, with little difference between isolated and bound tails. This consistency is a desirable feature that facilitates the formation of transient interactions, which are required for the liquid-like chromatin organisation. The existence of multiple, distinct structures on a multifunnel energy landscape is likely to be associated with multifunctionality, i.e. a set of evolved, distinct functions. Contrasting it with previously reported results for other disordered peptides, this type of landscape may be associated with a conformational selection based binding mechanism. Given the similarity to other systems exhibiting similar multifunnel energy landscapes, the disorder in histone tails might be better described in context of multifunctionality.
1 Introduction
Eukaryotic, genomic DNA is packed into chromatin, a high-order structure allowing for compact storage of genomic information in cells. Packing and unpacking of chromatin not only relates to storing the DNA in cells, but is also important in transcription, regulation and repair. Chromatin itself is formed by a large number of smaller structural units, nucleosomes, which are protein–DNA complexes where around 150 base pairs are wrapped around a protein core particle, formed by four histones (H2A, H2B, H3 and H4). The proteins contain positively charged, extended regions, the histone tails, which affect chromatin compaction by mediating interactions within and between nucleosomes.1–5 Furthermore, epigenetic changes of the histone tails have been associated with alterations in chromatin structure. Previous experimental6–9 and computational studies reveal a high degree of structural disorder in the histone tails. The resulting polymorphism is observed at local and global levels in experiment.10,11The H4 histone tail likely plays an important role in the internucleosome interaction,12 and deletion causes significant decompaction.13 Residues 16–20 (KRHRK) form the basic patch, a positively charged region, which can interact with the acidic patch in H2A and H2B, as well as DNA.14–16 Switching between the binding modes relies on ionic screening,14 but the interactions of the H4 tail are not simply electrostatic.17,18 The H4 tail likely regulates DNA-DNA interactions between nucleosomes.19,20 These observations have led to the suggestion that the H4 tail is responsible for the polymorphic nature of chromatin structure.21
The H4 tail itself exhibits a high degree of disorder in solution NMR22 and CD spectroscopy,23 even in highly condensed states.24 A small amount of α-helical content has been observed in the isolated tail, and acetylation of lysine residues in the tail increases the helical character significantly.23 Furthermore, it has been reported that the higher degree of helicity is conditional on DNA binding.25 In another combined CD and NMR study, isolated and hyperacetylated H4 tails were observed to exhibit flexible and elongated structures.26 A multiscale computational investigation, in combination with NMR-derived constraints, reported a large amount of disorder in the isolated, unmodified tail, with increased order observed after acetylation, in particular for K16Ac.27 The increased order leads to a larger persistence length (increase of 41%27), a similar increase in secondary structure, and altered structural ensembles. The folding of the histone tails in this computational study was associated with weaker internucleosome contact, and hence poorer chromatin compaction. More recently, this change has been further characterised through a direct link between acetylation of H4 K16 and the free energy surface.28 Super-resolution microscopy revealed the loss of compaction as a result of hyperacetylation in vivo.29
In contrast, other studies have reported enhanced binding30 and decreased helical content31 for H4 K16Ac. Metadynamics simulations suggested increased conformational diversity with loss of binding to the acidic patch, and the increased disorder upon acetylation may be related to loss of intramolecular hydrogen-bonding upon binding.32 Clearly, the highly dynamic structure of the H4 tail complicates analysis significantly in experimental and theoretical studies. The highly dynamic character persists in long time scale simulations.5 This behaviour differs significantly from globular proteins, and might be closer to the theoretical limits predicted by polymer theory.33 While a number of recent studies have focused on the mesoscale,34–38 the experimental and computational descriptions of the structural ensembles adopted by the H4 tail and the associated dynamics are incomplete, mainly due to the disorder.
The energy landscape of a molecular system contains all the information necessary to describe the kinetic, thermodynamic and structural properties of molecular systems.39 The energy landscape associated with intrinsically disordered proteins, are often described as rugged. However, previous work has shown that intrinsically systems may exhibit a large number of funnels relatively similar in energy,40 while a less structured energy landscape has been observed for Aβ monomers.41
Exploration of the potential energy landscape,42,43 can provide direct insight into the structural ensembles of the histone tail and in the organisation of the energy landscape. The computational framework exploits on geometry optimisation, and can therefore overcome the broken ergodicity associated with high energy barriers and long time scales.44 This approach has previously been applied to other intrinsically disordered peptides,40,41 and to analyse mutational changes.45
In this contribution, the potential and free energy landscape for the H4 histone tail are presented. In addition, the H4 histone tail was modified by removing a proton from K16 and K20, i.e. from the basic patch, to model binding to chromatin at a simplistic level. The energy landscapes for this modified system are presented alongside the isolated H4 tail, highlighting how chromatin binding modifies the structural ensembles and alters the energy barriers between different states. The key result of this study is that the energy landscape for the H4 histone tail exhibits a number of clearly defined states, with the associated funnels separated by large energy barriers. The hierarchy observed in the energy landscape is reminiscent of similar organisation observed in multifunctional systems, and offers an interpretation of the polymorphism of the histone tail in the context of an evolved, multifunctional system. These clearly defined states are observed to be even more stable in the bound model, indicating that the corresponding structures may play important and distinct roles.
2 Methodology
The energy landscapes were explored using discrete path sampling (DPS)46,47 to construct a kinetic transition network.48,49 Transition state candidates were located using the doubly-nudged elastic band (DNEB)50–53 algorithm. The quasi-continuous interpolation scheme (QCI)54,55 was employed, using the AMBER topology to create the percolating bonding network for the interpolation, with additional constraints on the peptide bonds to restrict cis–trans isomerism.55 For short end point distances of less than 10 Å, initial linear interpolations were used instead to reduce computational cost. Transition state candidates from DNEB were subsequently converged with hybrid eigenvector-following.56,57 The AMBER ff14SB force field58 with proper symmetrisation59,60 and an implicit General Born solvation (igb = 2) with implicit salt (M = 0.15) were used.61–63 Starting structures were obtained from previous work,27 with an H4 tail size of 26 residues.64 The sequence of the tail is SGRGK-GGKGL-GKGGA-KRHRK-VLRDN-Q, where the highlighted residues are the ones deprotonated in the bound-tail model. For these residues, the deprotonated lysine template from the standard AMBER libraries was used.The chosen force field retains some bias,65,66 in particular with disordered states that are too compact67 and secondary structure.68,69 Furthermore, a study of the effect of solvation models33 shows that the applied model does not represent the unfolded cases to a high accuracy. The representation of unfolded state requires accurate capture of entropic effects, but these will not affect the potential energy landscape. The discussion of the organisation of the potential energy landscape remains therefore valid within the error usually encountered in force fields, and has been applied previously.40,41 More caution needs to be applied when considering the free energy landscapes, and this point will be discussed in that context below.
The sampling was initiated by finding discrete paths between pairs of structures from previous work,27 using an improved version of the quasi-continuous interpolation scheme (details have been published elsewhere55). After discrete paths were found all databases were merged into one database for each system. Further sampling to remove unphysical barriers70,71 and kinetic traps,71 and to improve local connectivity within regions41 of the landscape was conducted. Convergence was judged by the disconnectivity graphs72,73 and convergence of the pathways between different funnels. Free energies were obtained using the superposition approach within a harmonic approximation,74 and a recursive regrouping scheme was employed to define free energy states.75
To analyse the resulting landscape, DSSP76 was employed for the secondary structure assignments, half-sphere exposure (HSE)77 for the solvent-accessibility, and the radius of gyration was considered as another structural measure. Further calculations included the solvent-accessible surface area (SASA)78 for the entire molecule and the contribution of the basic patch, the distance between the lysyl nitrogen atoms in residues 16 and 20, and the hydrogen-bonds formed, as well as the ϕ and ψ dihedrals for all residues. Apart from the HSE calculations, CPPTRAJ79 was used for all calculations.
3 Results
The energy landscapes analysed and presented here are containing 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 603 local minima and 43389 transition states for the isolated tail, and 56707 local minima and 71265 transition states for the bound model, in line with other energy landscapes published.41 The tight and complex structure of the tails led to a number of complications in the sampling. We observed regular unphysical chain crossing events and chirality and cis–trans inversions. To overcome these problems in the sampling an improved version of the quasi-continuous interpolation scheme was extensively used.55
603 local minima and 43389 transition states for the isolated tail, and 56707 local minima and 71265 transition states for the bound model, in line with other energy landscapes published.41 The tight and complex structure of the tails led to a number of complications in the sampling. We observed regular unphysical chain crossing events and chirality and cis–trans inversions. To overcome these problems in the sampling an improved version of the quasi-continuous interpolation scheme was extensively used.55
3.1 The potential energy landscape
The disconnectivity graphs for the isolated and bound H4 tail are shown in Fig. 1. Both landscapes exhibit a number of funnels with comparable energies and high potential energy barriers between these states. This organisation changes rather little upon binding. Comparing the two energy landscapes, we can identify a similar set of distinct funnels associated with particular structural features, although some of these subsets have more in common than others. The structural ensembles are labelled A to E, and the assignment, together with example structures, are illustrated in Fig. 1. Given the small change introduced by the addition or removal of protons, it is not unexpected that the landscape is largely conserved. Nonetheless, two sets of structures are unique to the individual landscapes, and they are highlighted with a red asterisk in Fig. 1.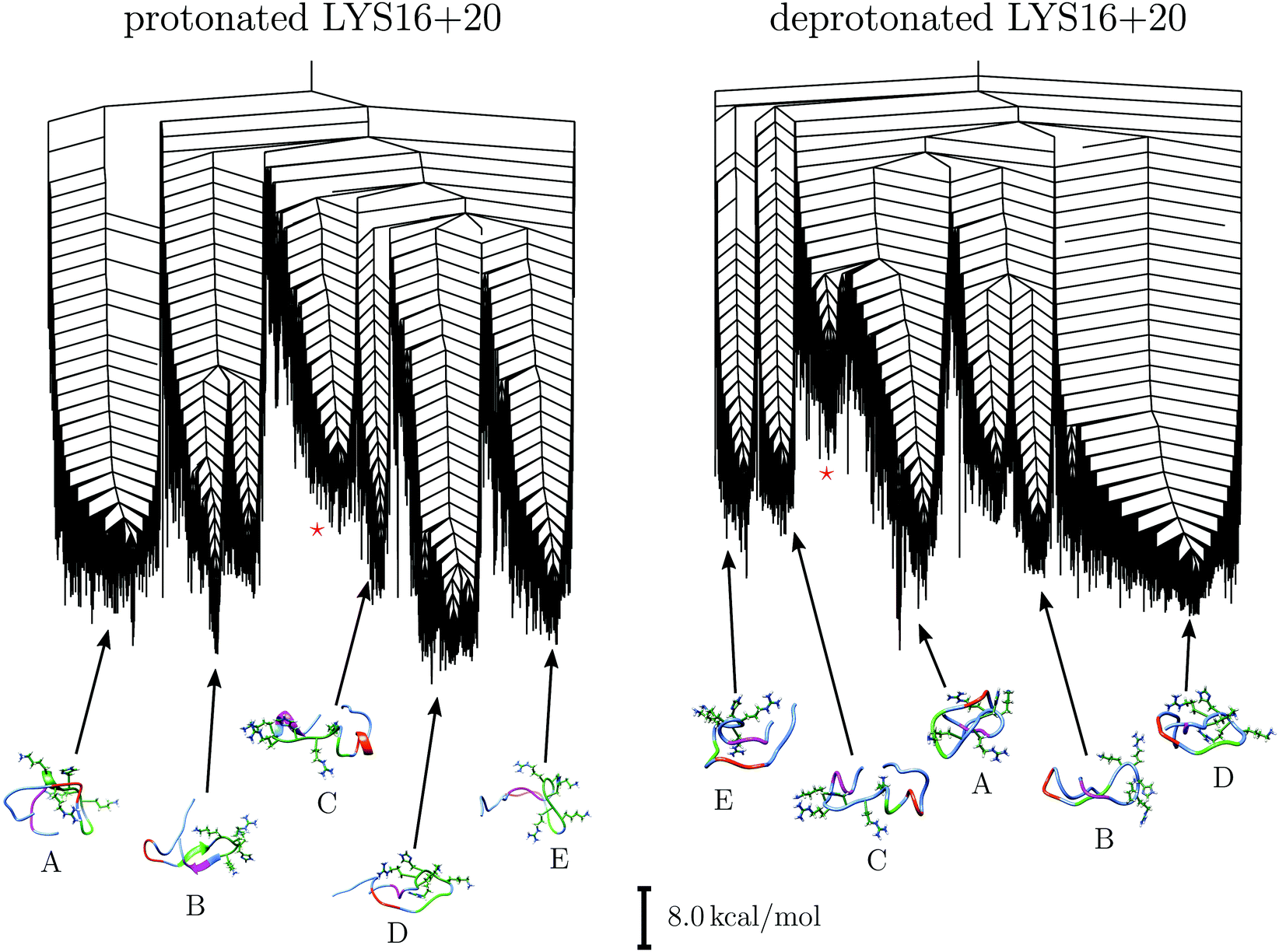 | ||
| Fig. 1 The potential energy landscapes for the isolated (left) and bound (right) H4 histone tail. Both landscapes exhibit a number of funnels, and high structural diversity. While the relative size of the funnels, their depths, and the barriers between them vary, the binding does not reduce the structural diversity, but biases towards different structural motifs. Besides the five shared set of structural ensembles, A to E, each of the landscapes exhibits an additional distinct set, highlighted with a red star. The structural representations show residues 16 to 20 (KRHRK) explicitly, and the regions forming important intramolecular contacts are highlighted as follows: Gly7 to Lu10 in red, Lys12 to Gly14 in green, and Leu22 to Asp24 in purple. The assignments of the corresponding funnels are based on the key structural features exhibited by structures in each funnel, as discussed in the text and the ESI.† UCSF Chimera80 was used for the structural representations. | ||
Four of the sets of structures, A, B, D and E, are based on hairpin like structures. The hairpins differ in size, but they all exhibit some free residues at the N-terminus, and there are distinct additional contacts for each set. Only one set, C, deviates and displays a different structure, which involves helices. The conservation of structures between the landscapes is strongly related to the formation of key interactions between residues. In Fig. 2, the lowest energy structures in the five sets for the isolated tail are shown. For each structure, important regions are highlighted, hydrogen-bonds between residues are shown, and a surface map highlighting the basic patch orientation is displayed. For set B, there are two slightly different subsets. A detailed description of all the sets, and some key properties are provided in the ESI.†
 | ||
| Fig. 2 Detailed structures for the sets A to E in the isolated H4 tail. For set B, the two subfunnels are represented in the second and third row. The first column shows a simplified cartoon representation with intramolecular hydrogen bonds between residues. The structural representations show residues 16 to 20 (KRHRK) explicitly, and the regions forming important intramolecular contacts are highlighted as follows: Gly7 to Lu10 in red, Lys12 to Gly14 in green, and Leu22 to Asp24 in purple. The structures in the second column shows the solvent accessible surface, with the basic patch (residues 16 to 20) highlighted in dark blue. UCSF Chimera80 was used for the structural representations in the first column, and PyMOL87 for the surface representations in the second column. | ||
A key observation for the potential energy landscape is their organisation. The existence of multiple, stable funnels gives this system a multifunnel character. Such character has been associated with multifunctionality before,81 and hints at an evolved minimisation of frustration in the system.82–85 Indeed, when comparing the frustration index86 for the histone tail with the one derived form the potential energy landscape for Aβ monomers,41 we observe an order of magnitude lower frustration at low temperatures for the histone tail (see ESI†).
3.2 The free energy landscapes
The free energy landscapes (Fig. 3 and 4) exhibit more differences than the potential free energies. This fact is not entirely surprising, as different charges will affect entropy directly, and the altered interaction patterns will have further effects. In both cases, the D set is the lowest in energy. While for the isolated tail there is a clear difference between this funnel and the other four sets, after binding sets A and B display comparable free energy. The free energies of the lowest local minima for all five sets are presented in Table 1.| Set | ΔG/kcal mol−1 isolated | ΔG/kcal mol−1 bound |
|---|---|---|
| A | 8.8 | 1.9 |
| B | 6.5 | 2.5 |
| C | 10.7 | 10.1 |
| D | 0.0 | 0.0 |
| E | 7.9 | 8.3 |
The high energy barriers observed in the potential energy barriers are still observed in the free energy landscapes, but there are differences. The highest transition state in the landscape of the isolated tail is around 45 kcal mol−1, while the largest barrier for the bound molecule is about 33% larger. Even the smallest barrier between sets, the free energy barrier between set C and set E in the isolated molecule, is over 20 kcal mol−1.
4 Discussion
This investigation aimed to study the structural ensembles of the H4 histone tail by mapping the energy landscape. A key observation from this work is the emerging organisation of the energy landscape and its relationship to various concepts of how disorder arises. In addition, a simple binding model was introduced to understand the changes upon binding. In the following sections, these points will be discussed, with a focus on the organisation of the energy landscape. Finally, some considerations are presented regarding the validity of the applied models.4.1 The structural ensembles
In our analysis of the energy landscape, we identify five key structural ensembles. Previous studies have described varying numbers of ensembles,27,64,88 with different characteristics. In a multiscale simulation effort,27 three ensembles were explicitly described for the H4 wild type tail. The reported structures featured a hairpin with antiparallel β-sheets characterised between residues 12 to 14 and 21 to 23. These structures correspond to ensemble B in the present work. A similar ensemble, though explicitly with a free tail end for residues 1 to 12, has been described elsewhere.64 The only helical content observed in our study was in set C. A similar structure was reported in previous work,27 with a helical region for residues 19 to 23, in agreement with our results.It has been reported that the structures observed for H4 are quite rigid, based on the formation of stable hairpins, and the resulting ψ and ϕ dihedral angles are restricted to few values, particularly for the Lys-Arg pairs in the basic patch.64,88 In fact, we find that this rigidity is even more restrained in the local funnels corresponding to each set, each one exhibiting a distinct pair of values for most residues. The residues heavily involved in structure stabilisation and binding (residues 12 to 23) appear to be particularly constrained. This result is in agreement with the differences in organisation and order reported for the C- and N-tail for the histone tail.64
Another property that has been described for the H4 tail is the distribution of the end-to-end distance.64 It was reported that the tail features very short and very long end-to-end distances, rather than a higher propensity for mid-ranged distances. Again, our observation of structures with very close contacts between the termini (for example set C), and structures where strong interactions keep the termini apart (for example D) are consistent with this observation.
 | ||
| Fig. 3 Simplified free energy landscape for the isolated H4 tail at 310 K. The graph is coloured according to the surface area formed by the basic patch, with blue indicating large surface exposure, and red indicating a small contribution. Only the lowest 200 free energy minima are shown. This simplification was applied as the funnel for set A is very wide, and therefore the funnels formed by B to E are difficult to see clearly in full representations. A regrouping threshold75 of 2 kcal mol−1 was used. | ||
The overall structural ensemble is very diverse, in agreement with NMR experiments,22 as long as the system is at or close to equilibrium. The helical content observed in experiment23 and other simulations27 is best matched by set C. The fact that the diversity in the structures is preserved in condensed environments24 might be a result of the high energy barriers.
 | ||
| Fig. 4 Free energy landscape for the bound H4 histone tails at 310 K. The graph is coloured according to the surface area formed by the basic patch, with blue indicating large surface exposure of the basic patch, and red indicating a small contribution to the surface area. A regrouping threshold75 of 2 kcal mol−1 was used. | ||
4.2 Differences between the free energy and potential energy landscapes
A key observation of this study is the correspondence between the sets of minima from the potential energy landscape and the free energy landscape. The energy barriers between these funnels are also preserved. These properties indicate that the enthalpic contributions from the strong intramolecular contacts are the key factor determining this organisation. Although a number of free residues and structures with loosely bound tails exist, it is likely that the dynamic behaviour is confined to a particular sets, and the structural diversity arises from the large number of alternatives.The differences in the free energies of the lowest energy structures is fairly similar for the bound and unbound model, with D the lowest and C the highest energy set. A key difference arises in the energies of sets A and B. They are significant lowered by deprotonation, probably due to the fact that sidechain interactions are partly replaced by backbone interactions, allowing for a larger entropic contribution from the now flexible sidechains. Another key difference is observed for the barriers between states. In the bound model, barriers are increased, which we would expect from a system upon binding, as further constraints are introduced, making refolding less likely.
The kinetics of the system are a result of the free energy barriers we observe. Based on the presented energy landscape we expect the states to be stable and long-lived with slow transitions between states, exceeding those reported recently for reversible motion within nucleosomes.89 However, this picture is for an isolated tail, and does not take into account that other molecules may alter the energy landscape and facilitate faster transitions. Furthermore, the force field inaccuracies, as discussed earlier, will likely lead to an overestimate of the barriers, as disordered states are nor represented accurately. While such a correction will not merge all funnels, it will lower barriers between them. Unfortunately, from the data presented here, we cannot precisely describe this effect, and further work is needed to estimate the exact transition time scales between the ensembles. Similarly, further work is needed to study the impact of other molecules and crowding effects on the energy landscape.
4.3 Implications for binding
Previous studies on the interactions of the H4 tail with other histone tails and DNA14–16 identified the importance of the basic patch. Furthermore, it was reported that the Lys16 + Arg17 pair inserts into DNA grooves and binds in this fashion.20 A similar binding mode can be achieved from interactions with Arg19 and Lys20. It is conceivable that the interactions occur with a single DNA duplex, but at the same time, the suggestion that H4 mediates the interactions between DNA strands19,20 indicates interactions with multiple strands. This scenario would require binding of Lys16 and Arg17 to one duplex and of Arg19 and Lys20 to the other.To achieve DNA binding the key residues must be accessible, and not involved in strong intramolecular interactions. Set B fits this criterion perfectly, as all residues in the basic patch are accessible. The basic patch is also generally exposed in set E. For set D, only part of the basic patch is exposed. Finally, in ensembles A and C the residues are quite heavily involved in intramolecular binding. However, as the changes upon binding for A show, these residues can be somewhat more exposed if necessary. Similarly, in set C, Lys20 is somewhat exposed to the surroundings. However, even in these cases the exposure is limited. As a result, it is likely that structures of set A and D, if they bind, would likely only interact through one of the groups, and consequently only with one binding partner. For set D, the arginines are involved in intramolecular structure formation. However, the distance between the lysyl groups is reduced, such that they end up in close proximity, and a single binding site contact would be possible.
Both sets B and E could potentially interact with two duplexes at once, or more strongly with one duplex. An interesting point to notice here is the distances between the lysyl groups in these two sets (see Tables S1 and S2 in the ESI†). For set B, which is the ensemble of hairpin structures that matches the structures described in previous studies, this distance is around 14 Å and effectively unchanged upon binding. In contrast, for set E, the distance is around 8.2 Å in the isolated case, which is reduced to 4.5 Å after binding. This shorter distance might allow for a stronger binding pattern with a single duplex. The changes in lysyl distance based on charges might also underlie the variations in bonding observed in ionic screening.14
While it is not known what binding partners the ensembles described are bound to, the existence of a number of these well-defined structures seems unlikely to be a coincidental. Instead, the different structures present different options for binding to other molecules, and we suggest that this organisation may correspond to evolved multifunctionality.81,85 The apparent stability and robustness of the energy landscape would facilitate chromatin binding. Recently, work on the H1 N-terminal domain revealed that while the region is disordered in solution, a disorder-to-order transition increases its binding affinity.90 The increased binding-affinity after the formation of a ordered structure for H1 and the increased stability of the observed structural ensembles upon binding, indicate that multifunctionality may be an underlying feature of the histone tails in general. This relation between multiple funnels observed and intrinsic multifunctionality consistent with the notion of evolved minimal frustration for biomolecules.81
To achieve the liquid-like organisation of chromatin37,91 transient binding of the histone tails is necessary. Recent work92,93 showed that intrinsic liquid–liquid phase separation is a key factor in chromatin organisation. Nonspecific electrostatic contacts are thought to be one key feature involved, alongside the disordered behaviour of histone tails. As discussed above, all the structural ensembles of the H4 histone tail exhibit some exposure of the binding residues in the basic patch, but the resulting contacts, as they are likely based on electrostatic interactions, will be nonspecific, though they would lead to different structures.
The behaviour of intrinsically disordered proteins and their participation in protein–protein and protein–DNA complexes has also been described through the concept of fuzziness.94,95 The concept establishes that folding and binding are not necessarily coupled in intrinsically disordered proteins, and that conformational heterogeneity can be maintained in bound complexes. Fuzziness exists in DNA complexes,95 through non-specific contacts. It has been proposed that fuzziness is related to structural multiplicity.96 The prediction of distinct structural ensembles in the case of the histone tail described here, with the potential for non-specific interactions fits the picture of the formation of fuzzy complexes well.
The energy landscapes characterised in this work suggest that the organisation might be better described in terms of well-defined, competing, meta-stable structures, rather than disorder. One question that arises from this observation is how histone tails can adopt alternative structures. Clearly, these transitions will not readily occur due to the high energy barriers observed. This stability is a desirable feature for chromatin binding as described above, and consequently external stimulus may be needed to alter the adopted structure. Such a stimulus might be provided by other molecules or environmental conditions, but it would be controllable rather than random. Further, it could be hypothesised that various modifications, such as acetylation, will select structures based on shifting the relative energies of funnels, similar to processes that are observed for mutational changes.45 From this point of view the chemical modifications provide a control mechanism for chromatin organisation. This idea is supported by the previously observed alterations of binding properties as a result of various acetylation patterns.26,27,29 Furthermore, post-translational modifications have been described as one possibility to control fuzzy complexes,97 like those encountered in chromatin assembly.
Finally, the results of this work can be seen in relation to previous studies of intrinsically disordered proteins and disordered tails. It has been proposed that the disorder in tails allows for better capture of binding partners via the ‘fly-casting’ mechanism.98 While the original work proposing this mechanism was based on a single-funnel picture, the multifunnel landscapes presented here are compatible with this mechanism, with other constraints, such as the available space, alternative contacts or environmental condition, potentially determining the funnel. This suggestion also hints at a conformational selection mechanism99 as a means to obtain an ordered bound structure.100 The existence of multifunnel landscapes for conformational selection has been proposed previously,101 and a dependence of the mechanism, i.e. induced-fit versus conformational selection, has been reported based on structural propensities in the isolated molecules.102 We may hypothesise that a multifunnel energy landscape, which supports a number of stable structural ensembles, leads to a conformational selection mechanism, while a higher disorder in the landscape organisation would likely lead to an induced fit mechanism. As most intrinsically disordered peptides are found in well-ordered structures in bound complexes,100 the potential energy landscape, which effectively removes the local vibrational entropic contributions, may allow us insight into the available structures for a given peptide. In this context, it has also been proposed that the inherent lack of order in these biomolecules does not stem from a desirability of disorder itself, but is an efficient way of reducing size and consequently energetic cost.103 Within this interpretation, the peptides still fulfil distinct functions, and multifunnel energy landscapes may be seen as a pathway to obtain such evolved small peptides.
The target functionality in the case of the H4 histone tail is binding to DNA to achieve chromatin organisation. A searching process to locate the binding sites via the ‘monkey bar’ mechanism has been proposed for similar processes,104 and the structural ensembles available for this histone tail could provide the means to facilitate this mechanism. Evidence of optimisation in disordered tails for such DNA searches was previously reported,105 with sequence patterns evolved to allow for speedy DNA searches. The existence of the organised, multifunnel landscape for such a tail provides further evidence for such optimisations.
4.4 Validity of the computational model
The influence of the chosen force field and its effects on the potential and free energy landscapes has been discussed already. Another point that merits attention is the validity of the ‘binding’ model chosen in this work. The deprotonation represents binding to a charged DNA duplex, which effectively neutralises the lysyl group. As this model does not include DNA explicitly, it should be viewed as a suggestion of how binding might be achieved and how strong interactions will be. Furthermore, as we do not include geometric constraints for binding, we cannot comment on which structures will eventually bind. Further calculations will be required to address these questions. Nonetheless, there are important suggestions that arise from our simple model. Firstly, protonation has little effect on the structural ensembles and the large energy barriers observed in the potential and free energy landscapes. This observation highlights the structural integrity based on strong intramolecular contacts. Remodelling of structures will likely require significant external impulse, and the resulting structures when formed are likely to persist. This picture may explain certain, seemingly contradictory, findings in previous work, and give us a better idea of which structures can fulfil particular functions.5 Conclusion
In this contribution we have demonstrated again that the computational exploration of potential and free energy landscapes is capable of describing complex structural ensembles.Our key observation is the organisation of the landscapes, characterised by the existence of deep funnels, each containing a well-defined structural ensemble. While the histone tail has often been described as disordered, this organisation might be better interpreted in terms of a multifunctional system. In fact, the landscapes bear some resemblance to other multifunnel systems, such as G-quadruplexes.106 Hence it is possible that histone tails are evolved multifunctional molecules, where different ensembles fulfil distinct roles. While these roles cannot be assigned from the current calculations, the relationship between multiple funnels and multiple functions is established for other systems.81 The existence of a well-defined set of funnels supporting distinct structures is qualitatively different from the disorder characterised for structural glasses.107,108
The distinct ensembles may facilitate the formation of different bound complexes, helping to provide the necessary transience required for the liquid-like chromatin organisation. The stability of the chromatin-bound structures is a desirable feature in this context. A number of interesting further questions arise from this work. Other histone tails should be investigated to examine whether they also support multifunnel, and perhaps multifunctional, landscapes. Furthermore, modifications, such as acetylation, provide a potential mechanism for structural selection. Again, this hypothesis can be tested with further calculations. Finally, the energy landscape suggests a multifunctional structural ensemble, and further research on the potential bound complexes formed by each set of structures will be insightful.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks Prof. Wales for access to computational resources, and Dr Collepardo-Guevara and Prof. Wales for helpful discussions and comments on the manuscript. KR is funded by the Cambridge Philosophical Society.References
- G. Arya and T. Schlick, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16236–16241 CrossRef CAS.
- G. Arya and T. Schlick, J. Phys. Chem. A, 2009, 113, 4045–4059 CrossRef CAS.
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381–395 CrossRef CAS.
- M. Biswas, K. Voltz, J. C. Smith and J. Langowski, PLoS Comput. Biol., 2011, 7, e1002279 CrossRef CAS.
- A. K. Shaytan, G. A. Armeev, A. Goncearenco, V. B. Zhurkin, D. Landsman and A. R. Panchenko, J. Mol. Biol., 2016, 428, 221–237 CrossRef CAS.
- K. Luger, A. W. Mäder, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS.
- K. Luger and T. J. Richmond, Curr. Opin. Genet. Dev., 1998, 8, 140–146 CrossRef CAS.
- C. A. Davey, D. F. Sargent, K. Luger, A. W. Maeder and T. J. Richmond, J. Mol. Biol., 2002, 319, 1097–1113 CrossRef CAS.
- T. J. Richmond and C. A. Davey, Nature, 2003, 423, 145–150 CrossRef CAS.
- G. Li, M. Levitus, C. Bustamante and J. Widom, Nat. Struct. Mol. Biol., 2005, 12, 46–53 CrossRef CAS.
- J. S. Choy and T. H. Lee, Trends Biochem. Sci., 2012, 37, 425–435 CrossRef CAS.
- M. Shogren-Knaak, H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie and C. L. Peterson, Science, 2006, 311, 844–847 CrossRef CAS.
- B. Dorigo, T. Schalch, K. Bystricky and T. J. Richmond, J. Mol. Biol., 2003, 327, 85–96 CrossRef CAS.
- P.-Y. Kan, T. L. Caterino and J. J. Hayes, Mol. Cell. Biol., 2009, 29, 538–546 CrossRef CAS.
- S. Pepenella, K. J. Murphy and J. J. Hayes, J. Biol. Chem., 2014, 289, 27342–27351 CrossRef CAS.
- J. Erler, R. Zhang, L. Petridis, X. Cheng, J. C. Smith and J. Langowski, Biophys. J., 2014, 107, 2911–2922 CrossRef CAS.
- A. Allahverdi, R. Yang, N. Korolev, Y. Fan, C. A. Davey, C.-F. Liu and L. Nordenskiöld, Nucleic Acids Res., 2011, 39, 1680–1691 CrossRef CAS.
- Y. Liu, C. Lu, Y. Yang, Y. Fan, R. Yang, C. F. Liu, N. Korolev and L. Nordenskiöld, J. Mol. Biol., 2011, 414, 749–764 CrossRef CAS.
- N. Korolev, A. Allahverdi, Y. Yang, Y. Fan, A. P. Lyubartsev and L. Nordenskiöld, Biophys. J., 2010, 99, 1896–1905 CrossRef CAS.
- N. Korolev, H. Yu, A. P. Lyubartsev and L. Nordenskiöld, Biopolymers, 2014, 101, 1051–1064 CrossRef CAS.
- H. Ishida and H. Kono, Biophys. J., 2017, 113, 978–990 CrossRef CAS.
- B. R. Zhou, H. Feng, R. Ghirlando, H. Kato, J. Gruschus and Y. Bai, J. Mol. Biol., 2012, 421, 30–37 CrossRef CAS.
- X. Wang, S. C. Moore, M. Laszckzak and J. Ausió, J. Biol. Chem., 2000, 275, 35013–35020 CrossRef CAS.
- M. Gao, P. S. Nadaud, M. W. Bernier, J. A. North, P. C. Hammel, M. G. Poirier and C. P. Jaroniec, J. Am. Chem. Soc., 2013, 135, 15278–15281 CrossRef CAS.
- J. L. Banères, A. Martin and J. Parello, J. Mol. Biol., 1997, 273, 503–508 CrossRef.
- E. Bang, C. H. Lee, J. B. Yoon, D. W. Lee and W. Lee, J. Pept. Res., 2001, 58, 389–398 CrossRef CAS.
- R. Collepardo-Guevara, G. Portella, M. Vendruscolo, D. Frenkel, T. Schlick and M. Orozco, J. Am. Chem. Soc., 2015, 137, 10205–10215 CrossRef CAS.
- J. Moller, J. Lequieu and J. J. De Pablo, ACS Cent. Sci., 2019, 5, 341–348 CrossRef CAS.
- J. Otterstrom, A. Castells-Garcia, C. Vicario, P. A. Gomez-Garcia, M. P. Cosma and M. Lakadamyali, Nucleic Acids Res., 2019, 47, 8470–8484 CrossRef CAS.
- D. A. Potoyan and G. A. Papoian, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17857–17862 CrossRef CAS.
- D. Yang and G. Arya, Phys. Chem. Chem. Phys., 2011, 13, 2911–2921 RSC.
- R. Zhang, J. Erler and J. Langowski, Biophys. J., 2017, 112, 450–459 CrossRef CAS.
- P. S. Shabane, S. Izadi and A. V. Onufriev, J. Chem. Theory Comput., 2019, 15, 2620–2634 CrossRef CAS.
- G. D. Bascom, K. Y. Sanbonmatsu and T. Schlick, J. Phys. Chem. B, 2016, 120, 8642–8653 CrossRef CAS.
- K. Y. Sanbonmatsu, Curr. Opin. Struct. Biol., 2019, 55, 104–113 CrossRef CAS.
- A. Bendandi, S. Dante, S. R. Zia, A. Diaspro and W. Rocchia, Front. Mol. Biosci., 2020, 7, 15 CrossRef CAS.
- A. Sridhar, S. E. Farr, G. Portella, T. Schlick, M. Orozco and R. Collepardo-Guevara, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7216–7224 CrossRef CAS.
- S. Portillo-Ledesma and T. Schlick, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1434 CAS.
- D. J. Wales, Energy Landscapes, Cambridge University Press, Cambridge, 2003 Search PubMed.
- Y. Chebaro, A. J. Ballard, D. Chakraborty and D. J. Wales, Sci. Rep., 2015, 5, 10386 CrossRef.
- K. Röder and D. J. Wales, J. Am. Chem. Soc., 2018, 140, 4018–4027 CrossRef.
- J. A. Joseph, K. Röder, D. Chakraborty, R. G. Mantell and D. J. Wales, Chem. Commun., 2017, 53, 6974–6988 RSC.
- K. Röder, J. A. Joseph, B. E. Husic and D. J. Wales, Adv. Theory Simul., 2019, 2, 1800175 CrossRef.
- D. J. Wales and P. Salamon, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 617–622 CrossRef CAS.
- K. Röder and D. J. Wales, J. Chem. Theory Comput., 2017, 13, 1468–1477 CrossRef.
- D. J. Wales, Mol. Phys., 2002, 100, 3285–3305 CrossRef CAS.
- D. J. Wales, Mol. Phys., 2004, 102, 891–908 CrossRef CAS.
- F. Noé and S. Fischer, Curr. Opin. Struc. Biol., 2008, 18, 154–162 CrossRef.
- D. J. Wales, Curr. Opin. Struc. Biol., 2010, 20, 3–10 CrossRef CAS.
- S. A. Trygubenko and D. J. Wales, J. Chem. Phys., 2004, 120, 2082–2094 CrossRef CAS.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- D. J. Wales and J. M. Carr, J. Chem. Theory Comput., 2012, 8, 5020–5034 CrossRef CAS.
- K. Röder and D. J. Wales, J. Chem. Theory Comput., 2018, 14, 4271–4278 CrossRef.
- L. J. Munro and D. J. Wales, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3969–3980 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- E. Małolepsza, B. Strodel, M. Khalili, S. Trygubenko, S. N. Fejer and D. J. Wales, J. Comput. Chem., 2010, 31, 1402–1409 Search PubMed.
- E. Małolepsza, B. Strodel, M. Khalili, S. Trygubenko, S. N. Fejer and D. J. Wales, J. Comput. Chem., 2012, 33, 2209 CrossRef.
- A. Onufriev, D. Bashford and D. A. Case, J. Phys. Chem. B, 2000, 104, 3712–3720 CrossRef CAS.
- A. Onufriev, D. A. Case and D. Bashford, J. Comput. Chem., 2002, 23, 1297–1304 CrossRef CAS.
- A. Onufriev, D. Bashford and D. A. Case, Proteins, 2004, 55, 383–394 CrossRef CAS.
- D. A. Potoyan and G. A. Papoian, J. Am. Chem. Soc., 2011, 133, 7405–7415 CrossRef CAS.
- S. Rauscher, V. Gapsys, M. Gajda, M. Zweckstetter, B. de Groot and H. Grubmüller, J. Chem. Theory Comput., 2015, 11, 5513–5524 CrossRef CAS.
- F. Palazzesi, M. Prakash, M. Bonomi and A. Barducci, J. Chem. Theory Comput., 2015, 11, 2–7 CrossRef CAS.
- S. Piana, J. Klepeis and D. Shaw, Curr. Opin. Struct. Biol., 2014, 24, 98–105 CrossRef CAS.
- R. Best, D. de Sancho and J. Mittal, Biophys. J., 2012, 102, 1462–1467 CrossRef CAS.
- A. Perez, J. MacCallum, E. Brini, C. Simmerling and K. Dill, J. Chem. Theory Comput., 2015, 11, 4770–4779 CrossRef CAS.
- J. M. Carr and D. J. Wales, J. Chem. Phys., 2005, 123, 234901 CrossRef.
- B. Strodel, C. S. Whittleston and D. J. Wales, J. Am. Chem. Soc., 2007, 129, 16005–16014 CrossRef CAS.
- O. M. Becker and M. Karplus, J. Chem. Phys., 1998, 106, 1495–1517 CrossRef.
- D. J. Wales, M. A. Miller and T. R. Walsh, Nature, 1998, 394, 758–760 CrossRef CAS.
- B. Strodel and D. J. Wales, Chem. Phys. Lett., 2008, 466, 105–115 CrossRef CAS.
- J. M. Carr and D. J. Wales, J. Phys. Chem. B, 2008, 112, 8760–8769 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS.
- T. Hamelryck, Proteins, 2005, 59, 38–48 CrossRef CAS.
- J. Weiser, P. Shenkin and W. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- D. Roe and T. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
- E. Pettersen, T. Goddard, C. Huang, G. Couch, D. Greenblatt, E. Meng and T. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- K. Röder and D. J. Wales, J. Phys. Chem. B, 2018, 122, 10989–10995 CrossRef.
- J. D. Bryngelson, J. N. Onuchic, N. D. Socci and P. G. Wolynes, Proteins, 1995, 21, 167–195 CrossRef CAS.
- J. N. Onuchic and P. G. Wolynes, Curr. Opin. Struct. Biol., 2004, 14, 70–75 CrossRef CAS.
- E. D. Nelson and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10682–10686 CrossRef CAS.
- P. G. Wolynes, Biochimie, 2015, 119, 218–230 CrossRef CAS.
- V. K. de Souza, J. D. Stevenson, S. P. Niblett, J. D. Farrell and D. J. Wales, J. Chem. Phys., 2017, 146, 124103 CrossRef CAS.
- The PyMOL Molecular Graphics System, Version 1.6.x, Schrödinger, LLC, New York, 2013.
- D. Winogradoff, I. Echeverria, D. A. Potoyan and G. A. Papoian, J. Am. Chem. Soc., 2015, 137, 6245–6253 CrossRef CAS.
- A. Gansen, S. Felekyan, R. Kühnemuth, K. Lehmann, K. Tóth, C. Seidel and J. Langowski, Nat. Commun., 2018, 9, 4628 CrossRef.
- A. Sridhar, M. Orozco and R. Collepardo-Guevara, Nucleic Acids Res., 2020, 48, 5318–5331 CrossRef CAS.
- S. Ashwin, T. Nozaki, K. Maeshima and M. Sasai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19939–19944 CrossRef CAS.
- S. Sanulli, M. Trnka, V. Dharmarajan, R. Tibble, B. Pascal, A. Burlingame, P. Griffin, J. Gross and G. Narlikar, Nature, 2019, 575, 390–394 CrossRef CAS.
- B. Gibson, L. Doolittle, M. Schneider, L. Jensen, N. Gamarra, L. Henry, D. Gerlich, S. Redding and M. Rosen, Cell, 2019, 179, 470–484 CrossRef CAS.
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS.
- M. Fuxreiter, J. Mol. Biol., 2018, 430, 2278–2287 CrossRef CAS.
- M. Miskei, A. Gregus, R. Sharma, N. Duro, F. Zsolyomi and M. Fuxreiter, FEBS Lett., 2017, 591, 2682–2695 CrossRef CAS.
- R. Sharma, Z. Raduly, M. Miskei and M. Fuxreiter, FEBS Lett., 2015, 589, 2533–2542 CrossRef CAS.
- B. Shoemaker, J. Portman and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS.
- C. Tsai, S. Kumar, B. Ma and R. Nussinov, Protein Sci., 1999, 8, 1181–1190 CrossRef CAS.
- C. Baker and R. Best, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 182–198 CAS.
- M. Kjaergaard, L. Andersen, L. Nielsen and K. Teilum, Biochemistry, 2013, 52, 1686–1693 CrossRef CAS.
- M. Arai, K. Sugase, H. Dyson and P. Wright, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9614–9619 CrossRef CAS.
- K. Gunasekaran, C. Tsai, S. Kumar, D. Zanuy and R. Nussinov, Trends Biochem. Sci., 2003, 28, 81–85 CrossRef CAS.
- D. Vuzman and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21004–21009 CrossRef CAS.
- D. Vuzman, A. Azia and Y. Levy, J. Mol. Biol., 2010, 396, 674–684 CrossRef CAS.
- T. Cragnolini, D. Chakraborty, J. Šponer, P. Derreumaux, S. Pasquali and D. Wales, J. Chem. Phys., 2017, 147, 152715 CrossRef.
- V. de Souza and D. Wales, J. Chem. Phys., 2009, 130, 194508 CrossRef.
- S. Niblett, V. de Souza, J. Stevenson and D. Wales, J. Chem. Phys., 2016, 145, 024505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the structural ensembles for the bound and unbound tail; radii of gyration, percentage of the sovlent-accessible surface area formed by the basic patch, and the distance between the lysyl groups in Lys16 and Lys20. Frustration indices. See DOI: 10.1039/d0cp05405d |
| This journal is © the Owner Societies 2021 |