
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe electronic structure of carbones revealed: insights from valence bond theory†
Remco W. A.
Havenith
 *abc,
Ana V.
Cunha
d,
Johannes E. M. N.
Klein
a,
Francesca
Perolari
b and
Xintao
Feng
a
*abc,
Ana V.
Cunha
d,
Johannes E. M. N.
Klein
a,
Francesca
Perolari
b and
Xintao
Feng
a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: r.w.a.havenith@rug.nl
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cGhent Quantum Chemistry Group, Department of Inorganic and Physical Chemistry, Ghent University, Krijgslaan 281 (S3), B-9000 Gent, Belgium
dScientific Computing Group, Oak Ridge National Laboratory, Oak Ridge, TN 37831-6373, USA
First published on 27th January 2021
Abstract
In this contribution, we studied the OC–C bond in carbon suboxide and related allene compounds using the valence bond method. The nature of this bond has been the subject of debate, whether it is a regular, electron sharing bond or a dative bond. We compared the nature of this bond in carbon suboxide with the gold–CO bond in Au(CO)2+, which is a typical dative bond, and we studied its charge-shift bond character. We found that the C–CO bond in carbon suboxide is unique in the sense that it cannot be assigned as either a dative or electron sharing bond, but it is an admixture of electron sharing and dative components, together with a high contribution of ionic character. These findings provide a clear basis for distinguishing the commonly found dative bonds between ligands and transition metals and the present case of what may be described as coordinative bonding to carbon.
Introduction
The nature of the chemical bond has been occupying chemists from the dawn of chemistry. The notions of electron sharing to form a covalent bond, and the octet rule have been and still are very successful concepts to explain structure and reactivity of molecules. However, even small molecules, that, at first sight, look straightforward and follow the rules, show a richer chemistry than expected based on the simplest Lewis structures.Frenking and coworkers studied carbon suboxide (1), related allenenic compounds, and carbodiphosphoranes (Scheme 1).1–6 Using various computational techniques, they studied the nature of the OC–C bond.3,7 A main question in this research was to explain the bonding in 1, as 1 shows a very shallow C–C–C bending potential8–10 and it is argued that it is slightly bent (156°) in the gas phase.11 In the solid state, it is on average linear, but large vibrational ellipsoids of the oxygen and central carbon atoms are visible.12 This very flexible C–C–C bending mode and the non-linear structure was also confirmed with coupled cluster calculations.13 Frenking and coworkers performed extensive energy decomposition analyses (EDA),14 using different fragments that can be envisioned to compose the bond, such as singlet/triplet CO that forms a bond with the 1D/5S of carbon, respectively.3 They not only looked at the orbital interaction terms, but also at the excitation energies for the fragments to access the so-called ‘reactive state’ from their ground state i.e., the electronic state that the fragments should have to form the bonds. This extensive analysis showed that the CO–C σ-bond could be best described as a dative bond from CO to the central C atom. Thus, they proposed that the C atom in 1 is in an oxidation state of 0, and that it formally has two lone pairs and accepts two dative bonds from the two CO moieties, and termed it a carbone.15 This conclusion was not only based on EDA, but was further substantiated with NBO analyses, proton affinities and reactivity studies, confirming the formal oxidation state of 0, that would be derived from the existence of dative bonds towards carbon.7
 | ||
| Scheme 1 Structures of the molecule studied in this contribution. | ||
Not only 1 exhibits a central carbon atom that can accept dative bonding: the allenes, such as 3 and 4, can also be viewed as a central carbon(0) atom that accepts two dative bonds from a carbene CL2 ((CL2)C(CL2)). Different allenes (with different ligands L) have been the subject of a study towards the nature of the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and similar conclusions were drawn: these allenenic structures have carbone character.2,3,5,7,16,17 Carbodiphosphorane 2 was also computationally intensively studied,1,18 and a more conclusive picture emerged: carbon is able to accept dative bonds, while being in a formal oxidation state of 0. It was found that if the singlet/triplet splitting of the ligand is high and the singlet is favored, the carbone character is higher.18 This view of dative bonding to carbon (for an example of using the term coordination chemistry in this context see ref. 19) was able to explain the non-linear geometry of the aforementioned allenes. The dative bonding and the existence of the two lone-pairs on the central carbon atom would lead to bent geometries, whereas the traditional viewpoint of two double, electron sharing, bonds would suggest a linear geometry.
C bond, and similar conclusions were drawn: these allenenic structures have carbone character.2,3,5,7,16,17 Carbodiphosphorane 2 was also computationally intensively studied,1,18 and a more conclusive picture emerged: carbon is able to accept dative bonds, while being in a formal oxidation state of 0. It was found that if the singlet/triplet splitting of the ligand is high and the singlet is favored, the carbone character is higher.18 This view of dative bonding to carbon (for an example of using the term coordination chemistry in this context see ref. 19) was able to explain the non-linear geometry of the aforementioned allenes. The dative bonding and the existence of the two lone-pairs on the central carbon atom would lead to bent geometries, whereas the traditional viewpoint of two double, electron sharing, bonds would suggest a linear geometry.
However, this view was disputed in the literature.20–22 Discussions erupted whether it was really necessary to introduce more ‘arrow drawing’ in chemistry, and it was questioned if terming these bonds to carbon as dative added a valuable perspective. It was argued that the traditional view with ionic resonance structures could equally well explain the bent geometry of 1 and related compounds and their observed chemistry. These two viewpoints remain to co-exist and have not been unified into one “final” concept.
In an attempt to provide a unified view, valence bond (VB) theory23 is applied to interpret the wavefunctions of these molecules, where we provide the basis for differentiating dative bonding in carbones when compared to classical transition metal complexes. The heavier homologues of 1 have been previously studied using VB theory,24 but 1 itself was left out. VB can offer a view to chemical bonding much closer to the commonly used Lewis structures than traditional, orthogonal, molecular orbital theories. In VB theory, a bond is represented by the spin-coupling of two electrons in two, mutually non-orthogonal, singly occupied orbitals. These singly occupied orbitals are atomic in nature. Using spin-coupled bonds, a wavefunction for a VB structure can be built that resembles one of the possible Lewis structures. The final VB wavefunction is written as a linear superposition of the possible VB structures. Each VB structure is then a linear combination of Slater determinants with fixed spin-coupling coefficients, resembling one particular binding motif, or Lewis structure. The expansion coefficients for the VB structures in the final VB wavefunction are variationally optimized, and the orbitals that comprise the determinants in each structure are simultaneously optimized. During the orbital optimization, the orbitals may be kept localized on atoms or particular molecular fragments, or may be allowed to delocalize over the entire molecule.25–27 It is also possible to use different orbitals for different VB structures (breathing orbital method28–31). The VB treatment offers a wavefunction that can directly be interpreted from a Lewis structure perspective, and provides insight into the importance of a particular structure/representation for the total description of the molecule. It also provides the set of optimized orbitals that form the chemical bonds in the molecule, which will aid in the interpretation of the bonding motifs in a molecule, by aiding in the interpretation of the VB structure. Furthermore, it provides energies of the individual structures, and an energy for the final wavefunction, thus a stabilization energy due to resonance between VB structures is unambiguously defined.32,33 Different flavours of VB theory exists, e.g., the spin-coupled VB method34–37 and the Generalized VB method,38–40 but we opt here for the valence bond self-consistent field (VBSCF) method.25,26
In this contribution, we performed VBSCF calculations and an interpretation of the VBSCF wavefunctions in terms of different bonding motifs for selected carbones, together with reference molecules containing traditional electron sharing and dative bonding (Scheme 1). The molecules 1–3 are reported to have high degrees of carbone character, whereas 4 is considered as a traditional allene with two traditional electron sharing bonds. The gold(I)(CO)2+ complex 5 is a typical complex with two dative bonds from CO to Au as commonly found in coordination chemistry; there is little π-backbonding from the gold towards CO, making the Au–CO bonds clean dative bonds.41–43
In our study, we considered the VB structures as depicted in Scheme 2. Structure A represents the traditional electron sharing bond, structure B is the carbone structure with two dative bonds, and structure C is a carbene-like model, with one dative and one covalent double bond. The structures D and E represent the ionic structures, which are invoked to explain the bonding in 2. We have chosen to keep the orbitals in the valence bond structures localized on the ligands (L) and on the central carbon atom. The electron pairs depicted in Scheme 2 are described by spin-coupled bonds, while the remaining bonds of the ligands are described by doubly occupied, orthogonal bonds.
 | ||
| Scheme 2 The five types of valence bond structures considered in this work. | ||
Computational details
Geometries were optimized with ADF,44–46 using the TZ2P basis set and BP86 functional (no symmetry constraints). Energy decomposition analyses were also performed with ADF.14 Hessian calculations were performed to confirm that all optimized structures are local minima at this level of theory. For 5, scalar relativistic effects were included via the ZORA method.47–49For 1, CCSD/cc-pVTZ geometry optimization and CCSD angle scan were done using Gaussian16,50 using the cc-pVTZ basis set.51
Valence bond SCF calculations25,26 were performed with TURTLE,52,53 as implemented in GAMESS-UK.54 For the Valence Bond calculations, the cc-pVDZ (gold: cc-pVDZ-PP) basis set51,55–57 was used. Basis sets were taken from the BasisSet Exchange Library.58–60 The molecules were divided in three fragments, viz. the central carbon atom, and each of the two ligands. The orbitals were kept localized on each fragment (for a justification of this approach, see the ESI†). Two sets of valence bond calculations were performed: (1) Only the neutral valence bond structures depicted in Scheme 2 were used: this resulted in 7 structures for 1 (2 × A, 1 × B, 4 × C), 3 for 2 (1 × B, 2 × C), 4 for 3 (1 × A, 1 × B, 2 × C), 4 for 4 (1 × A, 1 × B, 2 × C), and 7 for 5 (2 × A, 1 × B, 4 × C), and (2) both the neutral and ionic valence bond structures were used: this resulted in 12 structures for 1 (2 × A, 1 × B, 4 × C, 1 × D, 4 × E), 6 for 2 (1 × B, 2 × C, 1 × D, 2 × E), 7 for 3 (1 × A, 1 × B, 2 × C, 1 × D, 2 × E), 7 for 4 (1 × A, 1 × B, 2 × C, 1 × D, 2 × E), and 12 for 5 (2 × A, 1 × B, 4 × C, 1 × D, 4 × E). Note that for the linear molecules 1 and 5, the number of structures A, C, and E is doubled, due to degeneracies of x and y. The Gallup and Norbeck scheme61 is used to calculate the weights of the individual, non-orthogonal, VB structures (see also Table S9 (ESI†) for the Chirgwin and Coulson62 weights). The advantage of these weights is that they are always positive, and sum to 1.
Results and discussion
Energy decomposition analysis
For comparison with existing literature, we also performed energy decomposition analyses for the molecules 1–5 (Table 1 and Table S8, ESI†). For 1, the linear structure was used in these calculations. Two different fragment sets were used: (1) in which the central carbon atom is prepared in its 5S state, and triplet states for the ligands (referred to as open-shell), and (2) in which the carbon atom is prepared in a 1D state with singlet states for the ligands (referred to as closed-shell). In Table 1, the orbital interaction terms for the different fragments are listed for 1–5. According to Frenking and co-workers,3 the bonding situation is best described for fragments with the smallest orbital interactions. Note that in this study, we have chosen for three fragments in contrast to the earlier work,3 as we also have three fragments in our VB study. The reference molecule 4, allene, has the smallest orbital interaction energy (ΔEorb) for the open-shell fragments, while the orbital interaction energy is considerably larger for the closed-shell fragments. This result further confirms that 4 can be considered as having two electron sharing bonds. The gold(I) complex 5, that is typically described as having dative bonds,41–43 shows the smallest orbital interaction for the closed-shell fragments. It has a significantly higher ΔEorb for the open-shell fragments. Again, this result is in line with chemical intuition and corroborates the assignment of two dative bonds from the CO ligands to a central atom, here gold.| Compound | ΔEorb open shell | ΔEorb closed shell | ΔEorb(OS) − ΔEorb(CS) |
|---|---|---|---|
| 1 | −609.91 | −603.80 | −6.11 |
| 2 | −508.84 | −537.26 | 28.42 |
| 3 | −531.65 | −566.94 | 35.29 |
| 4 | −514.27 | −697.34 | 183.07 |
| 5 | −609.71 | −108.70 | −501.01 |
Now, we turn to the carbone compounds 1–3. The obtained differences between the open-shell and closed-shell ΔEorbs in these cases is considerably smaller. This suggests that the assignment of covalent or dative bonding is not straightforward in these cases. The open-shell fragment case is favored for molecules 2 and 3, whereas the closed-shell fragment case is favored for 1. This might lead to the temptation to interpret these results in a way that would assign 1 as a carbone, while 2 and 3 could be best described as having covalent, electron sharing, bonds. However, the differences between the open and closed-shell fragment ΔEorb is rather small (around 30 kcal mol−1), so these EDA results leave some ambiguity, and it can be expected that 2 and 3 can have (considerable) carbone character.
Valence bond self consistent field calculations
To analyze the bonding situation in the molecules 1–5 in a direct manner, we have performed valence bond self-consistent field (VBSCF) calculations using the structures depicted in Scheme 2. The Gallup and Norbeck (GN) weights for the two sets of calculations (set one with only the neutral VB structures and set two that also includes the ionic structures) are listed in Table 2.| Compound | W covalent (A) | W carbone (B) | W carbene (C) | W ionic (D) | W ionic (E) |
|---|---|---|---|---|---|
| 1 | 0.601 | 0.013 | 0.385 | ||
| 1* | 0.747 | 0.026 | 0.085 | 0.001 | 0.141 |
| 2 | — | 0.157 | 0.843 | ||
| 2* | — | 0.096 | 0.064 | 0.242 | 0.598 |
| 3 | 0.720 | 0.022 | 0.258 | ||
| 3* | 0.779 | 0.029 | 0.078 | 0.015 | 0.099 |
| 4 | 0.894 | 0.003 | 0.103 | ||
| 4* | 0.960 | 0.002 | 0.031 | 0.000 | 0.007 |
| 5 | 0.006 | 0.753 | 0.241 | ||
| 5* | 0.004 | 0.440 | 0.056 | 0.441 | 0.058 |
We consider the allene 4 first. The VBSCF calculation with only the neutral structures shows a high weight for the covalent structure (A), a negligible weight for the carbone structure (B) and some importance for structure C, which has a lone-pair on the central carbon (similar to a carbene), and one electron sharing bond with one of the ligands, and a dative bond with the other. Upon inclusion of the ionic components to the bonding, the importance of the covalent structure A increases, whereas the combined weight of VB structure C decreases; the weight of the carbone structure B is not affected and the weights of the ionic structures are also insignificant. In line with the EDA analysis, 4 can be considered as having two covalent, double bonds.
The situation for reference molecule 5 is different: when only the neutral VB structures are taken into consideration, the dominant structure is the VB structure with dative bonds from CO to gold. The carbene-like structure C also has a considerable contribution to the wavefunction. Upon including the ionic structures, this picture changes drastically: the weight of structure B decreases, and it gets as important as ionic structure D. The weight of structures C declines considerably, and its weight becomes equal to that of the ionic structures E. The weight of the pure covalent structure A is negligible in both sets of calculations. Hence, the OC–Au bond in 5 can be described as a hybrid between dative bonding B and the ionic structure D that possesses two covalent bonds as well. This picture is also in line with the EDA, although the information of the contributing ionic structure D is missing from the EDA.
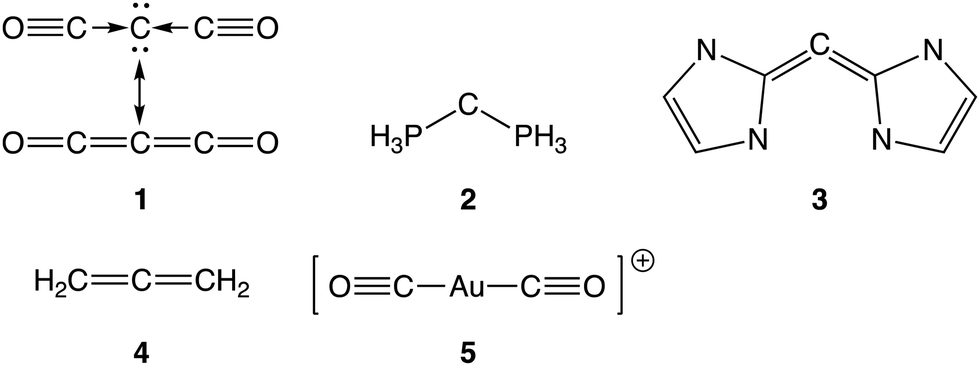
After the discussion of the reference compounds 4 and 5, where it was seen that the bonding in 4 can be assigned in a clear-cut way as being an electron sharing bond, and the bonding in 5 as dative B with ionic/covalent contributions D, we can turn our attention to the carbones 1 and 3, as they behave rather similarly. For both compounds, the covalent structure A is dominant, and it gains in weight upon including the ionic structures. Unexpectedly, the weight of the carbone structure B is rather low, and it is clear from the VBSCF point of view that both 1 and 3 cannot be described solely by a Lewis structure that has two dative bonds. When the ionic structures are not included in the calculation, the structures C with one dative bond are important for the description of the molecules. However, as soon as the ionic structures are added to the wavefunction, the weights of structures C decreases, and the ionic structures E get a non-negligible weight. Thus, 1 and 3 can be described as covalent structures with important contributions from the ionic structures and mixed dative/electron sharing structures C. The latter have one dative bond to the central carbon atom each. The VBSCF orbitals of 1 (Fig. 1) do not show any abnormalities and can be considered as regular σ and π orbitals localized on the CO fragment and the central C atom.
 | ||
| Fig. 1 The symmetry unique optimized VBSCF orbitals for 1. | ||
Geometry dependence of the Valence Bond wavefunctions
One can wonder whether the VBSCF results obtained for 1 are biased by the chosen linear geometry of 1, while the equilibrium geometry of 1 is bent (for the discussion of bond angles in carbones and related compounds, see ref. 3, 13 and 63–68). Therefore, we performed a scan of the ∠C–C–C angle at the CCSD/cc-pVTZ level of theory (keeping ∠C–C–C frozen and optimized the rest of the molecule) and did VBSCF calculations on the obtained geometries. The energy and the weights as a function of ∠C–C–C are depicted in Fig. 2. The results show that the VBSCF and the CCSD calculations agree that 1 is not linear, and that the linear structure is a transition state connecting to the two bent minima. However, the barrier calculated using VBSCF theory is considerably higher than the CCSD one. One might consider the bond angle to be quasi-linear when using CCSD.3 Also, the degree of bending is substantially different: VBSCF theory predicts a considerably smaller ∠C–C–C than CCSD. The inclusion of ionic structures in the VBSCF calculations has only a small effect on the height of the transition state and the C–C–C angle. The composition of the wavefunction, however, remains constant on the reaction path. Structure A remains the dominant structure on the whole path, and structure B, the carbone structure remains to contribute rather little. This means that VB results obtained for the linear geometry are also valid for the bent structure of 1 and that the geometry has hardly any influence on the composition of the wavefunction. | ||
| Fig. 2 (a) The CCSD, VB-I (only neutral structures), VB-II (neutral and ionic structures) and DFT (BP86/TZ2P) energies (kcal mol−1), relative to the lowest point, as a function of the C–C–C angle (°); (b) the weights of the VB structures as a function of the C–C–C angle (°). | ||
Full geometry optimizations are also possible with VBSCF theory.52,69,70 When all structures are included a bent geometry is obtained (Table 3, entry 1). The obtained OC–C bond length is a bit elongated with respect to the reference CCSD structure, and the angle is more bent. Omission of the ionic structures in the VBSCF wavefunction does not lead to any significant changes in the geometry of 1 (entry 2). Surprisingly, if only the covalent structure A is included in the VBSCF wavefunction (entry 3), a more bent structure is found, instead of the expected linear geometry. The carbone structure B alone does not lead to bonded minimum (entry 4), and dissociation of the CO moieties is observed. The dative bonding is not strong enough to keep the molecule bonded. A resonance hybrid of structures C leads to a similarly bent structure as has been obtained in the optimization of structure A only (entry 5).
| Entry | E VBSCF | E rel | Structures | ∠C–C–C | R C–C |
|---|---|---|---|---|---|
| 1 | −263.324650 | 0.00 | A + B + C + D + E | 133.0 | 1.324 |
| 2 | −263.296732 | 17.52 | A + B + C | 133.5 | 1.313 |
| 3 | −263.158396 | 104.33 | A | 124.6 | 1.307 |
| 4 | −263.097006 | 142.85 | B | 48.9 | 6.152 |
| 5 | −263.182792 | 89.02 | C | 124.1 | 1.316 |
| 6 | −263.144968 | 112.75 | A | 180.0 | 1.289 |
| 7 | −262.744954 | 363.77 | B | 180.0 | 1.334 |
| 8 | −264.256849 | CCSD | 146.3 | 1.285 |
When a linear geometry is enforced, and structure A is only used in the wavefunction (entry 6), the energy obtained is higher than the bent structure A geometry, as expected. If only carbone structure B is taken (entry 7), while enforcing linearity, a bonded situation is found. However, the energy is considerably higher than the dissociated products.
A last issue to address is whether the OC–C bond has some degree of charge-shift bond character;71,72 dative bonding in metal–ligand complexes has a high degree of charge-shift bond character,72 hence, the dative part of the OC–C bond in 1 may also have a high degree of charge-shift bond character. Therefore, we elongated one of the OC–C bonds and calculated the VBSCF energy, including the ionic structures. The covalent structure A is the structure with the lowest energy (Fig. 3) in the region of the equilibrium bond length. On the dissociation path, this structure has a minimum that nearly coincides with the minimum in the total energy curve. Ionic structures E are next higher in energy: this curve also possesses a minimum near the equilibrium structure. Although interactions between the structures considerably lowers the energy, the dissociation curves do not resemble the curves that are obtained for typical charge-shift bonds like the one in F2,73 hence, the OC–C bond cannot be assigned to the charge-shift bond family.
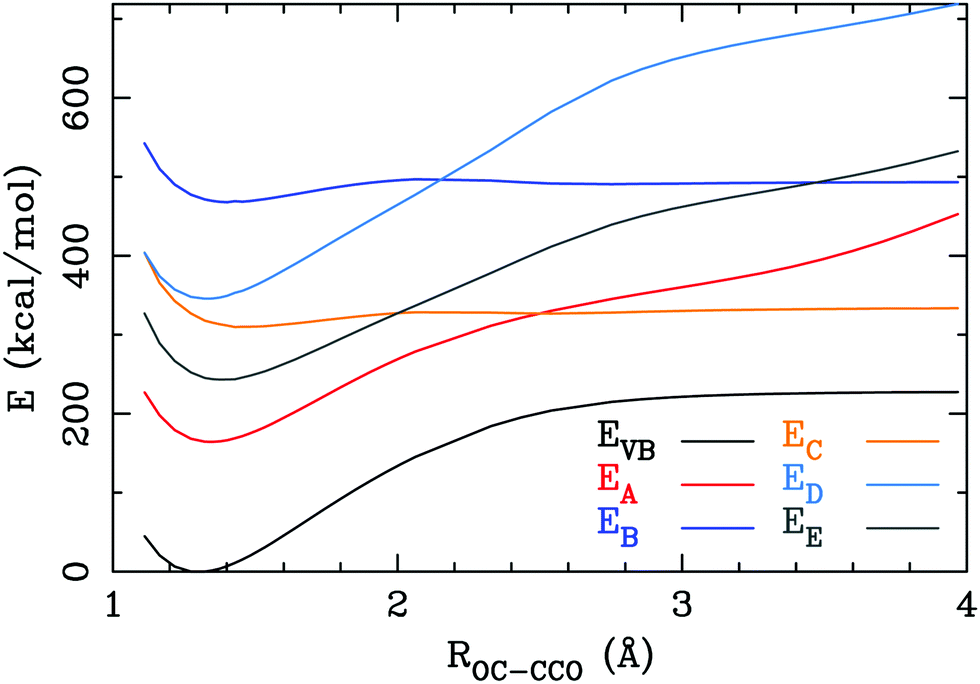 | ||
| Fig. 3 The total VBSCF energy (EVB), and the energy of the covalent structures (EA), the carbone structure (EB), the carbene structures (EC) and the ionic structures (ED) and (EE) as a function of the OC–CCO distance. | ||
Conclusions
Here we have reported a valence bond study of typical molecules that were assigned to possess dative bonding towards a carbon atom, and therefore termed as carbones. We have shown that, in comparison with archetypical molecules classified with electron sharing (H2CCCH2) and dative bonds ([OC–Au–CO]+), the carbone VB structure is not important in the description of the bond. The covalent structure prevails with relatively high contributions of ionic structures. The importance of the mixed dative/electron sharing bond structures shows that the bond in the so called carbone molecules do have dative bond character, but these bonds cannot be assigned to purely dative. The carbone structure itself proves to be unstable, and does not lead to a bonded molecule. The OC–C bond also does not show any significant sign of charge-shift bond character.
Based on these results, we see that indeed the OC–C bond in 1 and 3 are distinct from “normal” electron sharing bonds and from dative bonding. As bonding in molecules covers a large spectrum, it might be best classified to be placed in between these two extremes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research used resources (Summit and SummitDev) of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725 (DD and ESP). This work was sponsored by NWO Exact and Natural Sciences for the use of supercomputer facilities (contract no. 17197 7095). We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster. J. E. M. N. K. acknowledges funding from the Netherlands Organisation for Scientific Research (NWO START-UP grant (740.018.014)).References
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Carbodiphosphoranes: The chemistry of divalent carbon(0), Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS.
- R. Tonner and G. Frenking, C(NHC)2: Divalent Carbon(0) Compounds with N-Heterocyclic Carbene Ligands—Theoretical Evidence for a Class of Molecules with Promising Chemical Properties, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS.
- R. Tonner and G. Frenking, Divalent Carbon(0) Chemistry, Part 1: Parent Compounds, Chem. – Eur. J., 2008, 14, 3260–3272 CrossRef CAS.
- R. Tonner, G. Heydenrych and G. Frenking, First and second proton affinities of carbon bases, ChemPhysChem, 2008, 9, 1474–1481 CrossRef CAS.
- S. Klein, R. Tonner and G. Frenking, Carbodicarbenes and Related Divalent Carbon(0) Compounds, Chem. – Eur. J., 2010, 16, 10160–10170 CrossRef CAS.
- W. Petz and G. Frenking, Carbodiphosphoranes and Related Ligands, in Transition Metal Complexes of Neutral η1-Carbon Ligands, ed. R. Chauvin and Y. Canac, 2010, vol. 30, pp. 49–92 Search PubMed.
- G. Frenking and R. Tonner, Divalent carbon(0) compounds, Pure Appl. Chem., 2009, 81, 597–614 CAS.
- A. Clark and H. M. Seip, The potential function for the CCC bending in carbon suboxide, Chem. Phys. Lett., 1970, 6, 452–456 CrossRef CAS.
- R. L. Livingston and C. N. Ramachandra Rao, The molecular structure of carbon suboxide, J. Am. Chem. Soc., 1959, 81, 285–287 CrossRef CAS.
- H. Mackle and L. E. Sutton, The structure of carbon suboxide by the method of electron diffraction, Trans. Faraday Soc., 1951, 47, 937–942 RSC.
- P. Jensen and J. W. C. Johns, The Infrared Spectrum of Carbon Suboxide in the ν6 Fundamental Region: Experimental Observation and Semirigid Bender Analysis, J. Mol. Spectrosc., 1986, 118, 248–266 CrossRef CAS.
- A. Ellern, T. Drews and K. Seppelt, The Structure of Carbon Suboxide, C3O2, in the Solid State, Z. Anorg. Allg. Chem., 2001, 627, 73–76 CrossRef CAS.
- J. Koput, An ab initio study on the equilibrium structure and CCC bending energy levels of carbon suboxide, Chem. Phys. Lett., 2000, 320, 237–244 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 1–86 Search PubMed.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, New bonding modes of carbon and heavier group 14 atoms Si–Pb, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- C. Esterhuysen and G. Frenking, Distinguishing Carbones from Allenes by Complexation to AuCl, Chem. – Eur. J., 2011, 17, 9944–9956 CrossRef CAS.
- T. H. Wang, W. C. Chen and T. G. Ong, Carbodicarbenes or Bent Allenes, J. Chin. Chem. Soc., 2017, 64, 124–132 CrossRef CAS.
- G. Frenking and M. Hermann, Gilbert Lewis and the Model of Dative Bonding, in Chemical Bond I: 100 Years Old and Getting Stronger, ed. D. M. P. Mingos, 2016, vol. 169, pp. 131–156 Search PubMed.
- M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Coordination chemistry at carbon, Nat. Chem., 2009, 1, 295–301 CrossRef CAS.
- G. Frenking, Dative Bonds in Main-Group Compounds: A Case for More Arrows!, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 CrossRef CAS.
- D. Himmel, I. Krossing and A. Schnepf, Dative Bonds in Main-Group Compounds: A Case for Fewer Arrows!, Angew. Chem., Int. Ed., 2014, 53, 370–374 CrossRef CAS.
- H. Schmidbaur, Réplique: A New Concept for Bonding in Carbodiphosphoranes?, Angew. Chem., Int. Ed., 2007, 46, 2984–2985 CrossRef CAS.
- S. Shaik and P. C. Hiberty, A Chemist's Guide to Valence Bond Theory, John Wiley & Sons, Inc., Hoboken, New Jersey, 2008 Search PubMed.
- J. Turek, B. Braïda and F. De Proft, Bonding in Heavier Group 14 Zero-Valent Complexes—A Combined Maximum Probability Domain and Valence Bond Theory Approach, Chem. – Eur. J., 2017, 23, 14604–14613 CrossRef CAS.
- J. H. van Lenthe and G. G. Balint-Kurti, The valence-bond scf (VB SCF) method: Synopsis of theory and test calculation of OH potential energy curve, Chem. Phys. Lett., 1980, 76, 138–142 CrossRef CAS.
- J. H. van Lenthe and G. G. Balint-Kurti, The valence-bond self-consistent field method (VB–SCF): Theory and test calculations, J. Chem. Phys., 1983, 78, 5699–5713 CrossRef CAS.
- B. J. Duke and R. W. A. Havenith, Implications of the complete basis set limit in valence bond theory: a case study of molecular hydrogen, Theor. Chem. Acc., 2016, 135, 82 Search PubMed.
- P. C. Hiberty, Reconciling simplicity and accuracy: compact valence bond wave functions with breathing orbitals, J. Mol. Struct., 1997, 398–399, 34–43 Search PubMed.
- P. C. Hiberty, J. P. Flament and E. Noizet, Compact and accurate valence bond functions with different orbitals for different configurations: application to the two-configuration description of F2, Chem. Phys. Lett., 1992, 189, 259–265 CrossRef CAS.
- P. C. Hiberty, S. Humbel, C. P. Byrman and J. H. van Lenthe, Compact valence bond functions with breathing orbitals: Application to the bond dissociation energies of F2 and FH, J. Chem. Phys., 1994, 101, 5969–5976 CrossRef CAS.
- P. C. Hiberty and S. Shaik, Breathing-orbital valence bond method – a modern valence bond method that includes dynamic correlation, Theor. Chem. Acc., 2002, 108, 255–272 Search PubMed.
- M. Zielinski, R. W. A. Havenith, L. W. Jenneskens and J. H. van Lenthe, A comparison of approaches to estimate the resonance energy, Theor. Chem. Acc., 2010, 127, 19–25 Search PubMed.
- Z. Rashid, J. H. van Lenthe and R. W. A. Havenith, Resonance and Aromaticity: An Ab Initio Valence Bond Approach, J. Phys. Chem. A, 2012, 116, 4778–4788 CrossRef CAS.
- J. Gerratt, General Theory of Spin-Coupled Wave Functions for Atoms and Molecules, in Advances in Atomic and Molecular Physics, ed. D. Bates and I. Esterman, Academic Press, 1971, vol. 7, pp. 141–221 Search PubMed.
- J. Gerratt and M. Raimondi, The spin-coupled valence bond theory of molecular electronic structure. I. Basic theory and application to the 2Σ+ states of BeH, Proc. R. Soc. London, Ser. A, 1980, 371, 525–552 CAS.
- D. L. Cooper, J. Gerratt and M. Raimondi, Studies of molecular states using spin-coupled valence-bond theory, Faraday Symp. Chem. Soc., 1984, 19, 149–163 RSC.
- D. L. Cooper; J. Gerratt and M. Raimondi, Modern Valence Bond Theory, in Advances in Chemical Physics: Ab initio Methods in Quantum Chemistry – II, ed. K. P. Lawley, John Wiley & Sons, London, 1987, vol. LXIX, pp. 319–397 Search PubMed.
- W. J. Hunt, P. J. Hay and W. A. Goddard III, Self-Consistent Procedures for Generalized Valence Bond Wavefunctions. Applications H3, BH, H2O, C2H6, and O2, J. Chem. Phys., 1972, 57, 738–748 CrossRef.
- P. J. Hay, W. J. Hunt and W. A. Goddard III, Generalized valence bond description of simple alkanes, ethylene, and acetylene, J. Am. Chem. Soc., 1972, 94, 8293–8301 CrossRef CAS.
- W. A. Goddard III, T. H. Dunning Jr., W. J. Hunt and P. J. Hay, Generalized valence bond description of bonding in low-lying states of molecules, Acc. Chem. Res., 1973, 6, 368–376 CrossRef.
- H. V. R. Dias, C. Dash, M. Yousufuddin, M. A. Celik and G. Frenking, Cation Gold Carbonyl Complex on a Phosphine Support, Inorg. Chem., 2011, 50, 4253–4255 CrossRef CAS.
- M. A. Celik, C. Dash, V. A. K. Adiraju, A. Das, M. Yousufuddin, G. Frenking and H. V. R. Dias, End-On and Side-On π-Acid Ligand Adducts of Gold(I): Carbonyl, Cyanide, Isocyanide, and Cyclooctyne Gold(I) Complexes Supported by N-Heterocyclic Carbenes and Phosphines, Inorg. Chem., 2013, 52, 729–742 CrossRef CAS.
- L. Nunes dos Santos Comprido, J. E. M. N. Klein, G. Knizia, J. Kästner and A. S. K. Hashmi, Gold(I) Vinylidene Complexes as Reactive Intermediates and Their Tendency to π-Backbond, Chem. – Eur. J., 2016, 22, 2892–2895 CrossRef CAS.
- E. J. Baerends, ADF2017.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com, 2017 Search PubMed.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Towards an order-N DFT method, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, Relativistic regular two-component Hamiltonians, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, Relativistic total energy using regular approximations, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Relativistic regular two-component Hamiltonians, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- D. E. Woon and T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- J. H. van Lenthe, F. Dijkstra and R. W. A. Havenith, TURTLE – A gradient VBSCF Program. Theory and Studies of Aromaticity. In Valence Bond Theory, ed. D. L. Cooper, Elsevier, Amsterdam, 2002, vol. 10, pp. 79–112 Search PubMed.
- J. Verbeek, J. H. Langenberg, C. P. Byrman, F. Dijkstra, R. W. A. Havenith, J. J. Engelberts, M. Zielinski, Z. Rashid and J. H. van Lenthe, TURTLE, an ab initio VB/VBSCF program, Utrecht, The Netherlands, 1988–2016 Search PubMed.
- M. F. Guest, I. J. Bush, H. J. J. van Dam, P. Sherwood, J. M. H. Thomas, J. H. van Lenthe, R. W. A. Havenith and J. Kendrick, The GAMESS-UK electronic structure package: algorithms, developments and applications, Mol. Phys., 2005, 103, 719–747 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Energy-consistent pseudopotentials for group 11 and 12 atoms: adjustment to multi-configuration Dirac–Hartree–Fock data, J. Chem. Phys., 2005, 311, 227–244 CAS.
- K. A. Peterson and C. Puzzarini, Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- D. Feller, The role of databases in support of computational chemistry calculations, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibsom and T. L. Windus, A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, Basis Set Exchange: A Community Database for Computational Sciences, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS.
- G. A. Gallup and J. M. Norbeck, Population analyses of valence-bond wavefunctions and BeH2, Chem. Phys. Lett., 1973, 21, 495–500 CrossRef CAS.
- B. H. Chirgwin and C. A. Coulson, The electronic structure of conjugated molecules. VI, Proc. R. Soc. London, Ser. A, 1950, 201, 196–209 CAS.
- S. Böttger, M. Gruber, J. Eike Münzer, G. M. Bernard, N.-J. H. Kneusels, C. Poggel, M. Klein, F. Hampel, B. Neumüller, J. Sundermeyer, V. K. Michaelis, R. Tonner, R. R. Tykwinski and I. Kuzu, Solvent-Induced Bond-Bending Isomerism in Hexaphenyl Carbodiphosphorane: Decisive Dispersion Interactions in the Solid State, Inorg. Chem., 2020, 59, 12054–12064 CrossRef.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Synthesis and Reactivity of a CAAC–Aminoborylene Adduct: A Hetero-Allene or an Organoboron Isoelectronic with Singlet Carbenes, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, Main-Group Metallomimetics: Transition Metal-like Photolytic CO Substitution at Boron, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS.
- P. J. Quinlivan and G. Parkin, Flexibility of the Carbodiphosphorane, (Ph3P)2C: Structural Characterization of a Linear Form, Inorg. Chem., 2017, 56, 5493–5497 CrossRef CAS.
- R. Appel, U. Baumeister and F. Knoch, Darstellung und Molekülstruktur aminosubstituierter Carbodiphosphorane, Chem. Ber., 1983, 116, 2275–2284 CrossRef CAS.
- W.-L. Li, H.-T. Liu, T. Jian, G. V. Lopez, Z. A. Piazza, D.-L. Huang, T.-T. Chen, J. Su, P. Yang, X. Chen, L.-S. Wang and J. Li, Bond-bending isomerism of Au2I3−: competition between covalent bonding and aurophilicity, Chem. Sci., 2016, 7, 475–481 RSC.
- F. Dijkstra and J. H. van Lenthe, Gradients in Valence Bond theory, Chem. Phys. Lett., 1999, 310, 553–556 CrossRef CAS.
- F. Dijkstra and J. H. van Lenthe, Gradients in Valence Bond theory, J. Chem. Phys., 2000, 113, 2100–2108 CrossRef CAS.
- G. Sini, P. Maitre and P. C. Hiberty, Covalent, ionic and resonating single bonds, J. Mol. Struct., 1991, 229, 163–188 CrossRef.
- S. Shaik, D. Danovich, J. Morrison Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Charge-shift bonding: A new and unique form of bonding, Angew. Chem., Int. Ed., 2020, 59, 984–1001 CrossRef CAS.
- H. Zhang, D. Danovich, W. Wu, B. Braïda, P. C. Hiberty and S. Shaik, Charge-Shift Bonding Emerges as a Distinct Electron-Pair Bonding Family from Both Valence Bond and Molecular Orbital Theories, J. Chem. Theory Comput., 2014, 10, 2410–2418 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S5, containing the Cartesian coordinates of 1–5, a justification for the use of fragment restricted orbitals (including Table S6 (GN/CC weights of the structures) and Fig. S1 (VBSCF orbitals for 1 without any restrictions), Table S7 with all total energies (VB and CASSCF), Table S8 with all EDA components, Table S9, the combined CC weights for 1–5, and Table S10, containing the orbital overlaps. See DOI: 10.1039/d0cp05007e |
| This journal is © the Owner Societies 2021 |