
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe physical significance of the Kamlet–Taft π* parameter of ionic liquids†
Nadine
Weiß
 a,
Caroline H.
Schmidt
a,
Gabi
Thielemann
a,
Esther
Heid
b,
Christian
Schröder
*b and
Stefan
Spange
*a
a,
Caroline H.
Schmidt
a,
Gabi
Thielemann
a,
Esther
Heid
b,
Christian
Schröder
*b and
Stefan
Spange
*a
aChemnitz University of Technology, Straße der Nationen 62, 09111 Chemnitz, Germany. E-mail: stefan.spange@chemie.tu-chemnitz.de
bUniversity of Vienna, Faculty of Chemistry, Institute for Computational Biological Chemistry, Währingerstr. 17, A-1090 Vienna, Austria. E-mail: christian.schroeder@univie.ac.at
First published on 16th December 2020
Abstract
The Kamlet–Taft dipolarity/polarizability parameters π* for various ionic liquids were determined using 4-tert-butyl-2-((dicyanomethylene)-5-[4-N,N-diethylamino)-benzylidene]-Δ3-thiazoline and 5-(N,N-dimethylamino)-5′-nitro-2,2′-bithiophene as solvatochromic probes. In contrast to the established π*-probe N,N-diethylnitroaniline, the chromophores presented here show excellent agreement with polarity measurement using the chemical shift of 129Xe. They do not suffer from additional bathochromic UV/vis shifts caused by hydrogen-bonding resulting in too high π*-values for some ionic liquids. In combination with large sets of various ionic liquids, these new chromophores thereby allow for detailed analysis of the physical significance of π* and the comparison to quantum-mechanical methods. We find that π* correlates strongly with the ratio of molar refractivity to molar volume, and thus with the refractive index.
1 Introduction
The classification of organic solvents and ionic liquids (ILs) by empirical polarity scales is an established concept in physical organic chemistry.1–6 The most widely applied method to describe multiple polarity properties of such solvents is the Kamlet–Taft polarity scale:1,7–20| (XYZ) = (XYZ)0 + a·α + b·β + s·(π* + d·δ) | (1) |
Alternatively, the linear solvation energy concept of Catalán can be applied:21,22
| (XYZ) = (XYZ)0 + a·SA + b·SB + s·SP + e·SdP | (2) |
One may expect, that the solvent acidity and basicity correspond to the Kamlet–Taft hydrogen-bonding parameters, and that SP and SdP correlate with the Kamlet–Taft polarizability/dipolarity parameter. However, for organic solvents, the multiple square correlation analyses of π* as a function f(SP), f(SdP) or f(SP,SdP) lead to contradictory statements, especially when different organic solvent classes such as alcohols, alkanes, ketones, haloalkanes, esters, etc. are considered individually.23 In addition, we showed that π* correlates for organic solvents with SdP but not so much with SP using an extended record from literature.23 Nevertheless, SdP and SP are not really independent of each other and various correlations are recognized for particular solvent families.23 For this reason, we will only consider the Kamlet–Taft π* parameter in this paper.
In case of ILs, the physical interpretation of Kamlet–Taft α and β as well as Cataláns SA and SB is convincingly supported by various results of independent spectroscopic measurement methods.24–31 In contrast, the situation for interpretation of Kamlet–Taft π* is still not clear although several attempts were made to substantiate the π* data of ILs obtained by means of independent physico-chemical measurement data.13,15,17,19,32,33 Kobrak argued that π* is a linear function f(Vm) of the molar volume Vm.15 However, the predicted relationships of π* as a linear function of Vm is not convincingly demonstrated if all ILs are considered. For particular cation classes, these kind of correlations are indicated. Yoshida reported a linear relationship of π* with the inverse molar volume for dicyanimide-based ionic liquids,17i.e. f(1/Vm). In a recent publication, we discussed that π* = f(1/Vm) is due to the fact that the total dipolarity correlates with the number of dipoles per volume.23 However, when evaluating literature data on other types of ILs, a correlation of π* as a function of the inverse molar volume have not been recognized so far.16,18–20 Complementary to the correlation with the molar volume, the relationship of π* to the molar refractivity Am, π* = f(Am), and to the refractive index nD, π* = f(nD), have been discussed.32,33 A linear correlation of π* with the refractive index would make the disperse part of the polarizability responsible.34–36 On the other hand, Rani et al. stated that π* of the IL 1-butyl-1-methylpyrrolidinium bis-(trifluoromethanesulfonyl)imide strongly depends on the molecular structure of the solvatochromic probe.19 Indeed, various solvatochromic probe molecules resulted in different π*-values of particular ILs.12–19 Especially the hydrogen-bonding accepting ability of a probe may infer with measurements in solvents with large Kamlet–Taft α.37–40 However, the understanding of the dipolarity/polarizability uncoupled from hydrogen bonds is an important aspect to interpret photosensitization when different ILs are used.41
In order to contribute to this discussion, we measured Kamlet–Taft π*-values for a large and diverse set of ILs (see ESI† for the complete list) with two different solvatochromic probes, 4-tert-butyl-2((dicyanomethylene)-5-[4-N,N-diethylamino)-benzylidene]-Δ3-thiazoline (Th) and 5-(N,N-dimethylamino)-5′-nitro-2,2′-bithiophene (BT), which differ very much in size and functional groups. Experimental results are enhanced by computational determination of solvent polarizabilities, and together provide new insights into the significance and physical meaning of π* in ILs.
The remainder of this paper is organized as follows. Section 2 provides a short description of the properties polarizability, dipolarity, polarization and polarity as used throughout this article and summarizes important relations between them. Section 3 details the materials, experimental and computational setup of this study. Section 4 describes and discusses the obtained results, followed by concluding remarks in Section 5.
2 Theory
Since the terms (di)polarity and polarization are sometimes used in a different sense in literature, the following section gives definitions on what we perceive as polarizability, dipolarity, polarization and polarity.The term dipolarity is only vaguely defined as a measure of the dipolar character of the solvent.1 This indicates that the dipole moment ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) i = permi + indi of solvent molecule i should play an important role for this property. In principle, molecular dipole moments for single cations and anions can be computed quantum-mechanically. As each ion is not neutral, dipole moments are evaluated with respect to their center-of-masses.42 However, the problem for the current discussion of dipolarity of ionic liquids is how to combine the gas phase cationic and anionic dipole moments and compare them to the experimental π*-value in liquid phase.
i = permi + indi of solvent molecule i should play an important role for this property. In principle, molecular dipole moments for single cations and anions can be computed quantum-mechanically. As each ion is not neutral, dipole moments are evaluated with respect to their center-of-masses.42 However, the problem for the current discussion of dipolarity of ionic liquids is how to combine the gas phase cationic and anionic dipole moments and compare them to the experimental π*-value in liquid phase.
In liquid phase, molecular dipole moments are summed up to yield the collective rotational dipole moment ![[M with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_004d_20d1.gif) D which is not accessible by quantum-mechanics. The polarization density
D which is not accessible by quantum-mechanics. The polarization density ![[P with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0050_20d1.gif) (or often simply polarization) is defined by the ratio between this collective dipole moment and the occupied volume V:42,43
(or often simply polarization) is defined by the ratio between this collective dipole moment and the occupied volume V:42,43
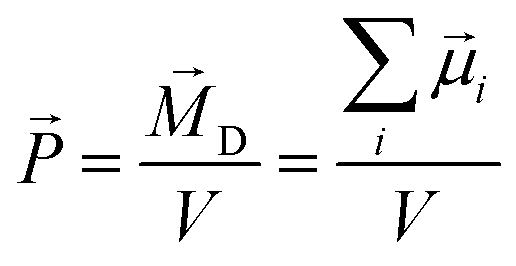 | (3) |
= ε0(εs − 1)![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) | (4) |
to the liquid will orientate the molecular dipoles. The emerging polarization density depends on the static dielectric constant εs and the dielectric permittivity of the vacuum ε0 = 8.85 × 10−12 A s V−1 m−1. In case of N nonpolar molecules,44 the polarization induced by the total electric field is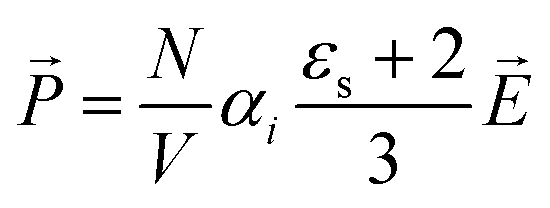 | (5) |
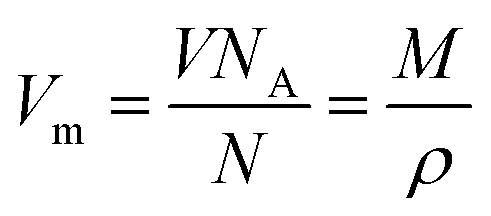 | (6) |
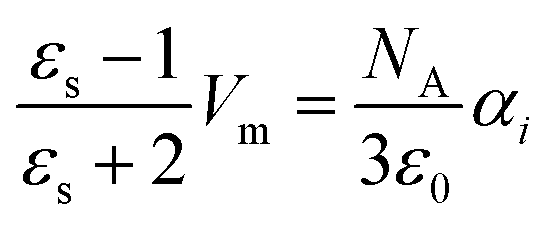 | (7) |
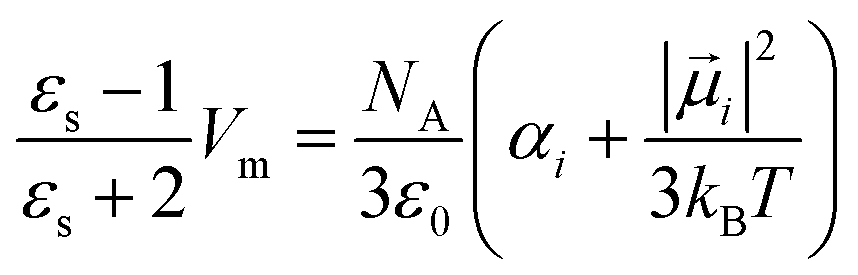 | (8) |
There is no clear definition of the term polarity. Usually it is used to empirically characterize the solvation capability of a particular solvent, but cannot be defined properly in terms of physico-chemical properties.1 Therefore, we skip this term for the discussion in this work.
The term polarizability αi defines the ease to deform the electron cloud and correlates the induced dipole indi with the electric field .
| indi = αi | (9) |
The experimental determination of π* uses a wave length in the UV/vis regime. If an electric field (ω) changes with the corresponding frequencies, translational and rotational motion of the molecules cannot follow. Consequently, we replace the static dielectric constant εs in eqn (7) with its high-frequency limit ε∞ which is the refractive index squared and obtain the Lorentz–Lorenz equation:
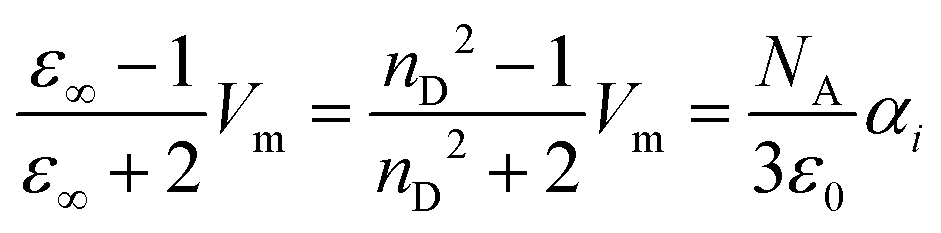 | (10) |
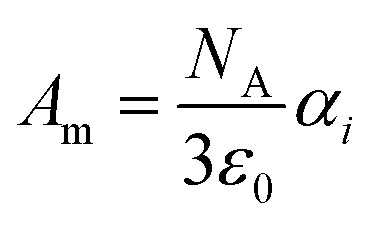 | (11) |
The usage of eqn (10) has several advantages: first, we do not have to consider the conductivity contribution to the dielectric constant from the IL ions. Second, strictly speaking, eqn (7) is only valid for nonpolar molecules which ILs are not. However, at this UV/vis frequencies only the molecular electron cloud and not the nuclei may react to the electric field.
3 Methods
3.1 Materials
The solvatochromic probe BT (see Fig. 1) was kindly provided by Prof. F. Effenberger, University Stuttgart, in highly purified form. It was dried over CaH2in vacuo before dissolved in the IL. For each measurement, only a very low quantity <1 × 10−5 mol L−1 is required. The dye Th (see Fig. 1) was synthesized and purified as described in the literature.45,46DENA was commercially purchased from Fluorochem in a purity of 97%.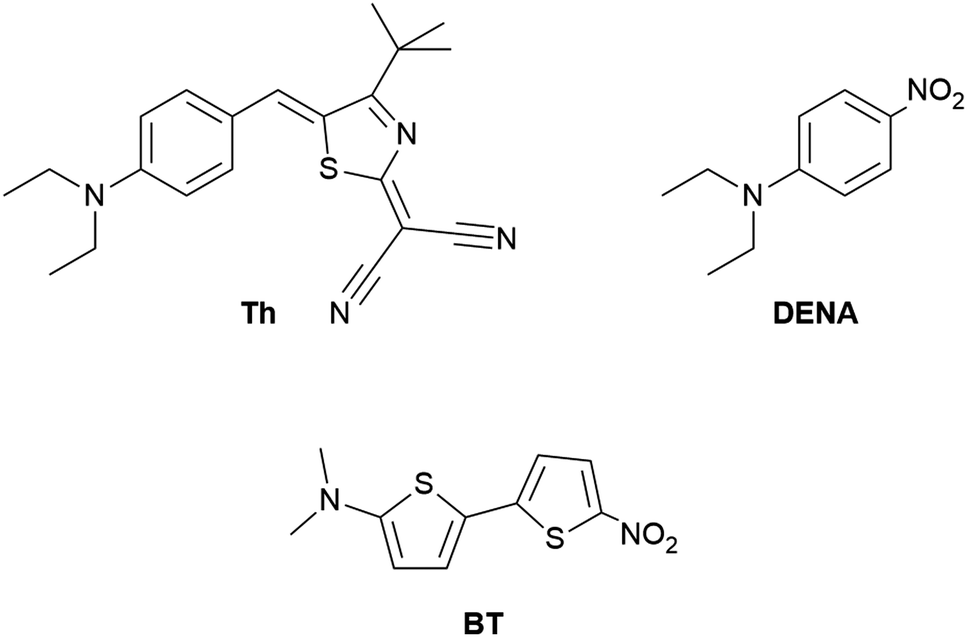 | ||
| Fig. 1 The solvatochromic probes 4-tert-butyl-2((dicyanomethylene)-5-[4-N,N-diethylamino)-benzylidene]-Δ3-thiazoline (Th), N,N-diethyl-4-nitroaniline (DENA) and 5-(N,N-dimethylamino)-5′-nitro-2,2′-bithiophene (BT) to determine the Kamlet–Taft π*-parameter. | ||
Dichloromethane (DCM) was freshly distilled over CaH2 before use under argon atmosphere. Then, the water content was below the detection limit of the Karl–Fischer titration.
The ionic liquids [C4mim]Cl, [C4mim]tosylate, [C8mim]Cl and [C10mim]Cl were purchased from ABCR; [C6mim]CF3SO3 from Acros; [C4mim]N(CN)2, [C6mim]Br, [C8mim]Br, [2C2mS]NTf2 and [3C8mN]NTf2 from IoLiTec; [C2mim]FAP, [C4mim]I, [C4mim]OctOSO3, [C4mim]CF3CO2, [C4mim]CF3SO3, [C4mim]BF4, [C4mim]C(CN)3, [C4mim]PF6, [C4mim]FAP, [C6mim]Cl, [C6mim]BF4 and [C8mim]CF3SO3 from Merck in highest purity grade. BASF kindly provided us [C4mim]Ac, [C4mim]CH3SO3, [C4mim]CH3OSO3 and [C4mim]SCN in high purity. Others 1-alkyl-3-methylimidazolium-based ILs were synthesized from the respective chloride salt according to established literature procedures.47–54 The tetraalkylammonium-based ILs were also synthesized from the respective chloride salt according to established literature procedures.55,56 In general, tri-N-butyl-methyl-ammonium chloride (5 g) was dissolved in water (150 mL) and a solution of equimolar silver salt (AgCF3CO2, AgN(CN)2 [freshly synthesized]) or lithium salt (LiNTf2) was added dropwise. For the synthesis of tri-n-octyl-methyl-ammonium-based ILs, [C8mim]Cl was dissolved in a H2O/DCM solution (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50% v/v) and the corresponding silver salt was added dropwise.
:50% v/v) and the corresponding silver salt was added dropwise.
Prior to use, the ILs were dried over molecular sieve (4 Å) from Roth, unless otherwise indicated. For this purpose, the respective IL was dried in vacuum at 300 °C for 2 h. After cooling to room temperature, dried DCM was added to the molecular sieve under argon atmosphere and the IL was dispersed in it. Volume ratio of DCM to IL was about 10. After 20 h, the mixture was filtered and the DCM was removed by a rotary evaporator. Subsequently the IL was dried for 24 h under high vacuum (0.5 mbar). [C4mim]Cl, [C4mim]BF4, [C4mim]FAP, [C6mim]FAP, [C8mim]Br, [C10mim]Cl and [3C6C14P]Cl were dried over silica. The versatile advantage of silica is that traces of water and anion impurities from precursors are completely removed. In case of many present impurities, the mass balance IL to silica can be adapted. The only downside of this method is the loss of some IL. However, the method is able to remove, for example, traces of fluoride ions (fluoride <1 × 10−5 mol L−1) from purchasable [C4mim]BF4 and other ILs. To dry these ILs, porous silica from Macherey–Nagel (BET surface area 500 m2 g−1) was first dried in vacuum at 200 °C for 2 h. After cooling to room temperature, dried DCM was also added to the silica under argon atmosphere and the IL was dissolved in it. Drying and subsequent reconditioning of the IL was carried out in the same way as drying using a molecular sieve. During the whole drying process steps, the temperature should be kept below 25 °C, especially when the dye is dissolved in the IL. For [C2mim]FAP, alkaline alumina from Sigma Aldrich (pore size 58 Å, pH = 9.5) was used instead of silica.
The purity of the IL was checked by measuring the diffraction index of the IL and compared with literature data. Effect of water on UV/vis spectra is weak as investigated by reference experiments with [C4mim]BF4 (see ESI†).
3.2 Experimental setup
For UV/vis spectroscopic investigations, the respective dye was first dissolved in 0.3 mL IL. Both solvatochromic probes, Th and BT, are soluble in the complete set of ionic liquids investigated in the current work. The UV/vis absorption spectra of this solution were recorded in cuvettes made of special optical glass with a light path of 2 mm at 25 °C on a Cary 60 UV/vis from Agilent Technologies.The Kamlet–Taft parameter 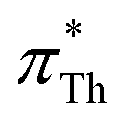 as reported by Th was calculated via
as reported by Th was calculated via
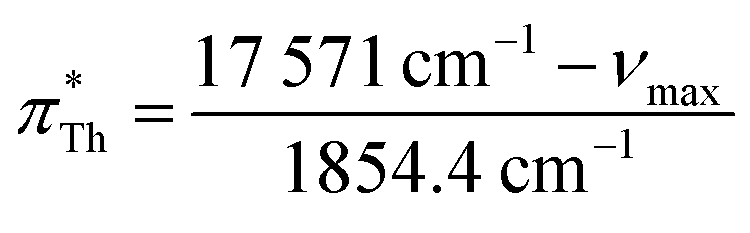 | (12) |
The Kamlet–Taft  -value gained from BT was determined according to Effenberger et al.,57via
-value gained from BT was determined according to Effenberger et al.,57via
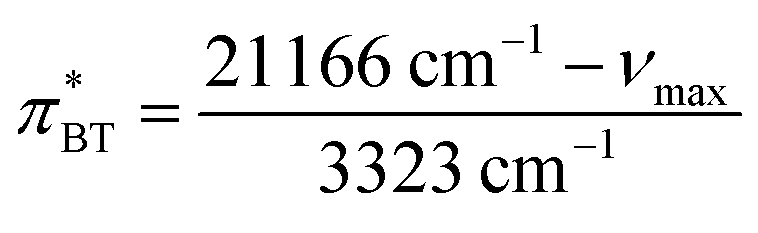 | (13) |
3.3 Computational approach
As the Kamlet–Taft π* is intended to characterize the polarizability, we computed the molecular polarizability of all ILs studied here quantum-mechanically. Based on eqn (9) we determine the isotropic molecular polarizability of ion as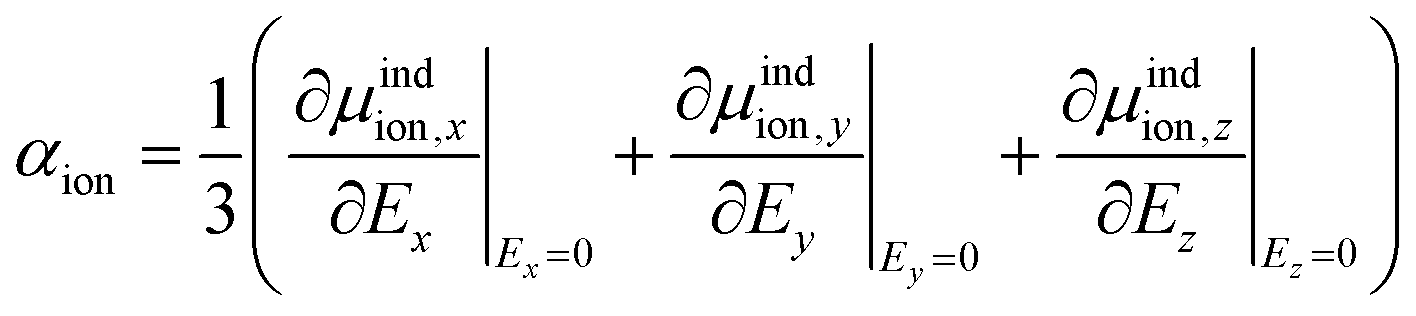 | (14) |
| αi = αcation + αanion | (15) |
in x-, y- and z-direction. The geometries of the respective cations and anions of each IL were optimized on a RI-MP2/6-31+G(d) level of theory in Psi458 starting from manually drawn starting configurations. Six single point energy evaluations employing RI-MP2 and the more extensive polarizable PVTZ basis set of Sadlej59 were calculated at electric fields of magnitude 0.0008 a.u. in the positive and negative x-, y- and z-direction. Møller–Plesset perturbation theory (MP2) with Sadlej's basis set was shown to yield accurate polarizabilities of ILs.60 Psi4 employs a density-fitting approximation of MP2 (Resolution-of-the-Identity MP2 or RI-MP2), which speeds up the calculations considerably, and was used in literature for the calculation of polarizabilities.61 A numerical differentiation of the molecular dipole moment with the electric field then yielded the molecular polarizability, as described in literature62,63 and eqn (14). These calculation are performed for each cation and anion separately in gas phase. In order to show their relevance for the current discussion we plot experimental molar refractivity Am = fLL(nD)·Vm (see eqn (10)) and the experimental molar volume Vm as a function of the molecular polarizability in Fig. 2. The experimental molar volumes Vm are determined by the experimental density ρ (see eqn (6)). Please note that we converted the molecular polarizability αi in units of A2 s4 kg−1 to the much more common molecular polarizability volume ![[small alpha, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dc.gif) i in units of Å3via = αi/(4πε0). The blue dotted line represents a linear regression with a slope of 2.56 cm3 mol−1 Å−3 and a R2 of 0.98 which agrees very well with the theoretical value of 2.52 cm3 mol−1 Å−3 based on eqn (11). This finding implies that the gas phase polarizabilities can be used as a measure for the molar refractivity in liquid phase. As the polarizabilities are computed for single ions, only negligible interactions between molecular polarizabilities are expected. Furthermore, the spatial heterogeneities present in ILs seem to be of minor importance for the molar refractivity Am. The linear relationship between the molecular polarizability and the molar volume in Fig. 2 confirms this finding.
i in units of Å3via = αi/(4πε0). The blue dotted line represents a linear regression with a slope of 2.56 cm3 mol−1 Å−3 and a R2 of 0.98 which agrees very well with the theoretical value of 2.52 cm3 mol−1 Å−3 based on eqn (11). This finding implies that the gas phase polarizabilities can be used as a measure for the molar refractivity in liquid phase. As the polarizabilities are computed for single ions, only negligible interactions between molecular polarizabilities are expected. Furthermore, the spatial heterogeneities present in ILs seem to be of minor importance for the molar refractivity Am. The linear relationship between the molecular polarizability and the molar volume in Fig. 2 confirms this finding.
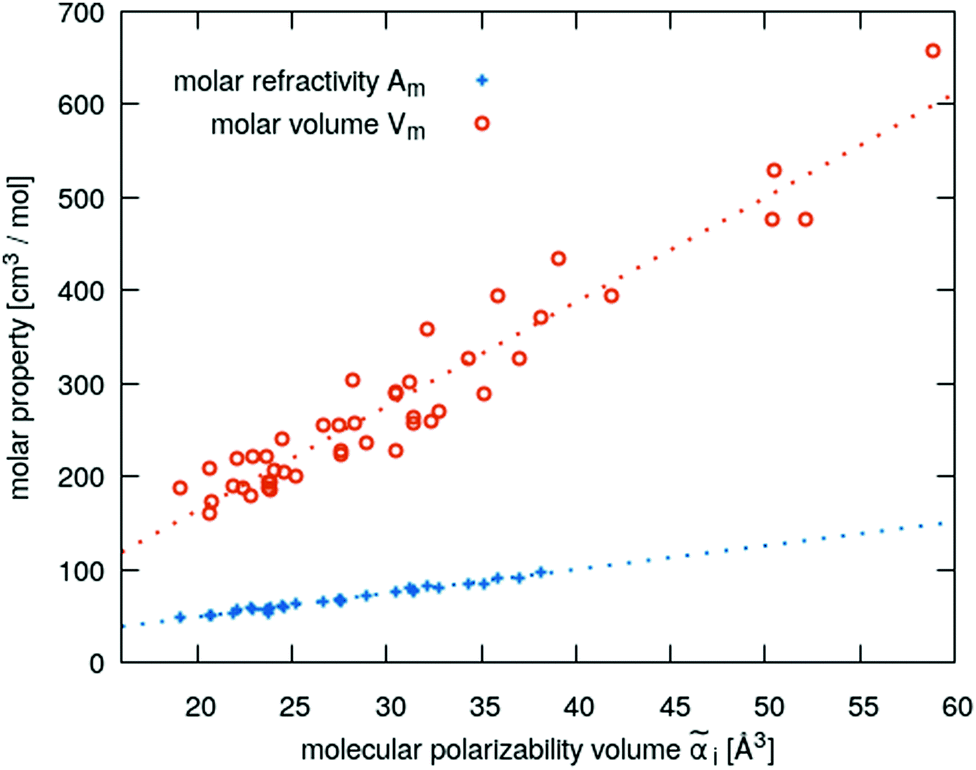 | ||
| Fig. 2 Correlation of the molecular polarizability volume i gained from gas phase quantum-mechanical calculations with the experimental molar refractivity Am (blue symbols) and molar volume Vm (orange symbols) of all ILs investigated here. | ||
We furthermore calculated the molecular dipole moments i = permi + indi of each cation and anion separately. Please note that these dipole moments are computed with respect to the center-of-mass as the molecules are charged species. In contrast to the molecular polarizabilities, molecular dipole moments cannot be summed up. Consequently, one has to fix a cationic or anionic species in order to correlate π* with the dipole moments of the unfixed counter-ions.
4 Results and discussion
The Kamlet–Taft π*-values of a set of 41 ILs (see ESI† for details) were measured via the solvatochromic probes Th and BT. In the following, we discuss the suitability of the chosen probes as means to determine π*, and investigate the physical meaning of π* in ionic liquids.4.1 Validity of the chosen solvatochromic probes
We agree with Welton and co-workers19 who state that inconsistent π*-values for ILs in literature are due to the inappropriate use of apparently established UV/vis probes. For example, N,N-dialkylnitroaniline derivatives like N,N-diethyl-4-nitroaniline (DENA, see Fig. 1) and other probes are routinely used for π* determination12–14,16–19 despite the fact that these probes also respond to hydrogen-bonding.37–40 The solvation of the nitro group by OH-dipoles induces an additional bathochromic UV/vis shift and likely results in too high π*-values.64 For example, DENA only provides useful π*-data if the hydrogen-bonding ability does not vary significantly within the solvent series under investigation.65 Thus, nitroaniline probes should actually only be used for non hydrogen bonding solvents. Particularly, N,N-dialkylnitroaniline compounds are of limited suitability for protic ILs.66–69 Hydrogen-bonding also plays a role for Stokes shift relaxation of indoline derivatives.41Hence, we recommended the use of Th (see Fig. 1) as π*-probe, because its UV/vis absorption energy (νmax) is insensitive to hydrogen bond donating of the solvent.13,45 However, this probe has not yet been used by other working groups to determine π* of ILs. Therefore, in this work we want to demonstrate again the usefulness of this special probe molecule. To independently prove the reliability of our π*-values, we use an alternative probe with similar solvatochromic properties as Th. For this purpose, 5-(N,N-dimethylamino)-5′-nitro-2,2′-bithiophene (BT, Fig. 1) is selected, whose position of the UV/vis absorption energy also does not depend on the specific hydrogen bond donating of the solvent.57,70 Furthermore, BT is one of the strongest positive solvatochromic probes.6 Surprisingly, in the literature only few studies on solvatochromic measurements by BT in ILs exist.71
The strong correlation between the π*-values gained from BT and Th is shown in Fig. 3a. The diagonal gray line corresponds to 100% agreement and almost all  (orange spheres) are close to that line. A linear regression of the
(orange spheres) are close to that line. A linear regression of the  as a function of
as a function of 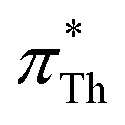 yields a slope of 0.96 and a R2 of 0.92.
yields a slope of 0.96 and a R2 of 0.92.
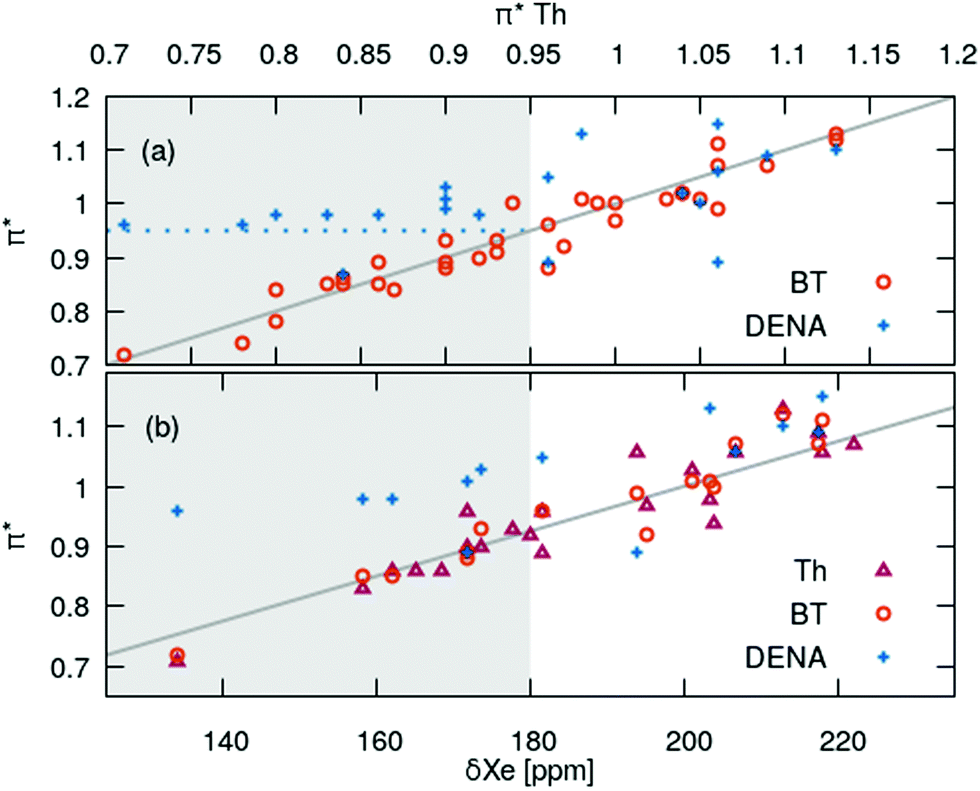 | ||
| Fig. 3 Comparison of the solvatochromic probes DENA, BT and Th. (a) Correlation between the probes. (b) Correlation of π* with 129Xe-NMR data at 25 °C.72,73 | ||
In contrast, π*-values evaluated from DENA (blue plus symbols) show a significant deviation from the other chromophores for 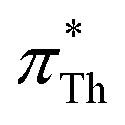 -values below 0.95 (gray shaded area) which is probably due to the additional bathochromic shift mentioned above. It seems that the solvatochromic behavior of DENA is not suitable to determine very low π*-values as they level-off sketched by the dotted blue line. This is particularly true for strong hydrogen bond donating ILs because νmax(DENA) is also a function of the Kamlet–Taft α-values.37,38 With increasing Kamlet–Taft α, the deviation between
-values below 0.95 (gray shaded area) which is probably due to the additional bathochromic shift mentioned above. It seems that the solvatochromic behavior of DENA is not suitable to determine very low π*-values as they level-off sketched by the dotted blue line. This is particularly true for strong hydrogen bond donating ILs because νmax(DENA) is also a function of the Kamlet–Taft α-values.37,38 With increasing Kamlet–Taft α, the deviation between  and
and 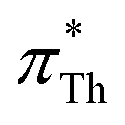 also increases.
also increases.
The IL influences the dipolar solvatochromic probe via dipole–dipole interactions. These interactions should be similar to those with the induced dipole of a Xe atom.74–79 Fortunately, for many of the ILs in this study also chemical shift data of 129Xe exist in literature.72,73 We restrict the correlation between the π*-values of the chromophores to δ129Xe at room temperature in Fig. 3b since they are known to be temperature sensitive.77–79 We find a significant correlation of the chemical shifts of 129Xe to the π*-values of BT and Th over the complete ppm range. In case of DENA similar problems (gray area in Fig. 3b) as discussed above occur which limits the validity of  -values of ILs to those above roughly 0.95.
-values of ILs to those above roughly 0.95.
As a result, we believe that π*-values obtained from the solvatochromic probes BT and Th have the quality to analyze the physical meaning of the Kamlet–Taft polarizability/dipolarity parameter. For the sake of clarity, we will display the correlations with other properties for Th only as more data exist for that chromophore. Nevertheless, similar quality of correlations always exist for the other chromophore BT.
4.2 Correlation of π* with physico-chemical properties
The Kamlet–Taft π*-values have been analyzed in terms of molar refractivity Am, molar volume Vm, molecular polarizability volumei and refractive index nD. Please note that the number of ILs measured for a particular property may differ as not all values were experimentally accessible or reported in literature (see ESI† for a complete list).
In order to compare the quality of the fits, all correlations are displayed as π* = f(…). In addition to classical coefficient of determination R2, we also apply the Akaike (AIC) and Bayesian80 (BIC) information criterion which are estimators of out-of-sample prediction error and consequently a measure of the correlation quality. Both, AIC and BIC, measure the relative amount of information loss by a given fit model.81,82 The less information a fit model loses, the lower the AIC/BIC value and the higher quality of the corresponding correlation. However, AIC and BIC are sensitive to the dimension of values of the function. Therefore, AIC and BIC cannot compare the quality of correlations like … = f(π*) where the function takes on different value ranges. Therefore, we report these measures only for π* = f(…). AIC and BIC also deals with the problem of overfitting as they take into account the number of fitting parameters as well as the likelihood function. In contrast to AIC, BIC also takes into account the number of data points. All statistical evaluations are performed in Mathematica 11.3.83
The correlations π* = f(…) are depicted in Fig. 4 and summarized in Table 1. In Fig. 4 the fit is shown as gray dashed line. The π*-values for Th and BT are depicted as red triangles and orange circles, respectively. Yoshida et al. reported for a small set of imidazolium dicyanamide based ILs a linear increase of π* with the concentration of the ionic liquid which corresponds to the inverse molar volume.17 This trend can be observed qualitatively for our set of ionic liquids as well as shown in Fig. 4a with an R2-value of 0.48 indicating many outliers, for example for imidazolium based tris(pentafluoroethyl)trifluorophosphate [Cnmim]FAP with n = 2, 4 and 6 possessing very low π*-values. This demonstrates the risk of finding correlation in a limited set of data. The inverse relationship π* = f(Vm) proposed by Kobrak15 does not show a linear trend. His threshold of 620 Å3 corresponds to a molar volume of 373 cm3 mol−1 and is shown as gray vertical dotted line in Fig. 4b. Left to this barrier π*-values decreases more or less linearly with increasing molar volume whereas right to this line one may find a positive slope. However, the FAP-based imidazoliums possess a positive slope (see gray shaded area in Fig. 4b) although [C2mim]FAP and [C4mim]FAP are on the wrong side of the threshold. Additionally, the π*-parameter is associated with a frequency (see eqn (12)) and consequently correlates with an absorption energy. As a result, one would expect a correlation of this energy parameter with the molar concentration 1/Vm but not with the molar volume.
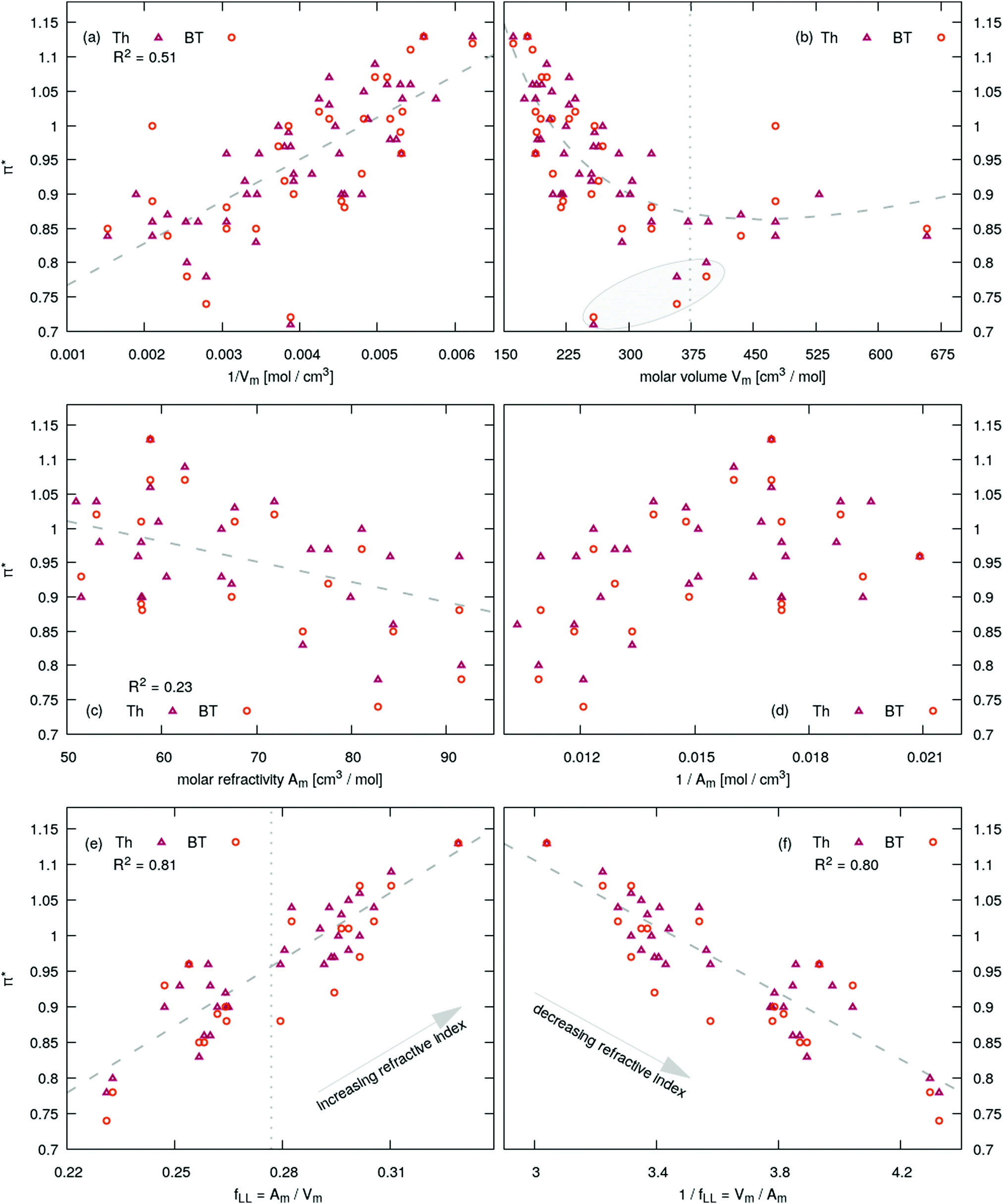 | ||
| Fig. 4 Dependence of the Kamlet–Taft π*-value measured by the probes Th and BT on the molar refractivity Am and volume Vm. R2 concerns the correlation with Th but the corresponding values for BT are very close. | ||
| π* = f(…) | #IL | R 2 | AIC | BIC |
|---|---|---|---|---|
| a C4mim based ILs. | ||||
| 1/Vm | 45 | 0.50 | −111.8 | −107.0 |
| V m | 45 | 0.35 | −100.1 | −95.2 |
| 1/Vm, Vm | 45 | 0.54 | −113.3 | −107.0 |
| A m | 30 | 0.23 | −66.4 | −63.1 |
| 1/Am | 30 | 0.19 | −64.8 | −61.5 |
|
i
|
51 | 0.18 | −103.7 | −98.4 |
| A m/Vm | 31 | 0.81 | −111.3 | −107.9 |
| V m/Am | 31 | 0.80 | −110.7 | −107.3 |
| n D | 31 | 0.81 | −111.2 | −107.8 |
| ΔEva | 11 | 0.58 | −23.6 | −25.9 |
In Fig. 5a the π*-values of the imidazolium subset of our ILs is shown as a function of the inverse molar volume. Ignoring some outliers, one may fit these values with a uniform slope (dotted lines) for Cnmim (n = 4, 6, 8 and 10). With increasing chain length n the overall offset increases, i.e. C10mim (blue symbols) has higher π*-values compared to C2mim (orange symbols) at a given inverse molar volume 1/Vm. Spatial heterogeneity cannot be blamed for the outliers as the molar volume in the liquid phase scales almost perfectly with the molecular polarizability volume obtained from gas phase quantum-mechanical calculations (see Fig. 2).
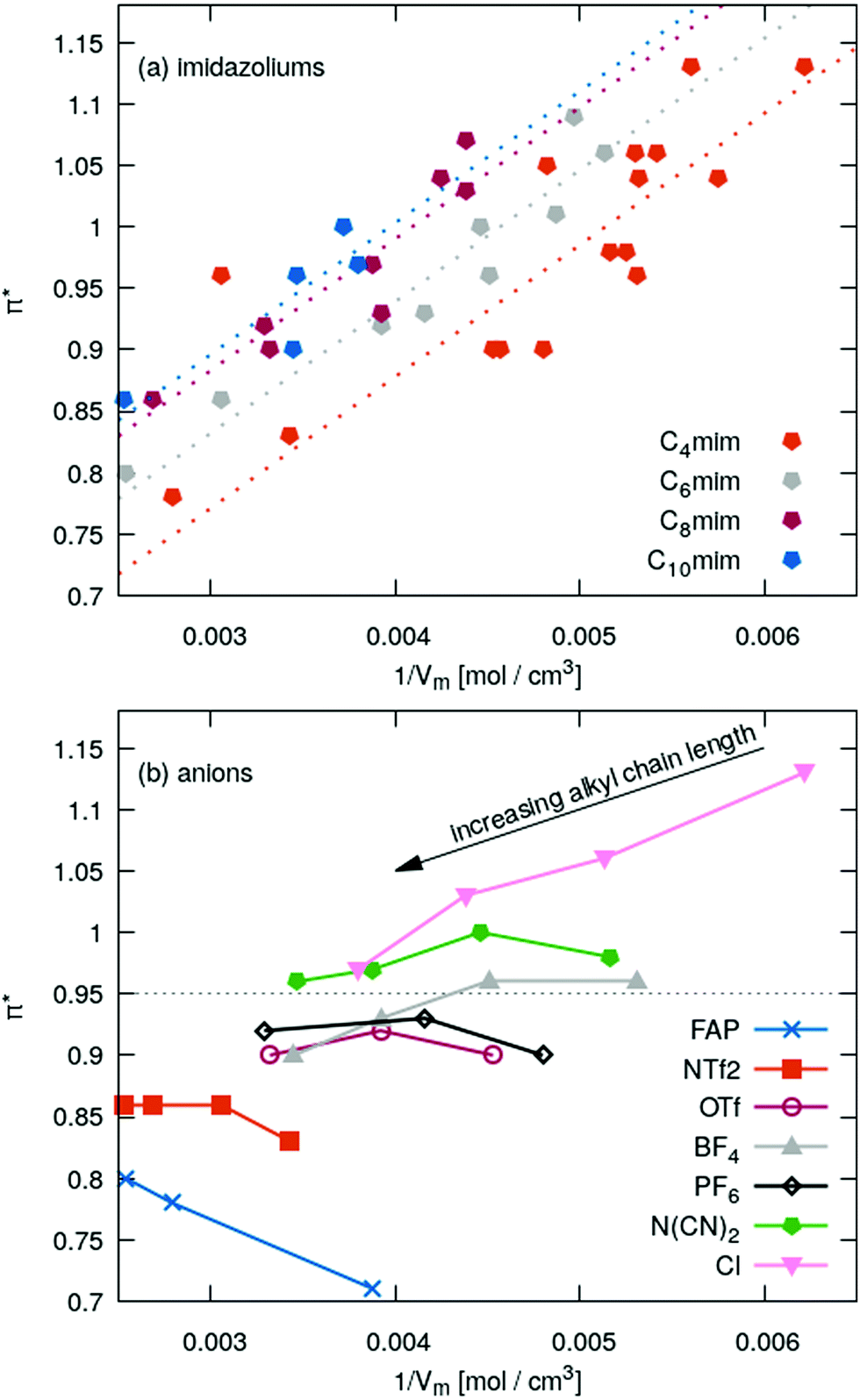 | ||
| Fig. 5 Kamlet–Taft π*-values as a function of inverse molar volume. (a) Restricting the set of ILs to a particular Cnmim. (b) Fixing the anion and follow the trend with increasing chain length of the imidazolium. | ||
For a fixed cation, for example C4mim (orange symbols in Fig. 5a), the π*-values obtained by Th correlate to some extent (see Table 1) to the vertical oxidation potential ΔEV of ref. 84 which characterizes the ionization potential. A similar correlation is found for π*-values obtained by BT which can be found in the ESI.† The correlation between π* and ΔEV seems interesting to investigate as ILs strongly interact with radicals.85–94 In particular, radical polymerizations are significantly influenced by ILs.85–91 Although the overall process of radical polymerization is rather complex and unpaired electrons undergo many types of interactions,89–91 Breuermann et al. could show that the gross reaction rate constant of the radical polymerization in ILs significantly increases with increasing π*.92 Additionally, solvent-dependent redox processes in ILs or organic solvents can be characterized by Kamlet–Taft parameters.93,94 In theory, the lower ΔEV-value, the easier the anion can be oxidized, i.e. an electron can be removed. This corresponds to the extreme case of electron cloud deformation. In fact, decreasing ΔEV of the IL anions results in increasing π*-values and indicate some correlation with the polarizability. However, tetrafluoroborate and hexafluorophosphate have the highest ΔEV of the anions investigated here. Nevertheless, their π*-values in Fig. 5b are close to the average value of 0.95. We also do not find a correlation between ΔEV and the molecular polarizability volume (see ESI†).
Given a particular anion as shown in Fig. 5b the π*-values approaches roughly 0.95 while increasing the alkyl chain length of the imidazoliums. In case of very small anions like chloride π* starts at high values and becomes smaller with increasing chain length. Medium sized anions, such as tetrafluoroborate, hexafluorophosphate, triflate and dicyanamide, show a marginal impact of the cation size on the π*-values. For the large anions like FAP and NTf2−, the π*-values increases with increasing chain length.
All these results indicate a dependency of π* on the polarizability: chloride has a very high polarizability whereas FAP and NTf2− contain many fluorine atoms with a small polarizability. This changes the ratio of polarizability per volume.
Highly polarizable molecules can be detected experimentally by the molar refractivity Am (see Lorentz–Lorenz eqn (10)). The corresponding correlation between Am and the Kamlet–Taft π* is shown in Fig. 4c. The overall correlation is not very strong indicated by the low R2-value of 0.23. In addition, there is a negative trend: with increasing molar refractivity, the corresponding π*-values decrease which is counter-intuitive on first sight. A direct plot of the π*-values as a function of molecular polarizability volume ãi is depicted in Fig. 6a. Again, there is no linear trend of the polarizability/dipolarity Kamlet–Taft parameter π* with the molecular polarizability or its respective volume. The π*-values are scattered. For example, for i of 28 Å3, the range of π*-values reaches from 0.7 to 1.1 which is the complete range of values investigated in this work. Furthermore, the trend is still negative. With increasing molecular polarizability, the π*-value roughly decreases. This can be rationalized by examining the nature of a rise in polarizability. The molecular polarizability arises from atomic contributions that are roughly additive. Adding more atoms to a molecule strictly increases the polarizability, but often decreases the polarizability density, since the volume of the molecule increases with the number of atoms as well. However, the ratio of polarizability change to volume change per added atom depends on the nature of the atom. For example, the addition of bromine or iodine atoms has a positive effect on the ratio, whereas an addition of carbon atoms has a neutral to negative effect, as shown recently for various ILs.60
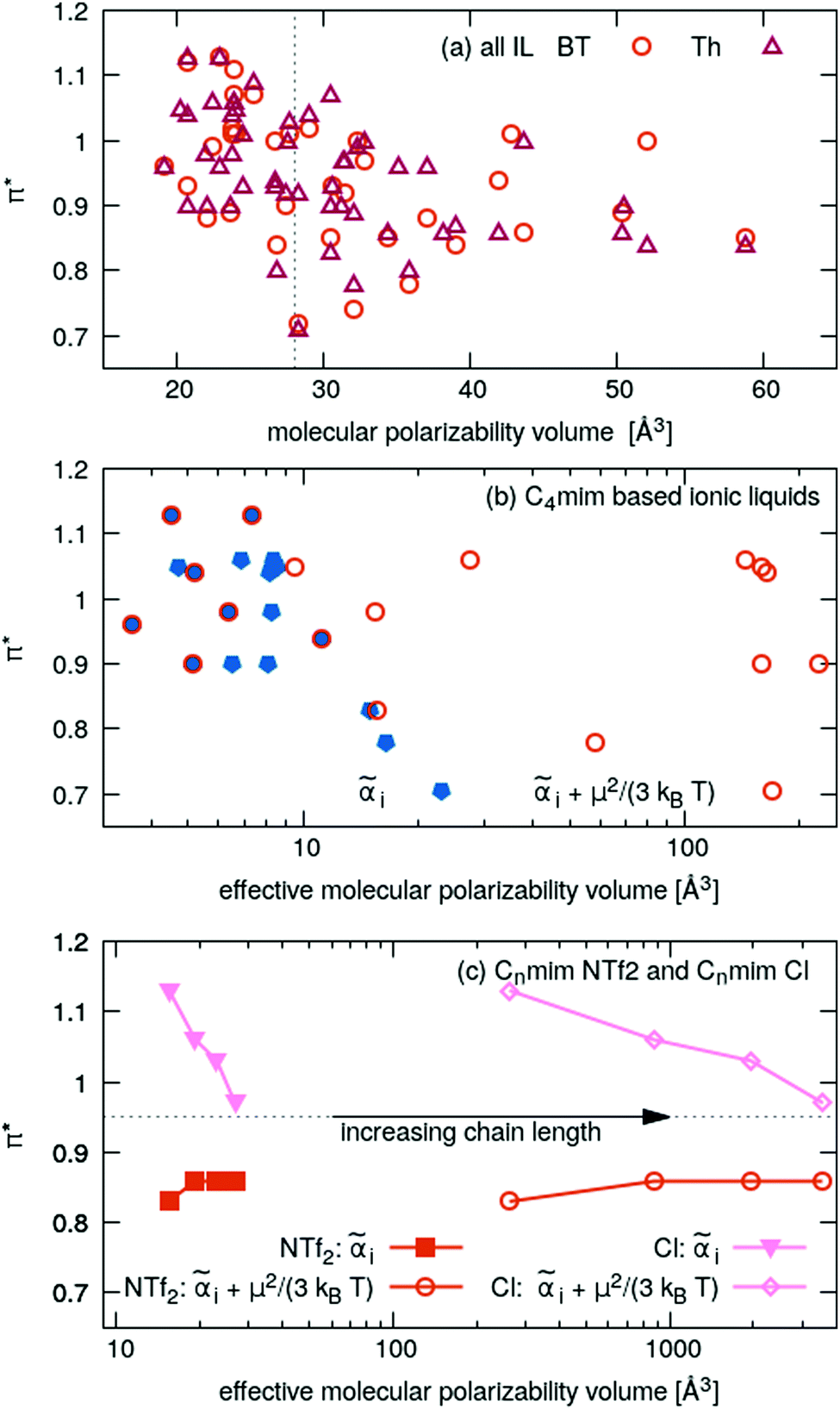 | ||
| Fig. 6 Polarizability/dipolarity parameter π* as a function of the molecular polarizability volume i: (a) all ILs without dipole correction, (b) C4mim-based ILs with (orange) and without (blue) dipole correction, (c) NTf2−- and Cl−-based imidazolium ILs with and without dipole correction. | ||
Ionic liquids are polar molecules. Consequently, the dipole moments may influence the π*-value. In eqn (8), the molecular polarizability αi was enhanced by a dipole-dependent term μ2/(3kBT). In Fig. 6b the molecular polarizability volumes (blue filled symbols) were augmented by this term using the anionic dipoles (orange circles). This results in a significant shift for very polar anions, such as acetate, trifluoroacetate, methylsulfonate, triflate and methylsulfate whereas halides and symmetrical anions like hexafluorophosphate, tetrafluoroborate, nitrate, are not shifted. However, the correlation is not improved taking the dipole correction into account. In Fig. 6c various imidazolium NTf2- and chloride-based ILs are displayed. Again, the dipole correction using the cationic dipoles does not improve the correlation but uncompresses the curve.
Also an inverse relationship between the π*-values and the molar refractivity as shown in Fig. 4d has no linear trend. Based on the results discussed so far, one may come to the conclusion that π* has nothing to do with the polarizability or refractivity. But this is not true. Interestingly, there is a strong correlation between the π*-values and the ratio between the molar refractivity and the molar volume Am/Vm as shown in Fig. 4e. This correlation is much better than π* = f(1/Vm) with an R2 of 0.81 (see Table 1 for comparison). The ratio Am/Vm is the Lorentz–Lorenz function fLL(nD). As the range of the refractive index nD of ILs is quite limited, the linear part of a Taylor-series fLL(nD) ≃ 0.278 + 0.510(nD − 1.468) with the expansion point of 〈nD〉 = 1.468 (which is the mean value of all ILs investigated in this work) is sufficient to reproduce fLL(nD) (see ESI†). A linear correlation between the refractive index and the polarizability per volume was already reported in literature.95,96
This ratio Am/Vm corresponds to a polarizability density as already proposed by Seddon and co-workers.32 They discriminated between two classes of ILs: the first class consisted of ILs with a higher polarizability density than a CH2-unit. This corresponds to ILs right of the gray dotted line in Fig. 4e and above the gray dotted line in Fig. 5b. The second class of ionic liquids has a lower polarizability density compared to the CH2-unit. It often consists of many low-polarizable atoms like fluorine.
Class I ionic liquids have π*-values higher than 0.95 where the linear relationship crosses the vertical line in Fig. 4e. Please note that the average refractive index 〈nD〉 and the theoretical value for a CH2-unit based on the calculations in ref. 32 are almost identical. This is expected as many ILs contain long alkyl chains driving the refractive index towards the theoretical values of a CH2-unit. Class I ionic liquids decrease their refractive index with increasing chain length and consequently their π*-values; class II ionic liquids increase their refractive index and hence their π*-values with increasing chain length. This effect was observed in Fig. 5b and 6c.
Fig. 4 unambiguously demonstrates that the correlation with a single quantity such as 1/Vm or Am as done by many authors in literature is insufficient to capture the complete picture. It is the combination of the molar refractivity and the molar volume which is important. Interestingly, the Kamlet–Taft π*-values show also a linear trend as a function of 1/fLL = Vm/Am as depicted in Fig. 4f. The quality of this linear trend is as good as for Am/Vm since no significant discrepancy exist between R2, AIC and BIC (see Table 1) for the two relationships. However, in both cases, π* = f(Am/Vm) and π* = f(Vm/Am), the Kamlet–Taft parameter increases with increasing refractive index nD. This is even more interesting as the single correlations π* = f(Vm) and π* = f(1/Am) show no linear trend emphasizing the importance of the combined property Vm/Amvs. its single components. In Fig. 4b the fit π* = f(Vm,1/Vm) results in the gray dashed line but show no significant improvement with respect to π* = f(Vm).
The molecular polarizability is an important parameter for the dynamics of ILs.97,98 With increasing polarizability the effective Coulomb interactions are reduced and the mobility of the ions is increased. On a collective level, the average impact of polarizability can also be mimicked by reduced partial charges and theoretically reasoned by a dielectric continuum model.97–99 The charge scaling factor in this case equals the inverse of the refractive index.97,98,100,101 In this sense, the Kamlet–Taft π* is a measure of the charge sharing and polarizable interactions in ILs, although it is not directly linked to molecular polarizability or dipole moments.
5 Conclusions
We have proven the applicability of Th and BT as solvatochromic probes for the measurement of the Kamlet–Taft parameter π*, where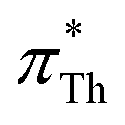 and
and  correlate well with alternative polarity measurements via the chemical shift of 129Xe. Both probes do not suffer from dependencies of the measured π* on the hydrogen bond donating abilities of the solvent, which makes them superior to conventional solvatochromic probes such as DENA. The accurate measurement of π* viaTh and BT then allowed us to examine the correlations of the dipolarity/polarizability parameter with other physico-chemical properties of the ionic liquids. We find that the correlation of π* on the inverse molar volume and molar refractivity which was found in a few studies is only of qualitative nature, and is not universally applicable between different classes of IL cations and anions. However, there is a strong correlation of π* on Am/Vm as well as Vm/Am, i.e. the ratio of molar refractivity and molar volume which is a function of the refractive index. We thus conclude that the Kamlet–Taft parameter π* is a collective parameter measuring both the volume and polarizable interactions in ionic liquids, which can be viewed as a polarizability density.
correlate well with alternative polarity measurements via the chemical shift of 129Xe. Both probes do not suffer from dependencies of the measured π* on the hydrogen bond donating abilities of the solvent, which makes them superior to conventional solvatochromic probes such as DENA. The accurate measurement of π* viaTh and BT then allowed us to examine the correlations of the dipolarity/polarizability parameter with other physico-chemical properties of the ionic liquids. We find that the correlation of π* on the inverse molar volume and molar refractivity which was found in a few studies is only of qualitative nature, and is not universally applicable between different classes of IL cations and anions. However, there is a strong correlation of π* on Am/Vm as well as Vm/Am, i.e. the ratio of molar refractivity and molar volume which is a function of the refractive index. We thus conclude that the Kamlet–Taft parameter π* is a collective parameter measuring both the volume and polarizable interactions in ionic liquids, which can be viewed as a polarizability density.
Our findings allow a more concise view on the meaning of π* in ionic liquids. We furthermore hope to encourage the reader to question the applicability of solvatochromic probes to their solvents of interest, especially for hydrogen bond donating solvents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. Heid is recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Computational Biological Chemistry. Financial support by the Deutsche Forschungsgemeinschaft grant number Sp 392/45-1 and 45-2 is gratefully acknowledged. We also thank Prof. Effenberger from the University of Stuttgart for kindly providing the solvatochromic probe BT in highly purified form.References
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- E. M. Kosower, J. Am. Chem. Soc., 1958, 80, 3253–3260 CrossRef CAS.
- E. Buncel and S. Rajagopal, Acc. Chem. Res., 1990, 23, 226–231 CrossRef CAS.
- C. Reichardt, Green Chem., 2005, 7, 339–351 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, Prog. Phys. Org. Chem., 1981, 13, 485–630 CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, Acta Chem. Scand., 1985, B39, 611–628 CrossRef CAS.
- D. M. Matyushew, R. Schmid and B. M. Ladanyi, J. Phys. Chem. B, 1997, 101, 1035–1050 CrossRef.
- J. P. Cerón-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, J. Phys. Org. Chem., 2014, 27, 512–518 CrossRef.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. I. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- A. Oehlke, K. Hofmann and S. Spange, New J. Chem., 2006, 30, 533–536 RSC.
- J. M. Lee, S. Ruckes and J. M. Prausnitz, J. Phys. Chem. B, 2008, 112, 1473–1476 CrossRef CAS.
- M. N. Kobrak, Green Chem., 2008, 10, 80–86 RSC.
- P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- Y. Yoshida, O. Baba, C. Larriba and G. Saito, J. Phys. Chem. B, 2007, 111, 12204–12210 CrossRef CAS.
- C. Chiappe, B. Melai, A. Sanzone and G. Valentini, Pure Appl. Chem., 2009, 81, 2035–2043 CAS.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- C. Chiappe, C. S. Pomelio and S. Rajamani, J. Phys. Chem. B, 2011, 115, 9653–9661 CrossRef CAS.
- J. Catalán, J. Phys. Chem. B, 2009, 113, 5951–5960 CrossRef.
- J. Catalán and H. Hopf, Eur. J. Org. Chem., 2004, 4694–4702 CrossRef.
- S. Spange, N. Friebe, C. Lienert and K. Schreiter, Chemistry-Methods, 2020 DOI:10.1002/cmtd.202000039.
- R. Lungwitz and S. Spange, New J. Chem., 2008, 32, 392–394 RSC.
- M. Holzweber, R. Lungwitz, D. Doerfler, S. Spange, M. Koel, H. Hutter and W. Linert, Chem. – Eur. J., 2013, 19, 288–293 CrossRef CAS.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS.
- K. Noack, P. S. Schulz, N. Paape, J. Kiefer, P. Wasserscheid and A. Leipertz, Phys. Chem. Chem. Phys., 2010, 12, 14153–14161 RSC.
- G. Thielemann and S. Spange, New J. Chem., 2017, 41, 8561–8567 RSC.
- H. V. Huynh, T. C. Lam and H. T. T. Luong, RSC Adv., 2018, 8, 34960–34966 RSC.
- S. Spange, R. Lungwitz and A. Schade, J. Mol. Liq., 2014, 192, 137–143 CrossRef CAS.
- A. Schade, N. Behme and S. Spange, Chem. – Eur. J., 2014, 20, 2232–2243 CrossRef CAS.
- K. Bica, M. Deetlefs, C. Schröder and K. R. Seddon, Phys. Chem. Chem. Phys., 2013, 15, 2703–2711 RSC.
- Y. Kayama, T. Ichikawa and H. Ohno, Chem. Commun., 2014, 50, 14790–14792 RSC.
- B. Linder, J. Chem. Phys., 1960, 33, 668–675 CrossRef CAS.
- I. Renge, J. Photochem. Photobiol., A, 2018, 353, 433–444 CrossRef CAS.
- A. Ghanadzadeh, A. Zeini, A. Kashef and M. Moghadam, Spectrochim. Acta, Part A, 2009, 73, 324–329 CrossRef CAS.
- M. J. Kamlet, E. G. Kayer, J. W. Eastes and W. H. Gilligan, J. Am. Chem. Soc., 1973, 95, 5210–5214 CrossRef CAS.
- L. P. Novaki and O. A. El Seoud, Ber. Bunsenges. Phys. Chem., 1996, 100, 648–655 CrossRef CAS.
- C. Laurence, P. Nicolet, M. T. Dalati, J. M. Abboud and R. Notario, J. Phys. Chem., 1994, 98, 5807–5816 CrossRef CAS.
- Y. Marcus, The Chemistry of Anilines Part I, Wiley-VCH, Weinheim, Germany, 2007, pp. 373–406 Search PubMed.
- Y. Smortsova, H. Oher, F.-A. Miannay, R. Vanel, J. Dubois, O. Kalugin and A. Idrissi, J. Mol. Liq., 2017, 245, 76–84 CrossRef CAS.
- C. Schröder, T. Rudas, G. Neumayr, W. Gansterer and O. Steinhauser, J. Chem. Phys., 2007, 127, 044505 CrossRef.
- F. Kremer and A. Schönhals, Broadband Dielectric Spectroscopy, Springer, Berlin, Heidelberg, 2003 Search PubMed.
- C. Böttcher, Theory of Electric Polarization, Elsevier Science, 1973 Search PubMed.
- S. Spange, R. Sens, Y. Zimmermann, A. Seifert, I. Roth, S. Anders and K. Hofmann, New J. Chem., 2003, 27, 520–524 RSC.
- S. Spange, S. Prause, E. Vilsmeier and W. R. Thiel, J. Phys. Chem. B, 2005, 109, 7280–7289 CrossRef CAS.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- J. Dupont, P. A. Z. Suarez, R. F. De Souza, R. A. Burro and J.-P. Kintzinger, Chem. – Eur. J., 2000, 6, 2377–2381 CrossRef CAS.
- P. Wasserscheid, M. Sesing and W. Karth, Green Chem., 2002, 4, 134–138 RSC.
- S. Rivera-Rubero and S. Baldelli, J. Phys. Chem. B, 2006, 110, 4756–4765 CrossRef CAS.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- A. J. Carmichael and K. R. Seddon, J. Phys. Org. Chem., 2000, 13, 591–595 CrossRef CAS.
- P. B. Webb, M. F. Sellin, T. E. Kunene, S. Williamson, A. M. Z. Slawin and D. J. Cole-Hamilton, J. Am. Chem. Soc., 2003, 125, 15577–15588 CrossRef CAS.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- R. K. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC.
- M. Fabris, V. Lucchini, M. Noe, A. Perosa and M. Selva, Chem. – Eur. J., 2009, 15, 12273–12282 CrossRef CAS.
- F. Effenberger and F. Würthner, Angew. Chem., Int. Ed. Engl., 1993, 32, 719–721 CrossRef.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince III, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. D. Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer III, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS.
- A. J. Sadlej, Theor. Chim. Acta, 1991, 79, 123 CrossRef CAS.
- E. Heid, M. Heindl, P. Dienstl and C. Schröder, J. Chem. Phys., 2018, 149, 044302 CrossRef.
- E. Heid, M. Fleck, P. Chatterjee, C. Schröder and A. D. MacKerell Jr., J. Chem. Theory Comput., 2019, 15, 2460 CrossRef CAS.
- E. Heid, P. Hunt and C. Schröder, Phys. Chem. Chem. Phys., 2018, 20, 8554 RSC.
- E. Heid and C. Schröder, Phys. Chem. Chem. Phys., 2018, 20, 10992 RSC.
- C. E. A. de Melo, L. G. Nandi, M. Dominguez and M. C. Rezende, J. Phys. Org. Chem., 2015, 28, 250–260 CrossRef CAS.
- W. J. Cheong and P. W. Carr, Anal. Chem., 1988, 60, 820–826 CrossRef CAS.
- S. Zhang, X. Qi, X. Ma, L. Lu and Y. Deng, J. Phys. Chem. B, 2010, 114, 3912–3920 CrossRef CAS.
- S. Kant Shukla, N. D. Khupse and A. Kumar, Phys. Chem. Chem. Phys., 2012, 14, 2754–2761 RSC.
- J. M. Padró and M. Reta, J. Mol. Liq., 2016, 213, 107–114 CrossRef.
- D. Yalcin, C. J. Drummond and T. L. Greaves, Phys. Chem. Chem. Phys., 2020, 22, 114–128 RSC.
- F. Effenberger, F. Würthner and F. Steybe, J. Org. Chem., 1995, 60, 2082–2091 CrossRef CAS.
- M. G. Bogdanov, I. Svinyarov, H. Kunkel, C. Steinle, M. Arkhipova, W. Kantlehner and G. Maas, Z. Naturforsch., 2010, 65b, 791–797 Search PubMed.
- F. Castiglione, R. Simonutti, M. Mauri and A. Mele, J. Phys. Chem. Lett., 2013, 4, 1608–1612 CrossRef CAS.
- P. Morgado, K. Shimizu, J. M. S. S. Esperanca, P. M. Reis, L. P. N. Rebelo, J. N. Canongia Lopes and E. J. M. Filipe, J. Phys. Chem. Lett., 2013, 4, 2758 CrossRef CAS.
- K. W. Mittler, N. V. Reo, A. J. M. Schoot Uiterkamp, D. P. Stengle, T. R. Stengle and K. L. Williamson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4946–4949 CrossRef.
- J. Jokisaari, Prog. Nucl. Magn. Reson. Spectrosc., 1994, 26, 1–26 CrossRef CAS.
- E. M. Arnett and P.-S. Wernett, J. Am. Chem. Soc., 1993, 115, 12187–12188 CrossRef CAS.
- T. R. Stengle, S. M. Hosseini and K. L. Williamson, J. Solution Chem., 1986, 15, 777–790 CrossRef CAS.
- M. Iihautala, J. Lounila and J. Jokisaari, J. Chem. Phys., 1999, 110, 6381–6388 CrossRef.
- J. Saunavaara and J. Jokisaari, J. Magn. Reson., 2006, 180, 58–62 CrossRef CAS.
- G. Schwarz, Ann. Stat., 1978, 6, 461–466 CrossRef.
- K. Aho, D. Derryberry and T. Peterson, Ecology, 2014, 95, 631–636 CrossRef.
- K. P. Burnham and D. R. Anderson, Sociol. Methods Res., 2004, 33, 261–304 CrossRef.
- Wolfram Research, Inc., Mathematica, Version 11.3, Champaign, IL, 2018 Search PubMed.
- E. Jónsson and P. Johansson, Phys. Chem. Chem. Phys., 2015, 17, 3697–3703 RSC.
- K. Hong, H. Zhang, J. W. Mays, A. E. Visser, C. S. Brazel, J. D. Holbrey, W. M. Reichert and R. D. Rogers, Chem. Commun., 2002, 1368–1369 RSC.
- H. Ma, X. Wan, X. Chen and Q.-F. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 143–151 CrossRef CAS.
- T. Biedron and P. Kubisa, Polym. Int., 2003, 52, 1584–1588 CrossRef CAS.
- V. Strehmel, A. Laschewsky, H. Krudelt, H. Wetzel and E. Görnitz, ACS Symp. Ser., 2005, 17–36 CrossRef CAS.
- G. Schmidt-Naake, I. Woecht and A. Schmalfuß, Macromol. Symp., 2007, 259, 226–235 CrossRef CAS.
- G. Schmidt-Naake, A. Schmalfuß and I. Woecht, Chem. Eng. Res. Des., 2008, 86, 765–774 CrossRef CAS.
- K. J. Thurecht, P. N. Gooden, S. Goel, C. Tuck, P. Licence and D. J. Irvine, Macromolecules, 2008, 41, 2814–2820 CrossRef CAS.
- A. Jeličić, N. Garcia, H. G. Löhmannsröben and S. Beuermann, Macromolecules, 2009, 42, 8801–8808 CrossRef.
- H. Svith, H. Jensen, J. Almstedt, P. Andersson, T. Lundbäck, K. Daasbjerg and M. Jonsson, J. Phys. Chem. A, 2004, 108, 4805–4811 CrossRef CAS.
- C. Bizzarri, V. Conte, B. Floris and P. Galloni, J. Phys. Org. Chem., 2011, 24, 327 CrossRef CAS.
- S. Seki, S. Tsuzuki, K. Hayamizu, Y. Umebayashi, N. Serizawa, K. Takei and H. Miyashiro, J. Chem. Eng. Data, 2012, 57, 2211–2216 CrossRef CAS.
- A. Bhattacharjee, J. A. P. Coutinho, M. G. Freire and P. J. Carvalho, J. Solution Chem., 2015, 44, 703–717 CrossRef CAS.
- C. Schröder, Phys. Chem. Chem. Phys., 2012, 14, 3089–3102 RSC.
- D. Bedrov, J.-P. Piquemal, O. Borodin, A. D. MacKerell Jr., B. Roux and C. Schröder, Chem. Rev., 2019, 119, 7940–7995 CrossRef CAS.
- I. V. Leontyev and A. A. Stuchebrukhov, J. Chem. Phys., 2009, 130, 085102 CrossRef CAS.
- H. Yu and W. F. van Gunsteren, Comput. Phys. Commun., 2005, 172, 69–85 CrossRef CAS.
- W. Zhao, H. Eslami, W. L. Cavalcanti and F. Mülller-Plathe, Z. Phys. Chem., 2007, 221, 1647–1662 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04989a |
| This journal is © the Owner Societies 2021 |