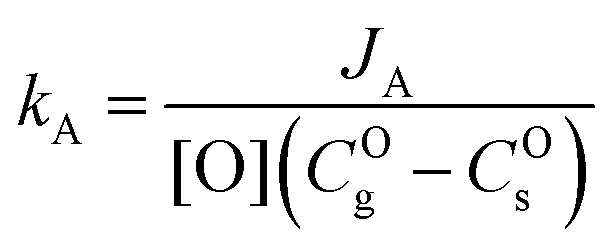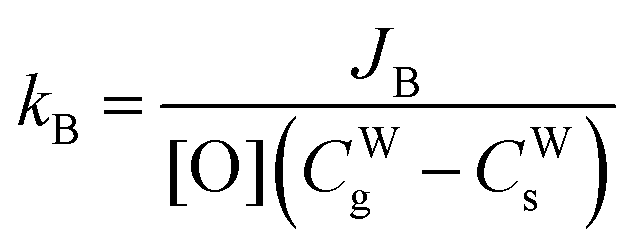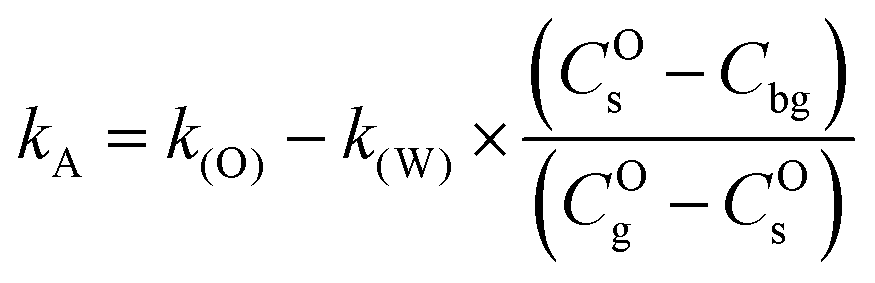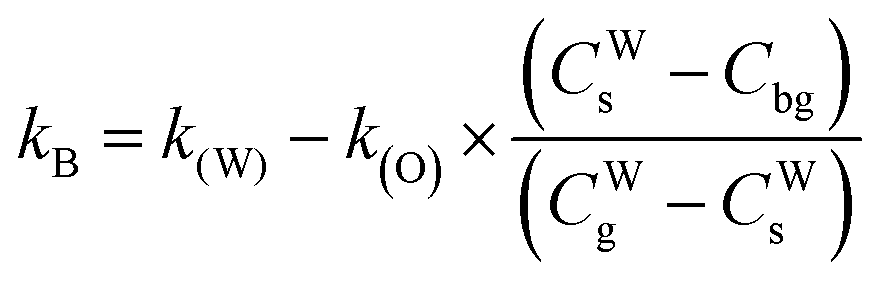Kinetics of competing exchange of oxygen and water at the surface of functional oxides†
Received
18th September 2020
, Accepted 14th January 2021
First published on 14th January 2021
Abstract
The presence of water vapour in the input gas stream influences the performance of air electrodes of solid oxide cells. In this work, the oxygen transport kinetics were determined by isotopic exchange depth profiling at 350 °C on polycrystalline La0.6Sr0.4Co0.2Fe0.8O3−δ samples in humidified oxygen, comparing the differences in tracer diffusion profile using either 18O2 or H218O as the labelling medium. The apparent surface exchange coefficients of oxygen were determined in each case and used together to estimate the oxygen surface exchange coefficients of molecular oxygen and water. It was found that, in humid conditions, the surface exchange coefficient of molecular oxygen is significantly decreased in comparison to a reference in dry conditions. In addition, the surface exchange coefficient of water is higher than that for molecular oxygen. This is in good agreement with the hypothesis that, water monopolises the active exchange sites at the material surface and thus oxygen from water exchanges faster than the one of molecular oxygen.
Introduction
The storage of intermittent renewable electrical energy from sources such as solar and wind is a topic of much current interest. There are several technologies under consideration and at different stages of maturity.1,2 A promising technological approach is to use high temperature, reversible, Solid Oxide Cells (SOCs) to electrolyse water, store the hydrogen produced, and reverse the process to generate electricity to meet demand. These cells, based on oxygen ion transport, operate at temperatures greater than 700 °C. The challenge is to reduce operating temperatures to the intermediate range of circa 500–600 °C, to reduce degradation rates and to recycle industrial waste heat that may be used to power the electrolysis process. Intermediate Temperature Solid Oxide Cells (IT-SOCs) require the stable performance of all of its components, particularly of its Mixed Ionic Electronic Conducting (MIEC) ceramic air electrode, where the operating conditions can change drastically from pure oxygen in electrolysis mode, to ambient air (including contaminants) in fuel cell mode. In order to operate efficiently the electrode must be optimised for the exchange of oxygen across the gas solid MIEC interface in all modes of operation and over the long timescales necessary for successful commercial operation of SOCs. This exchange process is fast but is susceptible to change together with the physical and chemical structure degradation of the gas solid interface that can take place over much longer timescales. As an example, operation in air may cause long-term performance degradation issues upon exposure to common atmospheric components such as H2O, as it has been detailed in a number of studies on perovskite materials employing aliovalent substitution on the A-site.3–8 The formation of secondary phases on the surface were mentioned to explain the passivation of the surface, either as a result of the transport by water of contaminants such as alkaline species or silicon6 or as a result of the decomposition of the electrode materials on their surface.7 This decomposition may be linked to an increased strontium segregation rate that can lead to the subsequent partial deactivation of the surface with the heterogeneously distributed SrOx/Sr(OH)2 phases.8–11 Overall, the performance degradation of the air electrode of SOC is not fully understood and may be due to the combination of several mechanisms, most of which operate over timescales of hours or thousands of hours.
As well as affecting degradation processes, oxygen bearing components (e.g. H2O and CO2) will also modify the exchange of oxygen across the gas MIEC solid interface, hence a better understanding of their influence on the surface exchange kinetics of oxygen is necessary. In the case of Intermediate Temperature Solid Oxide Fuel Cells (IT-SOFCs) cathode and electrolyte materials it has been reported from Isotopic Exchange Depth Profiling (IEDP) experiments that during dry 18O2 exchange, residual water could improve the exchange of molecular oxygen.12,13 It was also measured, on a variety of relevant materials, that the surface exchange coefficient is higher when using labelled water (H218O) as compared to dry molecular oxygen.12,14–19 One should note that the latter experiments were performed at a defined partial pressure of oxygen but with a relatively short exposure to water. Pietrowski et al.18 explain that the main reason why water exchange is faster than dry molecular oxygen exchange could be related to the fact that electrons are not formally required for oxygen incorporation from water vapour. It is also suggested that the faster exchange is due to facilitated adsorption and dissociation of water on surface oxygen vacancies, forming two surface hydroxyl groups.17,19,20 It is pertinent to point out that in an operating SOC, it is only the molecular oxygen that can contribute to the overall oxygen flux through the device. Water can only accelerate or impede this process. To this end, Nenning et al.19 highlighted that, from an electrochemical point of view, IEDP may provide a surface exchange coefficient k* that is not always meaningful, especially in the presence of water (even a trace amount).19 In addition, a consistent decrease of the chemical surface exchange coefficient kchem was observed by electrical conductivity relaxation (ECR) measurements in wet conditions on polycrystalline La0.6Sr0.4CoO3−δ (LSC) and La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF).4–6 Measurements of the electrical surface exchange coefficient kq by electrical impedance spectroscopy (EIS) in wet conditions were also performed. In the presence of water, Nenning et al. reported a slight decrease of kq on La0.6Sr0.4FeO3−δ (LSF) and SrTi0.3Fe0.7O3−δ (STF) thin films at 418 °C.19 Jong HoonJoo et al., identified two effects in humidified oxygen La0.6Sr0.4CoO3−δ (LSC) thin films: a short-term improvement of the oxygen surface exchange but also the increased degradation over longer time scales.21 Further investigation by means of gas-phase-analysis of isotope exchange allowed better discrimination between reactions involving oxygen-containing species on metal oxides. Wachsman et al. pointed out the active role of water in the oxygen reduction reaction (ORR), leading to enhancement or preclusion of the exchange of molecular oxygen with lattice oxygen, depending on the material.22,23 Specifically, the authors reported a decrease of the surface exchange rate of molecular oxygen by one order of magnitude in the temperature range from 350 °C to 450 °C for LSCF and an increase of the same exchange rate for (La0.8Sr0.2)0.95MnO3 (LSM) in the temperature range from 400 °C to 550 °C. The findings mentioned previously for various materials can be perplexing, considering their contrasting behaviours. It is thus important to understand the role of water in the exchange process for MIEC materials commonly used as air electrodes in IT-SOCs.
IEDP experiments are commonly used to characterise the oxygen transport kinetics of ceramic oxides. Most of these experiments focus on providing meaningful values of the surface exchange coefficient and are performed in dry labelled oxygen with a water content, in some cases, as low as <1 ppm. Recently, the back-exchange technique was developed to study the effect of different atmospheres on the oxygen transport kinetics while avoiding the use of expensive isotopes such as H218O or C18O2.24 However, IEDP experiments, back-exchange included, do not give any mechanistic information on the exchange reactions occurring at the surface with oxygen bearing molecules from the surrounding atmosphere and thus a different approach is necessary. In the present study, we applied a sequential double labelling approach. LSCF samples were exchanged, either with 18O2 or with H218O as the labelling medium, keeping all the other parameters the same. The apparent surface exchange coefficients of oxygen were determined in each case and used together to estimate the surface exchange coefficients specific to molecular oxygen and water vapour in humidified oxygen.
Choice of a model
In order to describe the exchange mechanisms of oxygen taking place at the surface of a material, one can consider many elementary reactions, involving dissociated species, charged adsorbates, migration of species over the surface and diverse catalytic effects from the cations, etc. In the case of LSCF and other oxygen deficient perovskites, it is commonly assumed that oxygen vacancies at the vicinity of the surface play a central role in this process, acting as exchange sites between the adsorbed oxygen bearing species and the solid.25 For the purpose of the paper we will simplify the problem and disregard the nature of the exchange sites as well as any reaction intermediate in the writing of the reactions. Further thoughts on the matter will be brought into the discussion. Let us consider then a MIEC material at chemical equilibrium, in the presence of a gas mixture of molecular oxygen (O2 or OO) and water vapour (H2O). We assume that the only two following competing, but independent, hetero-exchange reactions may happen simultaneously at the surface (expressed in Kröger–Vink notation):| | 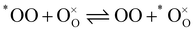 | (1) |
| | 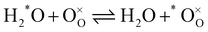 | (2) |
In the previous two reactions, one oxygen has been identified (*O) to facilitate following its transfer from one molecule to the solid. It does not necessarily correspond to a labelled oxygen (as labelled oxygen will be used later). Reactions (1) and (2) describe respectively the hetero-exchange from molecular oxygen and water with the solid. Also important is (for the prerequisite of independence) the assumption that, even if both reactions may take place through the intermediary of oxygen vacancies near the surface, the reactions are localised and far enough apart so that reaction intermediates do not interact.
Given the low oxygen substoichiometry of LSCF below 600 °C,26,27 this assumption is reasonable as one can expect low availability of active sites near the surface. Under these conditions, IEDP provides an apparent oxygen exchange coefficient resulting from concomitance of reactions (1) and (2).
Experimental procedure
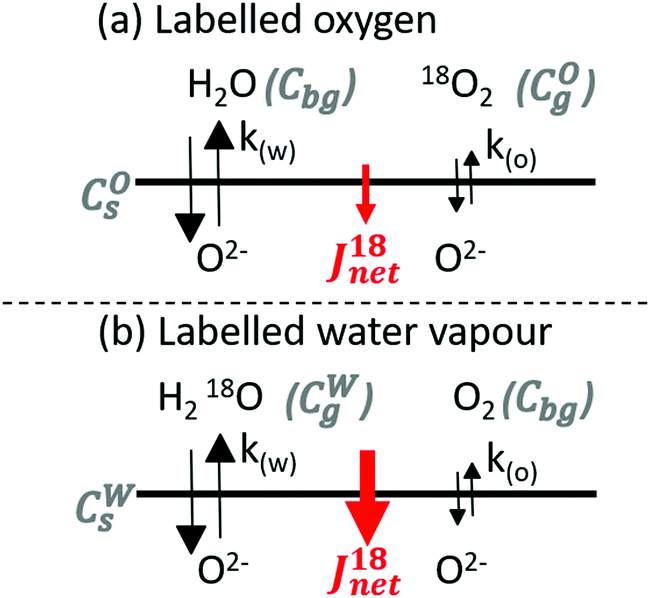
Two IEDP experiments were performed at 350 °C on polished (SiC paper abrasion then diamond paste polishing down to 1/4 μm, achieving a mean roughness Ra = 8(±2) nm) LSCF dense (drel = 99%) samples in humidified oxygen. The partial pressure of oxygen and water were respectively set to 0.20 and 0.10 bar. Samples were pre-annealed for 17.5 hours in a predetermined atmosphere with unlabelled gasses to attain thermodynamic equilibrium between the solid and the gas phase. They were then exchanged for 175 minutes in the same conditions but this time with either the oxygen or the water being enriched with 18O. In experiment A, labelled oxygen (enriched at 99%) was mixed with unlabelled water (Fig. 1a). In experiment B, labelled water vapour (H218O, 97%atom of 18O) was mixed with unlabelled oxygen (Fig. 1b). Care was taken to have no exposed platinum (in particular a Pt–Pt/Rh thermocouple was covered by a long and narrow quartz sleeve to limit the mass transport of products from isotope scrambling that may happen in the hot zone) or similar catalysts in the hot zone. It was made sure that the concentration of labelled species remained the same before and after the exchange by analysing the atmosphere by Residual Gas Analysis (RGA) with a quadrupole mass spectrometer (Hiden HAL 51). Diffusion profiles of the oxygen isotopes were obtained by Secondary Ion Mass Spectrometry (SIMS) of negative species using a IONTOF TOF-SIMS V instrument equipped with an Extended Dynamic Range (EDR) analyser. Two ion guns were used. A bunched 30 keV Bi+ (∼1.5 pA) beam was used for analysis, interlaced with a 2 keV Ar+ sputter (∼600 nA) beam used for sputtering. More information about the general procedure, and the use of EDR can be found respectively in ref. 28 and 29.
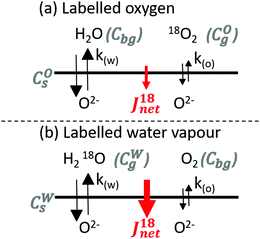 |
| | Fig. 1 Schematic of the isotopic exchange experiments with (a) labelled oxygen or (b) labelled water vapour. The net flux of labelled oxygen into the sample during each experiment is indicated, revealing the concomitant but unequal surface exchange kinetics of molecular oxygen and water observed here. | |
Theory
Isotope exchange with a single exchanging gas (18O2).
To establish what will occur with two exchanging gasses it is first salient to look at the case of a single exchanging gas to define some terms. We assume that we have a small sample and a large volume of exchanging gas such that the change of the isotopic fraction in the gas is negligible. At equilibrium, the net flux of oxygen across the surface is zero. In the case of an isotopic exchange involving only one gas phase molecule, i.e. dry labelled oxygen, the atomic fluxes, J18in, and J18out of labelled oxygen across the surface are defined by the surface exchange coefficient k, the volume concentration of oxygen in the solid [O], is the isotopic fraction of the gas phase Cg and the instantaneous isotopic fraction at the solid surface Cs:| | | J18in = k[O]Cg and J18out = k[O]Cs | (3) |
i.e.| | | J18net = J18in − J18out = k[O](Cg − Cs) | (4) |
An important part of this process is the instantaneous surface isotopic fraction, Cs. This surface isotopic fraction is dependent upon a dimensionless quantity h′ defined as 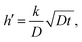 where D is the diffusion coefficient and t the annealing time. For any particular exchange experiment, the normalised surface isotopic fraction
where D is the diffusion coefficient and t the annealing time. For any particular exchange experiment, the normalised surface isotopic fraction 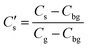 (where Cbg is the natural background concentration of 18O) increases with time:
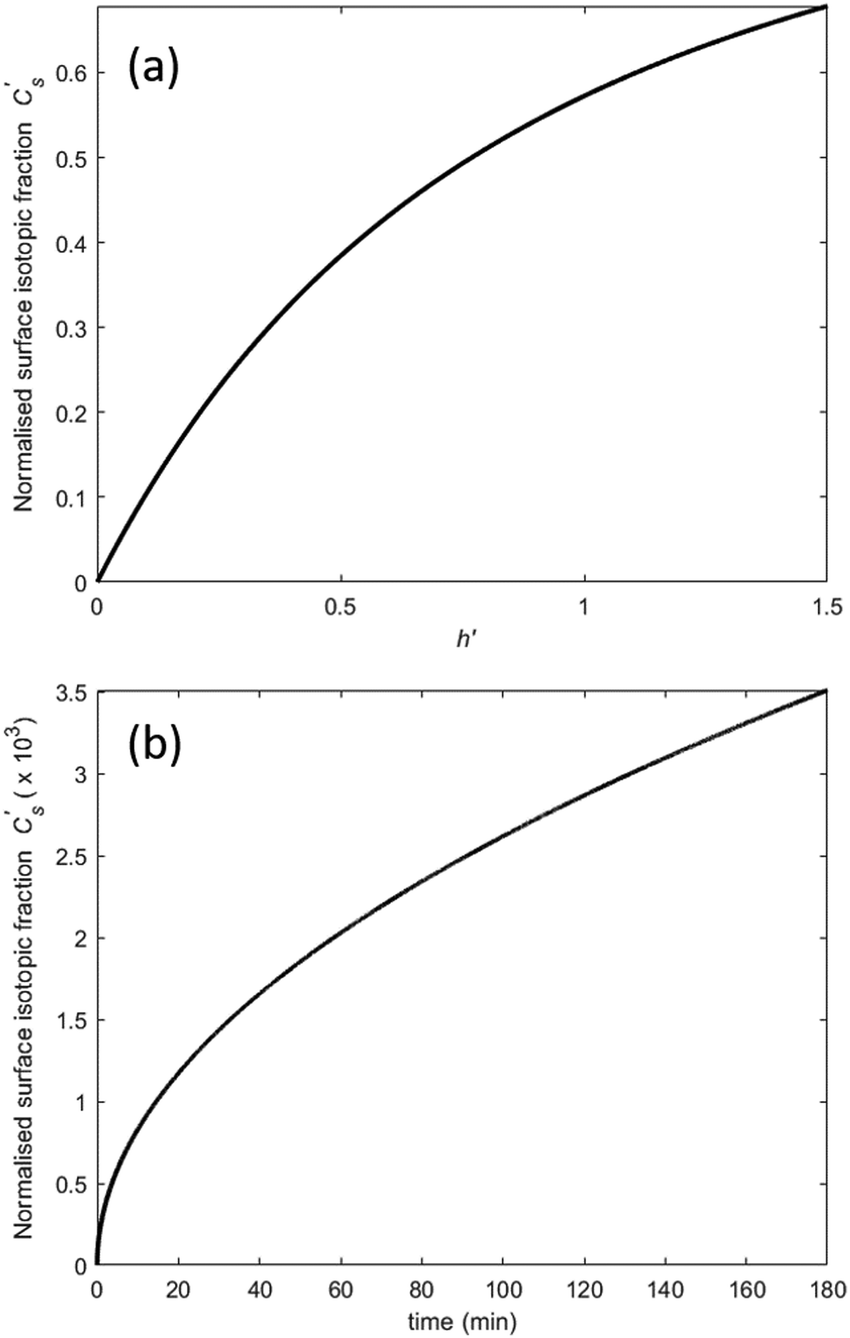
(where Cbg is the natural background concentration of 18O) increases with time: 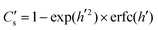 30,31 as illustrated in Fig. 2a. h′ is an important parameter in determining the rate at which the isotope surface concentration builds-up. Fig. 2b illustrates a concrete case related to experiment A.
30,31 as illustrated in Fig. 2a. h′ is an important parameter in determining the rate at which the isotope surface concentration builds-up. Fig. 2b illustrates a concrete case related to experiment A.
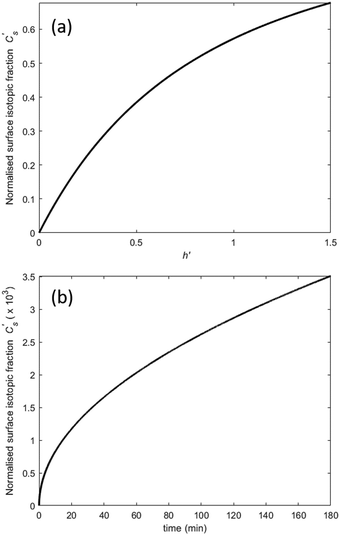 |
| | Fig. 2 Build-up of the surface isotope concentration at the surface 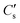 as a function of (a) the dimensionless quantity h′, (b) time according to a specific case where k = 3.0 × 10−10 cm s−1 and D = 1.0 × 10−10 cm2 s−1. as a function of (a) the dimensionless quantity h′, (b) time according to a specific case where k = 3.0 × 10−10 cm s−1 and D = 1.0 × 10−10 cm2 s−1. | |
Isotope exchange with two gases (labelled 18O2 or H218O)
In the case where we have two exchanging gas molecules, we must make some further assumptions. The first is that there is no isotopic mixing in the gas phase and the second that the surface exchange coefficients are unaffected by the labelling. The situation for this type of exchange is shown in Fig. 1. For such an experiment the fitting of the resultant isotopic depth profile to the solution to the diffusion equation will give an apparent exchange coefficient, kapp, which can be used to deconvolute the independent exchange coefficients for the individual exchanging molecules. Both experiments should be performed for the same time, t, and under identical thermodynamic conditions as then we can assume that value of D is the same and thus the value of h′ 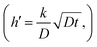 for each experiment is only dependent upon the apparent value of k in each case. If we consider now the conditions at the end of the diffusion exchanges, then we can obtain the surface isotopic fractions and the apparent exchange coefficients for the two exchange experiments by analysis of the isotopic depth profiles.
for each experiment is only dependent upon the apparent value of k in each case. If we consider now the conditions at the end of the diffusion exchanges, then we can obtain the surface isotopic fractions and the apparent exchange coefficients for the two exchange experiments by analysis of the isotopic depth profiles.
Exchange experiment A (18O2, H216O)
The net 18O flux in experiment A, JA, can be expressed as the combination of the flux from the labelled oxygen JOA and the flux to the unlabelled water JWA:| | | JA = JOA − JWA = k(O) × [O] × (COg − COs) − k(W) × [O] × (COs − Cbg) | (5) |
where k(O) and k(W) are the surface exchange coefficients respectively of oxygen and water, [O] is the oxygen concentration in the solid, COg the isotopic fraction of 18O in the labelled oxygen, COs the concentration of 18O at the surface of the solid, at the end of the experiment, and Cbg the natural isotopic fraction of oxygen in the water.
Exchange experiment B (16O2, H218O)
In the same way, the net 18O flux in experiment B, JB, can be expressed as the combination of the flux from the labelled water JWB and the flux to the unlabelled oxygen JOB:| | | JB = JWB − JOB = k(W) × [O] × (CWg − CWs) − k(O) × [O] × (CWs − Cbg) | (6) |
where CWg is the isotopic fraction of 18O in the labelled water and CWs the concentration of 18O at the surface of the solid.
Combination of experiments A and B
The apparent surface exchange coefficients of oxygen in experiments A and B, respectively kA and kB are defined as:| | 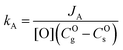 | (7) |
| | 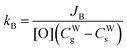 | (8) |
and thus:| | 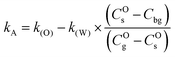 | (9) |
| | 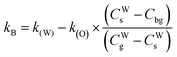 | (10) |
Then we can measure the surface isotopic fractions in the two experiments and hence make some estimation of the relative values of the surface exchange coefficients for the oxygen and the water molecules.
Results
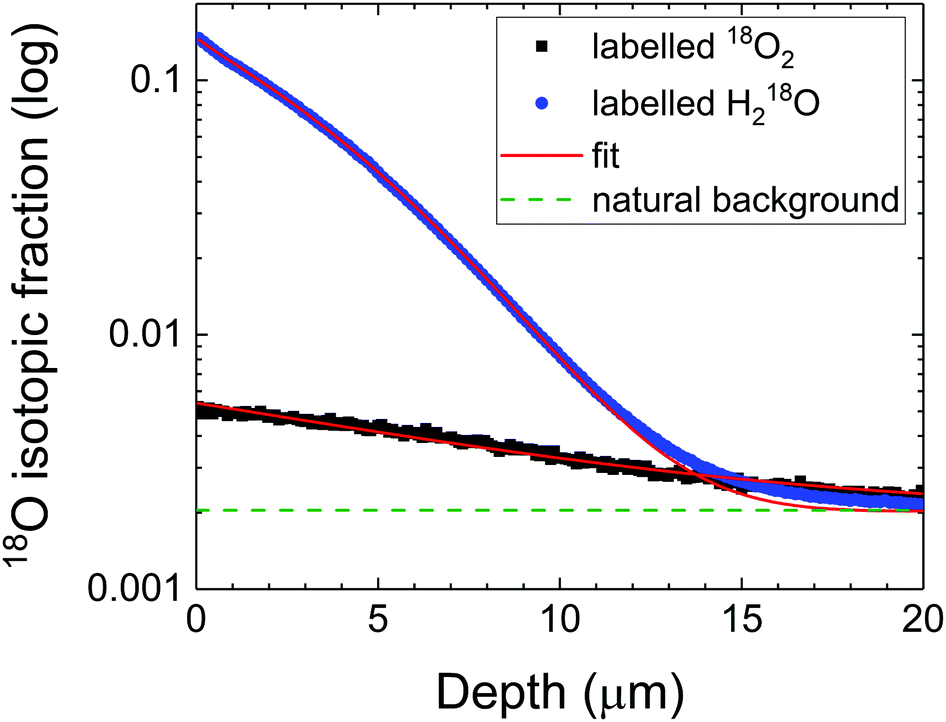
The diffusion depth profiles in LSCF from experiments A and B are compared in Fig. 3. The diffusion coefficients for both experiments were reasonably close (DA = 9 × 10−11 cm2 s−1 and DB = 2 × 10−11 cm2 s−1). The apparent surface exchange coefficients are compared to the one measured in dry oxygen (<1 ppm H2O) in Table 1. From Fig. 3, it is noticeable that the surface fraction of 18O supplied from 18O2 (experiment A) is low (significantly depleted by concomitant exchange with unlabelled water), in fact lower than would be expected for the same anneal time in dry oxygen. In contrast, the surface concentration of labelled oxygen is much higher (experiment B) when the labelled oxygen is sourced from the water molecule (although depleted by unlabelled oxygen). It is reasonable to assume that in eqn (10), k(W) is much larger than the following term, therefore that:
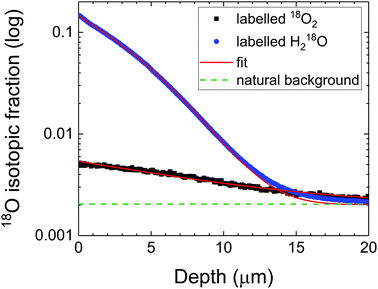 |
| | Fig. 3 Diffusion depth profiles in LSCF at 350 °C in humidified oxygen with different labelled species. Note that the isotopic fraction is displayed on a logarithmic scale, not the usual linear representation. | |
Table 1 Apparent surface exchange coefficient of LSCF depending of the atmosphere composition and of the total annealing time at 350 °C
| Experiment |
Atmosphere |
Labelled species |
Total annealing time (hours) |
Temperature (°C) |
Exchange time (minutes) |
Apparent k (cm s−1) |
| A |
Humidified oxygen |
18O2 |
17.5 |
347 |
174 |
3.0 × 10−10 |
| B |
Humidified oxygen |
H218O |
17.5 |
353 |
175 |
5.7 × 10−9 |
| From ref. 33 |
Dry oxygen |
18O2 |
13 |
350 |
166 |
2.2 × 10−9 |
From our previous theoretical considerations, we can benefit from experiment A. The final maximum value of COs was taken (Fig. 3) and used within eqn (9) to estimate the surface exchange coefficient specific to molecular oxygen in humid conditions: k(O) ≈ 3.2 × 10−10 cm s−1. The specific surface exchange coefficients of molecular oxygen and water in dry or humidified oxygen at 350 °C are reported in Table 2.
Table 2 Specific surface exchange coefficient of LSCF of molecular oxygen (0.20 bar) and water (0.10 bar) in dry and humidified oxygen at 350 °C
| Atmosphere |
k
(O) (cm s−1) |
k
(W) (cm s−1) |
| Dry oxygen |
2.2 × 10−9 |
— |
| Humidified oxygen |
3.2 × 10−10 |
5.7 × 10−9 |
Discussion
Among IT-SOFC cathode materials, LSCF is at the state-of-the-art and commonly used in real IT-SOFC systems. LSCF was chosen for this experiment as its exchange and diffusivity properties have been studied extensively, particularly by IEDP analysis.11,24,32,33 Most of the previous studies were performed at intermediate to high temperatures (500–1000 °C), however, the properties of LSCF at low temperatures have not been studied. As such, 350 °C was chosen for this experiment. An additional advantage of using low temperature is the ability to use short diffusion profiles (around 20 μm) to obtain high quality depth profiles using ION-TOF's EDR.
The current work relies on the assumption that only reactions (1) and (2) happen independently at the surface. In other words, we disregard other reactions such as the homo-exchange of the adsorbed species (i.e. water and molecular oxygen) at the surface:
| | | *OO + H2O ⇌ OO + H2*O | (12) |
The low concentration of active sites available at the surface (oxygen vacancies in the current case) supports this independence prerequisite.
26,27 One could argue that the number of active sites available at the surface may vary in the presence of a space charge layer. However, this should go along with further decrease of the concentration of active sites near the surface,
34 therefore a lower chance of interaction between the adsorbed species. The nature of the point defects contributing to the oxygen transport has
a priori no influence on the model. For instance, the approach should hold for materials for which oxygen transport is ensured by oxygen interstitials. One should notice that it is also possible to describe the homo-exchange of water and oxygen by combination of
reactions (1) then -(2). In any case, it is out of the scope of this work to differentiate the homo-exchange from the succession of two hetero-exchange. However, one must keep in mind that the concentration of the labelling medium must be kept constant while performing IEDP experiments.
35 This consideration becomes even more crucial in a situation where isotope scrambling can happen (
e.g. by homo-exchange catalysed at the surface of quartz, of platinum or of the sample), typically in the presence of two different oxygen bearing molecules such as water and molecular oxygen. In this case it can become impossible to know from which molecule the labelled oxygen is sourced. For instance, during
18O
2 isotope exchange, residual water might remove labelled species from the solid at a higher rate than it gets incorporated from
18O
2, or might produce H
218O that exchanges faster with the solid.
12 In any case, isotope scrambling will distort the measurement of
k. We made sure that in experiments A and B, the concentrations of the different isotopologues measured by RGA remained the same contrary to what could be observed at higher temperature (see Fig. S1 in the ESI
†), which was also observed by Sha
et al.36 From this consideration, the sequential double labelling approach presented in this paper should effectively be applicable at temperatures up to ∼500–550 °C.
Over the relatively short timescale of experiments A and B, the surface exchange coefficient specific to water, k(W), is found to be higher than the surface exchange coefficient from the dry molecular oxygen. This is in good agreement with the result obtained from back-exchange experiments.33 Incidentally, the use of labelled water has been used advantageously to enhance the incorporation of 18O in fast diffusing materials, allowing a more accurate determination of their tracer diffusion coefficient.12,15 The facilitated exchange of oxygen from the water molecule has been explained in term of dissociation chemistry17–20 and can also be explained by the higher energy required to dissociate oxygen compared to water.37 It is still unclear, from one single exchange with H218O and 16O2, (experiment B) what the effect of water is on the exchange of molecular oxygen. It would be wrong to interpret the increase of the apparent oxygen exchange coefficient, kB, also in term of increase of the exchange rate of molecular oxygen, as it has been highlighted by Nenning et al.19 As a matter of fact, we found that, in humid conditions, the surface exchange coefficient attributed to molecular oxygen, k(O), is about 1 order of magnitude lower than in dry conditions, showing that the surface exchange rate of molecular oxygen decreases in the presence water vapour.
This is in good agreement with the work from Huang et al. on LSCF.23 The authors explain it by the preferential coverage of the surface by water in this temperature range. The same explanation was suggested for other materials such as SrTiO338 or Y-doped BaZrO3;39 thus water may block the access to the few active exchange sites for molecular oxygen. However, this cannot be generalised. In other cases, the surface exchange coefficient of molecular oxygen increased in the presence of water. A catalytic role was generally attributed to water in the case of Ba0.7La0.3CoO3 (BLC)16 and more specifically to the hydroxide species in the case of LSC.21 In the case of LSM, it was suggested that slow incorporation could favour the homo-exchange and invalidate the boundary condition at the surface, resulting in an incorrect measurement.23 From DFT calculations on SrTiO3, it has been proposed that the water molecule could catalyse the dissociation of molecular oxygen above a certain temperature.38
Overall, there is no clear consensus how water may increase or decrease the surface exchange kinetics of molecular oxygen with MIEC materials. Any effect is most likely specific to the material and dependent upon the temperature and gas compositions involved and thus the results given above for this experiment must be viewed in this context. If water could lead to a long-term improvement of the surface exchange of oxygen (electrochemically) useful to applications, the question remains to find out with what material, combined to what type of electrolyte, in what atmosphere, at which temperature, at what partial pressure or with what kind of electrode's morphology and microstructure. The fact that water is a versatile reactant40,41 means that it may also take part in several reactions over different timescales. It should be kept in mind that these reactions may have contradictory effects.
Conclusions and future perspectives
In this work, we demonstrated that it is possible to use two complementary IEDP experiments and simple approximations to estimate the surface exchange kinetics specific to both molecular oxygen and water, in humidified oxygen. We observed that, at the temperature of the study (350 °C) the surface exchange rate of molecular oxygen at the surface of a dense polycrystalline sample of LSCF is hindered by water. This is likely due to preferential coverage of the surface by water, thus blocking the access of molecular oxygen to active exchange sites. In the meantime, oxygen from water exchanges more effectively than that from dry molecular oxygen. This behaviour may change as a function of temperature and for different materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the support of World Premier International Research (WPI)-I2CNER and funding from the EPSRC-JSPS Core-to-Core Program, Advance research Network (EP/P026478/1). The help from Dr Richard Chater and Dr Andrea Cavallaro is also warmly acknowledged. VT would also like to acknowledge support of the FRINATEK project 262393 “Fundamentals of Surface Kinetics in High Temperature Electrochemistry” (FUSKE) of the Research Council of Norway.
Notes and references
- M. Aneke and M. Wang, Energy storage technologies and real life applications – a state of the art review, Appl. Energy, 2016, 179, 350–377 CrossRef.
- T. M. Gür, Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Degradation Mechanisms of La–Sr–Co–Fe–O3 SOFC Cathodes, Electrochem. Solid-State Lett., 2006, 9, A478 CrossRef CAS.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Oxygen exchange kinetics of La0.58Sr0.4Co0.2Fe0.8O3 at 600 °C in dry and humid atmospheres, Solid State Ionics, 2011, 191, 61–67 CrossRef CAS.
- E. Bucher and W. Sitte, Long-term stability of the oxygen exchange properties of (La,Sr)1−z(Co,Fe)O3−δ in dry and wet atmospheres, Solid State Ionics, 2011, 192, 480–482 CrossRef CAS.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Impact of humid atmospheres on oxygen exchange properties, surface-near elemental composition, and surface morphology of La0.6Sr0.4CoO3−δ, Solid State Ionics, 2012, 208, 43–51 CrossRef CAS.
- R. R. Liu, S. H. Kim, S. Taniguchi, T. Oshima, Y. Shiratori, K. Ito and K. Sasaki, Influence of water vapor on long-term performance and accelerated degradation of solid oxide fuel cell cathodes, J. Power Sources, 2011, 196, 7090–7096 CrossRef CAS.
- A. Egger, M. Perz, E. Bucher, C. Gspan and W. Sitte, Effect of Microstructure on the Degradation of La0.6Sr0.4CoO3−δ Electrodes in Dry and Humid Atmospheres, Fuel Cells, 2019, 19, 458–471 CAS.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chemical Heterogeneities on La0.6Sr0.4CoO3–δ Thin Films—Correlations to Cathode Surface Activity and Stability, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Surface termination and subsurface restructuring of perovskite-based solid oxide electrode materials, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- M. Niania, R. Podor, T. Ben Britton, C. Li, S. J. Cooper, N. Svetkov, S. Skinner and J. Kilner,
In situ study of strontium segregation in La0.6Sr0.4Co0.2Fe0.8O3–δ in ambient atmospheres using high-temperature environmental scanning electron microscopy, J. Mater. Chem. A, 2018, 6, 14120–14135 RSC.
- R. N. Vannier, S. J. Skinner, R. J. Chater, J. A. Kilner and G. Mairesse, Oxygen transfer in BIMEVOX materials, Solid State Ionics, 2003, 160, 85–92 CrossRef CAS.
- S. P. Waldow, B. J. Statham, H. F. Wardenga, T. E. Weirich, A. Klein and R. A. D. Souza, Oxygen Surface Exchange and Tracer Diffusion in Differently Oriented Thin Films of Gd-Doped CeO2, ACS Appl. Mater. Interfaces, 2020, 12, 36768–36777 CrossRef CAS.
- N. Sakai, K. Yamaji, T. Horita, Y. P. Xiong, H. Kishimoto and H. Yokokawa, Effect of Water on Oxygen Transport Properties on Electrolyte Surface in SOFCs: I. Surface Reaction Mechanism of Oxygen Isotope Exchange on Solid Oxide Electrolytes, J. Electrochem. Soc., 2003, 150, A689–A694 CrossRef CAS.
- S. Georges, S. J. Skinner, P. Lacorre and M. C. Steil, Oxide ion diffusion in optimised LAMOX materials, Dalton Trans., 2004, 3101–3105 RSC.
- T. Ishihara, S. Fukui and H. Matsumoto, Effects of Water Coexisting on the Cathode Activity for the Solid Oxide Fuel Cells Using LaGaO3 -Based Perovskite Oxide Electrolyte, J. Electrochem. Soc., 2005, 151, A2035–A2039 CrossRef.
- M. Kessel, R. A. De Souza, H.-I. Yoo and M. Martin, Strongly enhanced incorporation of oxygen into barium titanate based multilayer ceramic capacitors using water vapor, Appl. Phys. Lett., 2010, 97, 21910 CrossRef.
- M. J. Pietrowski, R. A. D. Souza, M. Fartmann, R. T. Veen and M. Martin, Oxygen isotope transport properties of yttria-stabilized zirconia (YSZ) in O2- and H2O-containing atmospheres, Fuel Cells, 2013, 13, 673–681 CAS.
- A. Nenning, E. Navickas, H. Hutter and J. Fleig, Water-Induced Decoupling of Tracer and Electrochemical Oxygen Exchange Kinetics on Mixed Conducting Electrodes, J. Phys. Chem. Lett., 2016, 7, 2826–2831 CrossRef CAS.
- R. Merkle and J. Maier, How Is Oxygen Incorporated into Oxides? A Comprehensive Kinetic Study of a Simple Solid-State Reaction with SrTiO3 as a Model Material, Angew. Chem., Int. Ed., 2008, 47, 3874–3894 CrossRef CAS.
- J. H. Joo, R. Merkle and J. Maier, Effects of water on oxygen surface exchange and degradation of mixed conducting perovskites, J. Power Sources, 2011, 196, 7495–7499 CrossRef CAS.
- E. D. Wachsman, Y. Huang, C. Pellegrinelli, J. A. Taillon and L. G. Salamanca-Riba, Towards a Fundamental Understanding of the Cathode Degradation Mechanisms, ECS Trans., 2014, 61, 47–56 CrossRef CAS.
- Y.-L. Huang, C. Pellegrinelli and E. D. Wachsman, Oxygen Dissociation Kinetics of Concurrent Heterogeneous Reactions on Metal Oxides, ACS Catal., 2017, 7, 5766–5772 CrossRef CAS.
- S. J. Cooper, M. Niania, F. Hoffmann and J. A. Kilner, Back-exchange: a novel approach to quantifying oxygen diffusion and surface exchange in ambient atmospheres, Phys. Chem. Chem. Phys., 2017, 19, 12199–12205 RSC.
- M. Mosleh, M. Sógaard and P. V. Hendriksen, Kinetics and Mechanisms of Oxygen Surface Exchange on La0.6Sr0.4FeO3−δ Thin Films, J. Electrochem. Soc., 2009, 156, B441 CrossRef CAS.
- M. Kuhn, Y. Fukuda, S. Hashimoto, K. Sato, K. Yashiro and J. Mizusaki, Oxygen Nonstoichiometry and Thermo-Chemical Stability of Perovskite-Type La0.6Sr0.4Co1−yFeyO3−δ (y = 0, 0.2, 0.4, 0.5, 0.6, 0.8, 1) Materials, J. Electrochem. Soc., 2013, 160, F34–F42 CrossRef CAS.
- S. Upasen, P. Batocchi, F. Mauvy, A. Slodczyk and P. Colomban, Chemical and structural stability of La0.6Sr0.4Co0.2Fe0.8O3–δ ceramic vs. medium/high water vapor pressure, Ceram. Int., 2015, 41, 14137–14147 CrossRef CAS.
- J. A. Kilner, S. J. Skinner and H. H. Brongersma, The isotope exchange depth profiling (IEDP) technique using SIMS and LEIS, J. Solid State Electrochem., 2011, 15, 861–876 CrossRef CAS.
- H. Téllez, J. Druce, J.-E. Hong, T. Ishihara and J. A. Kilner, Accurate and precise measurement of oxygen isotopic fractions and diffusion profiles by selective attenuation of secondary ions (SASI), Anal. Chem., 2015, 87, 2907–2915 CrossRef.
-
J. A. Kilner, Second International Symposium on Ionic and Mixed Conducting Ceramics, 1994, p. 174.
- A. Berenov, A. Atkinson, J. Kilner, M. Ananyev, V. Eremin, N. Porotnikova, A. Farlenkov, E. Kurumchin, H. J. M. Bouwmeester, E. Bucher and W. Sitte, Oxygen tracer diffusion and surface exchange kinetics in Ba0.5Sr0.5Co0.8Fe0.2O3−δ, Solid State Ionics, 2014, 268, 102–109 CrossRef CAS.
-
S. J. Benson, PhD thesis, University ofLondon, 1992.
-
V. Thoréton, et al., to be published.
- R. A. De Souza, Limits to the rate of oxygen transport in mixed-conducting oxides, J. Mater. Chem. A, 2017, 5, 20334–20350 RSC.
- J. A. Kilner, B. C. H. Steele and L. Ilkov, Oxygen self-diffusion studies using negative-ion secondary ion mass spectrometry (SIMS), Solid State Ionics, 1984, 12, 89–97 CrossRef CAS.
- Z. Sha, E. Cali, G. Kerherve and S. J. Skinner, Oxygen diffusion behaviour of A-site deficient (La0.8Sr0.2)0.95Cr0.5Fe0.5O3−δ perovskites in humid conditions, J. Mater. Chem. A, 2020, 8, 21273–21288 RSC.
- S. Barraza-Lopez and T. P. Kaloni, Water Splits To Degrade Two-Dimensional Group-IV Monochalcogenides in Nanoseconds, ACS Cent. Sci., 2018, 4, 1436–1446 CrossRef CAS.
- A. Staykov, S. Fukumori, K. Yoshizawa, K. Sato, T. Ishihara and J. Kilner, Interaction of SrO-terminated SrTiO3 surface with oxygen, carbon dioxide, and water, J. Mater. Chem. A, 2018, 6, 22662–22672 RSC.
- J. M. Polfus, B. Yildiz and H. L. Tuller, Origin of fast oxide ion diffusion along grain boundaries in Sr-doped LaMnO3, Phys. Chem. Chem. Phys., 2018, 20, 19142–19150 RSC.
- P. A. Thiel and T. E. Madey, The interaction of water with solid surfaces: fundamental aspects, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
- M. A. Henderson, The interaction of water with solid surfaces: fundamental aspects revisited, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04953k |
| ‡ Current address: University of Oslo, FASE, Gaustadalléen 21, 0349 Oslo, Norway. |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mathew
Niania
*a,
Mathew
Niania
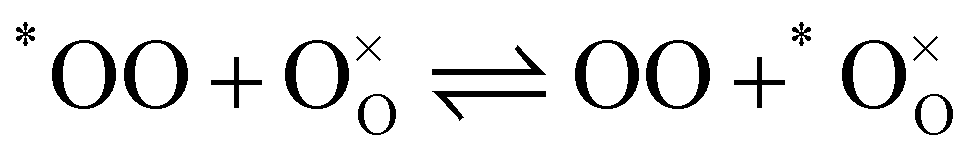
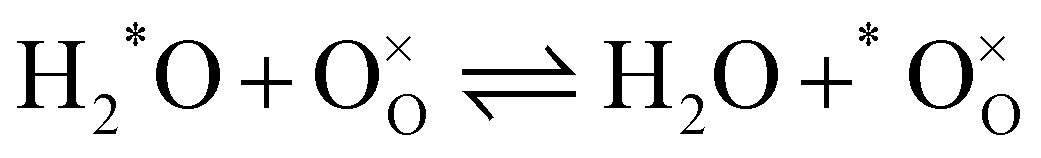
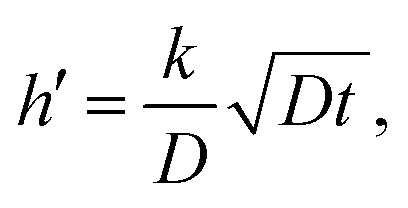 where D is the diffusion coefficient and t the annealing time. For any particular exchange experiment, the normalised surface isotopic fraction
where D is the diffusion coefficient and t the annealing time. For any particular exchange experiment, the normalised surface isotopic fraction 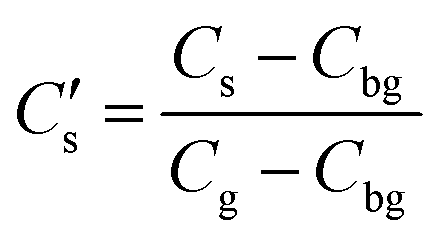 (where Cbg is the natural background concentration of 18O) increases with time:
(where Cbg is the natural background concentration of 18O) increases with time: 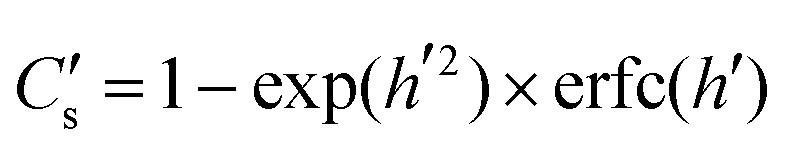 30,31 as illustrated in Fig. 2a. h′ is an important parameter in determining the rate at which the isotope surface concentration builds-up. Fig. 2b illustrates a concrete case related to experiment A.
30,31 as illustrated in Fig. 2a. h′ is an important parameter in determining the rate at which the isotope surface concentration builds-up. Fig. 2b illustrates a concrete case related to experiment A.
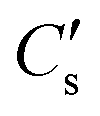 as a function of (a) the dimensionless quantity h′, (b) time according to a specific case where k = 3.0 × 10−10 cm s−1 and D = 1.0 × 10−10 cm2 s−1.
as a function of (a) the dimensionless quantity h′, (b) time according to a specific case where k = 3.0 × 10−10 cm s−1 and D = 1.0 × 10−10 cm2 s−1.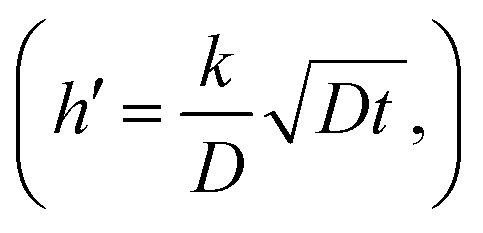 for each experiment is only dependent upon the apparent value of k in each case. If we consider now the conditions at the end of the diffusion exchanges, then we can obtain the surface isotopic fractions and the apparent exchange coefficients for the two exchange experiments by analysis of the isotopic depth profiles.
for each experiment is only dependent upon the apparent value of k in each case. If we consider now the conditions at the end of the diffusion exchanges, then we can obtain the surface isotopic fractions and the apparent exchange coefficients for the two exchange experiments by analysis of the isotopic depth profiles.