
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TD-DFT simulations of K-edge resonant inelastic X-ray scattering within the restricted subspace approximation†
Vinícius
Vaz da Cruz
 *a,
Sebastian
Eckert
a and
Alexander
Föhlisch
ab
*a,
Sebastian
Eckert
a and
Alexander
Föhlisch
ab
aHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Institute for Methods and Instrumentation for Synchrotron Radiation Research, 12489 Berlin, Germany. E-mail: vinicius.vaz_da_cruz@helmholtz-berlin.de
bUniversität Potsdam, Institut für Physik und Astronomie, 14476 Potsdam, Germany
First published on 19th October 2020
Abstract
A scheme for simulations of resonant inelastic X-ray scattering (RIXS) cross-sections within time-dependent density functional theory (TD-DFT) applying the restricted subspace approximation (RSA) is presented. Therein both occupied core and valence Kohn–Sham orbitals are included in the donor-space, while the accepting virtual orbital space in the linear response TD-DFT equations is restricted to efficiently compute both the valence- and core-excited states of the many electron system. This yields a consistent description of all states contributing to the RIXS scattering process within a single calculation. The introduced orbital truncation allows to automatize the method and facilitates RIXS simulations for systems considerably larger than ones accessible with wave-function based methods. Using the nitrogen K-edge RIXS spectra of 2-thiopyridone and its deprotonated anion as a showcase, the method is benchmarked for different exchange–correlation functionals, the impact of the RSA is evaluated, and the effects of explicit solvation are discussed. Improvements compared to simulations in the frozen orbital approximation are also assessed. The general applicability of the framework is further tested by comparison to experimental data from the literature. The use of TD-DFT core-excited states to the calculation of vibrationally resolved RIXS spectra is also investigated by combining potential energy scans along relevant coordinates with wave packet simulations.
Introduction
Resonant Inelastic X-ray Scattering1 (RIXS) is a widely used experimental probe for investigations of the local electronic structure in complex systems. RIXS is chemically appealing for its ability to probe the decay from the occupied orbitals, reaching valence excited states, often not accessible in the UV range. Its inherent dependence on the photon polarization also allows for examinations of the symmetry of occupied orbitals. Furthermore, since the spectral resolution is not limited by the large core-hole lifetime broadening, it can deliver information on the nuclear motion and associated potential energy surfaces through a vibrational substructure of electronic transitions. Recent studies ranging from isolated molecules,2–5 molecules in solution,6–9 fluctuating networks in liquid phase10 and to correlated materials11,12 demonstrate its versatility. RIXS also draws substantial interest for its applicability in pump–probe spectroscopies,13–17 specially in the context of modern X-ray Free Electron Laser experiments.Extracting chemical and physical insight from such intricate spectroscopies, however, often relies on an extensive computational effort in terms of electronic structure and spectral simulations. To that end, several theoretical approaches for simulating X-ray spectra have been developed, their capabilities and applications have been recently reviewed,18 and only a schematic overview is given here. Pertaining to wave function based methods, RIXS spectra have been computed via the restricted active space second order perturbation theory14,19,20 (RASPT2), multi-reference configurational interaction,21 and the complex polarization propagator approach implemented for both an ADC22 as well as an CCSD reference,23 to list a few examples. As for Kohn–Sham based methods, RIXS simulations have largely relied on the one-electron picture based on orbitals optimized via the transition-potential method24 or by the maximum-overlap-method (MOM).25 Also, the DFT based restricted open-shell configuration interaction singles (ROCIS) method has been developed for treating X-ray spectra of transition metal systems.26,27 In contrast, the application of standard linear-response time-dependent DFT (LR-TD-DFT) to RIXS has been largely unexplored and remains limited to a few isolated instances.28
In this work we investigate the applicability of LR-TD-DFT within the Restricted Subspace Approximation29 (RSA) to K-edge RIXS spectroscopy. A consistent description of both core- and valence-excited states is achieved by a pragmatic truncation of the orbital spaces, thus enabling computations of RIXS cross-sections in a near black-box fashion. Moreover, the efficiency of TD-DFT allows to tackle systems larger than those feasible with wave function based methods, while leading to a better description of valence excited states than one-electron DFT approaches.
We also demonstrate that although TD-DFT notoriously leads to very poor excitation energies for excited states in the X-ray region, often deviating by 10–20 eV, RIXS spectra of near quantitative accuracy can be obtained on the chemically relevant energy loss scale. More specifically, as long as TD-DFT produces a qualitatively correct intermediate core-state the quality of the RIXS spectrum can be expected to be comparable to that of the typical UV-Vis absorption spectrum. This stems from the fact that the final states in RIXS are valence excited states, which TD-DFT can often describe with reasonable accuracy.30
In the following sections, we briefly recollect the general theory of RIXS and RSA-TD-DFT, followed by a detailed exploration of the framework based on the RIXS spectrum of 2-thiopyridone and its conjugated base. Then a longer series of molecules is investigated where multiple experimental instances from the literature are used as reference. Furthermore, the applicability of TD-DFT to the simulation of high-resolution RIXS data is addressed by wave packet simulations of the spectra of H2O, O2 and liquid acetone. Lastly, the concluding remarks are presented.
General theory of resonant inelastic X-ray scattering
Resonant inelastic X-ray scattering, also called X-ray Raman spectroscopy, is a coherent scattering process where the system is excited by a X-ray photon into a short-lived core-excited state which continuously decays onto the manifold of valence excited states, as well as the ground state.1 A diagram is shown in Fig. 1 with a depiction of the process. Atomic units are used for all equations.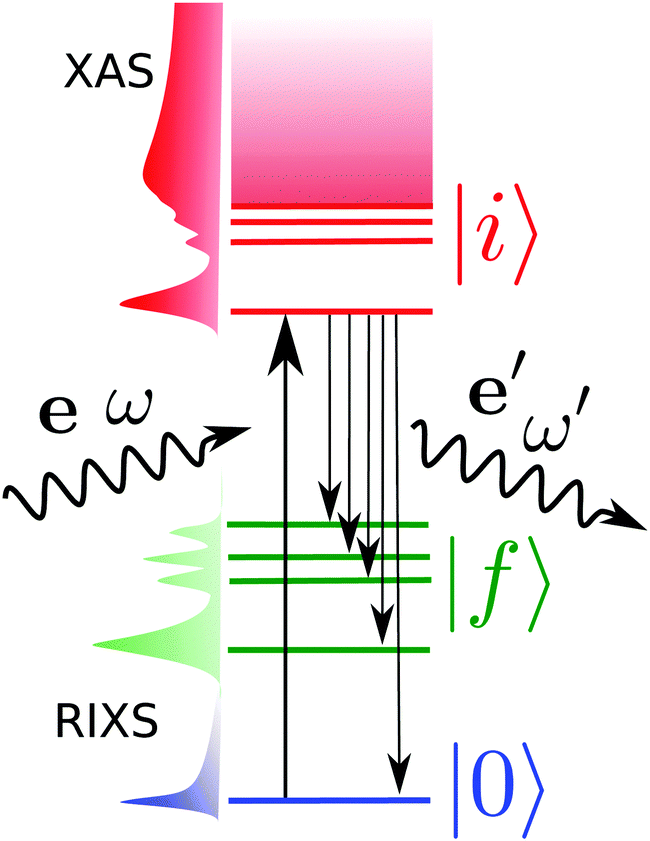 | ||
| Fig. 1 Diagram of the RIXS process showing the involved states. ω and ω′ are the incoming and outgoing photon frequencies, respectively. Pictorial representations of the cross-sections for the X-ray absorption spectrum (XAS) and RIXS are vertically shown. | ||
The general equation for the double differential RIXS cross-section reads1
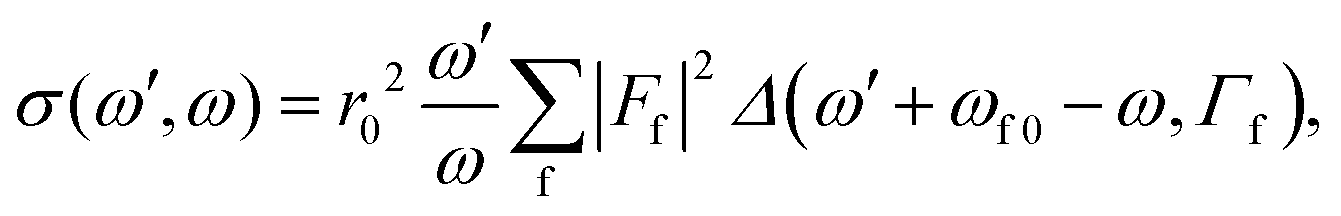 | (1) |
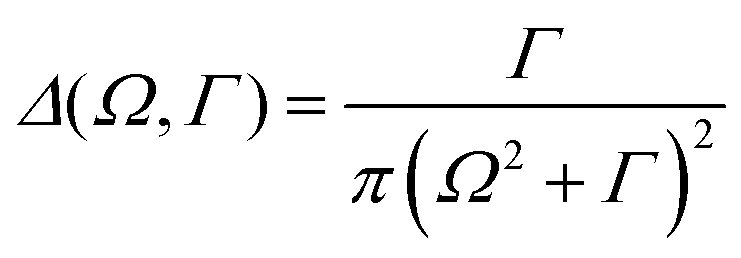 | (2) |
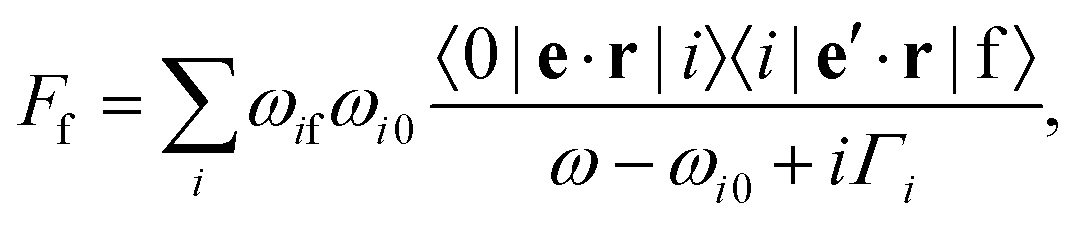 | (3) |
| σ(ω′,ω) ∝ |Ff|2 ∝ eα′eβ′eγ′eδ × σ(f)αβγδ, | (4) |
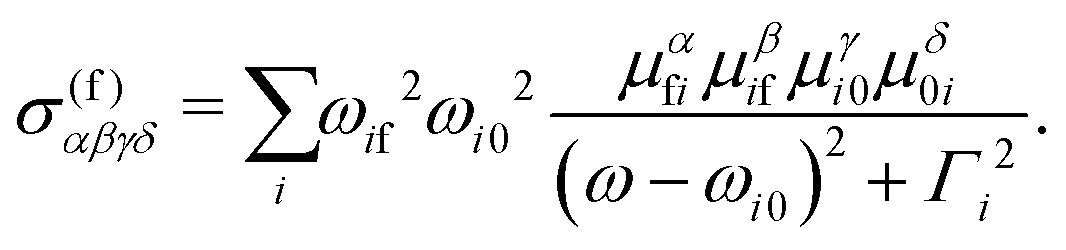 | (5) |
If we had a molecule fixed in space this would give the RIXS cross-section for any arbitrary relative angle between the transition dipole moments and the photons. However, in routine experiments we have fixed measuring angles and polarizations, along with randomly oriented molecules. Therefore, we need to average over orientations.
Using now the general RIXS average equation for a fixed scattering angle1 we may write
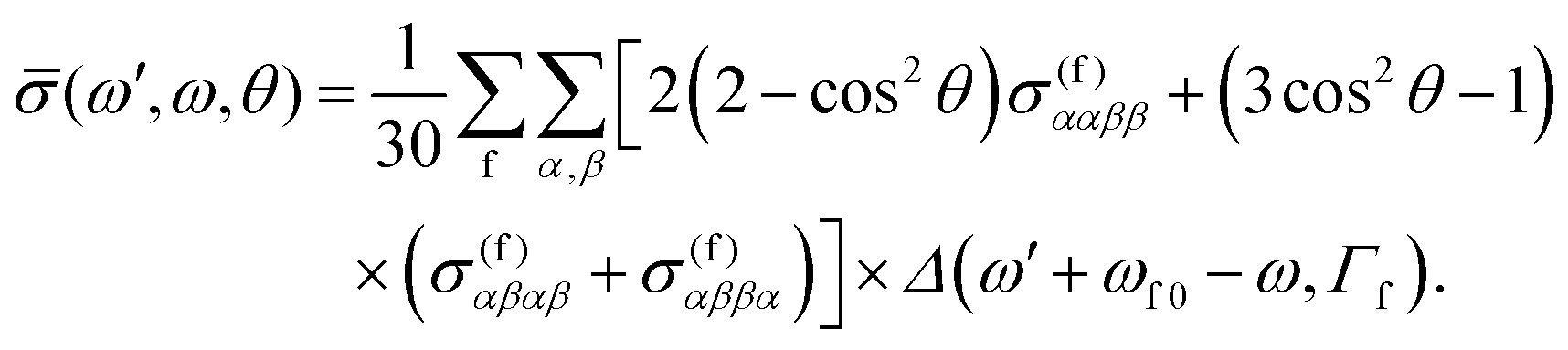 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ by
θ by 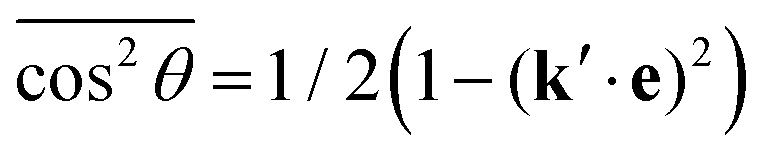 , defining cosχ = k′·e, where χ is the angle between the incoming polarization and the detection direction. This yields the final expression
, defining cosχ = k′·e, where χ is the angle between the incoming polarization and the detection direction. This yields the final expression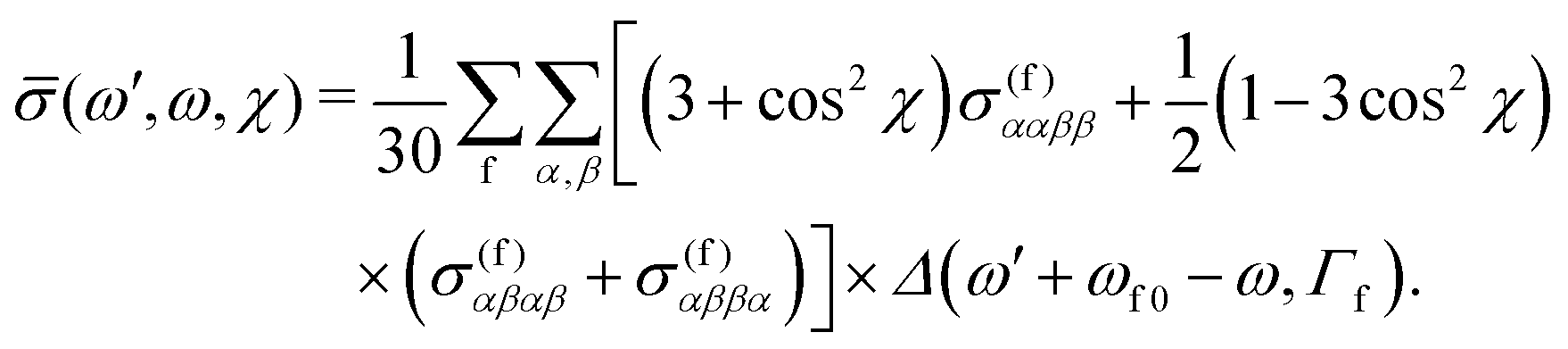 | (7) |
Subspace-restricted linear response TD-DFT
Time-dependent density functional theory is the most widely used theory to compute the excited states of molecular systems. In practice, the excited states are obtained by solving the linear response equations, based on a single Slater determinant constructed from the ground state Kohn–Sham orbitals in the presence of an infinitesimally small oscillatory electromagnetic perturbation with frequency ω. These equations, called the Casida equations, can be compactly written in matrix form as30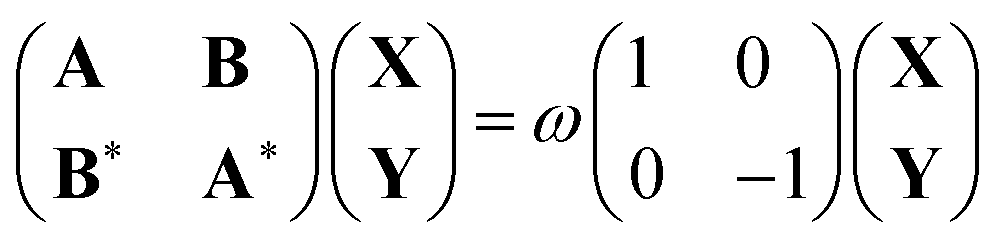 | (8) |
| Aia,jb = δijδab(εa − εi) + (ia|jb) − cHF(ij|ab) + (1 − cHF)(ia|fxc|jb) | (9) |
| Bia,jb = (ia|bj) − cHF(ib|aj) + (1 − cHF)(ia|fxc|bj) | (10) |
In order to reach the core-excited states, typically, one projects eqn (8) onto an orbital subspace,33–35 this procedure is also referred to as the core-valence separation (CVS) approximation,18,36 this approach entails the restriction of the indices i, j in eqn (9) to include only the core-hole orbitals, while the indices a, b run over the full set of virtual orbitals. The main motivation for this constraint is reducing the required number of roots that are needed to reach the core-excited states. This approach has been very successful in the description of the X-ray absorption spectra (XAS) of many systems,18 correctly capturing the bound core-excited states and the lowest resonances embedded in the continuum. It should be noted, however, that due to the self-interaction error and core-hole relaxation effects,34,37–40 the excitation energies provided by TD-DFT often deviate from the experiment in the order of tens of electronvolts.
Naturally, the approach outlined above does not yield the necessary final states for the simulation of RIXS spectra. To that end, one needs to compute the manifold of valence excited states as well. Therefore, we propose a different subspace restriction scheme, namely instead of restricting the donor orbital subspace, we shall constrain the acceptor orbital space in the response equations. This is done bearing in mind the goal of achieving a computationally-feasible number of roots for the description of both core- and valence-excited states. Such a seemingly drastic approximation can be motivated by the observation that the resonant scattering contribution to the total emission spectrum is only dominant for the lowest-lying core-excited states. As the excitation energy is tuned beyond the core-ionization threshold, the scattering cross-section is overtaken by the non-resonant emission process. Furthermore, restricting the virtual space in TD-DFT has been proposed early on as a means for tackling states for adsorbed molecules as well as molecules in solution,41–43 and it has been shown to also be a suitable route to obtaining excited states energies,29 geometries44 and vibrational frequencies45 of large systems. Virtual space restriction has also been successfully used in RIXS simulations at the L-edge of transition metal complexes within the multi-configurational Kohn–Sham ROCIS method.27
Hence, by solving the response equations with restricted donor and acceptor orbital spaces, one can obtain a full set of orthogonal valence- and core-excited states suitable for the calculation of the transition dipole moments necessary for specifying the Kramers–Heisenberg amplitudes in eqn (5). Fig. 2 shows a schematic of the orbital spaces used, where the donor space is made up by the core orbitals and the relevant occupied orbitals, and the acceptor space contains the lower lying unoccupied orbitals. To describe the orbital spaces, we define the array with the number of orbitals in the subspaces (Nc,No,Nu), where c, o, u denote core, occupied and unoccupied.
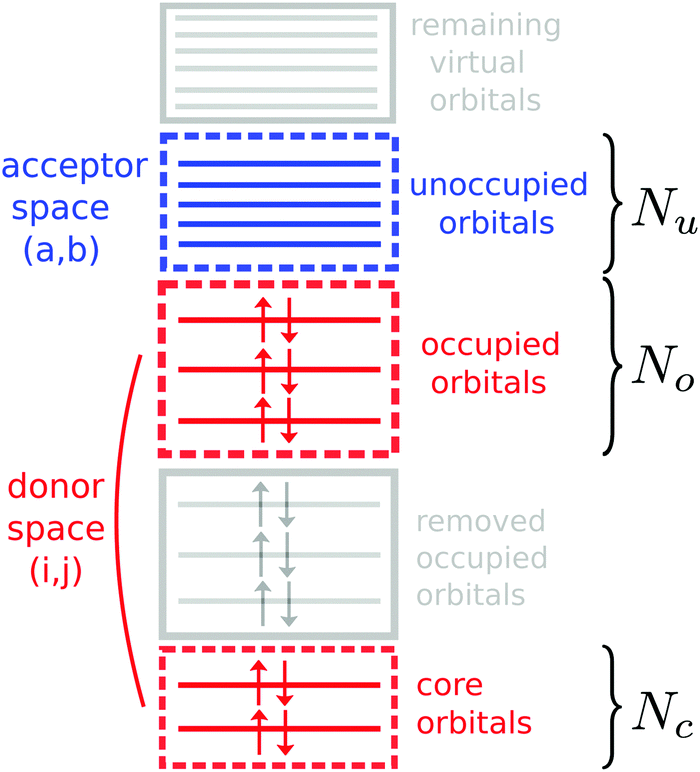 | ||
| Fig. 2 Diagram of the constrained orbital spaces used in the TD-DFT based RIXS simulations. | ||
Computational details
The proposed framework was tested by interfacing the Orca46 quantum chemistry package and the Multiwfn47 analysis suite. In this set-up, Orca was used for the solution of the LR-TD-DFT equations, while Multiwfn was employed to compute the necessary transition dipole moments from the Orca output. Manipulation of the orbital spaces was carried out using the native orbital window and the orbital rotation features of Orca. All calculations employed the RIJCOSX48 approximation with a fine integration grid GridX7, which greatly reduces the computational cost of the calculations. Geometries were optimized using the PBEh-3c49 compound method. The TD-DFT simulations were performed using the def2-TZVP(-f) basis set using the Coulomb fitting auxiliary basis def2/J. The PBE050 exchange–correlation functional on the GRID5 option was used, if not specified differently. Additional details and sample input files are shown in the ESI.† The spectra were computed using the core-hole lifetime broadenings (HWHM) for nitrogen Γ = 0.06 eV, oxygen Γ = 0.08 eV and carbon Γ = 0.05 eV. The lineshape of the spectra were modeled by a Gaussian function with HWHM of 0.8 eV for best comparison with the experimental data.Proof of principle: the RIXS of 2-thiopyridone
Nitrogen K-edge RIXS of the heterocyclic compound 2-thiopyridone (2-TP) will serve as the principal test case for the TD-DFT based method to simulate RIXS spectra presented here. Experimental data for scattering through the nitrogen 1s → π* resonance for different nitrogen protonation states in aqueous environments are available.14 Additionally, simulations of the nitrogen K-edge RIXS cross-sections on a RASPT2 level of theory were performed in previous studies. Hence, 2-TP allows to investigate the applicability of our method and will let us determine the impact of subspace restriction and choice of exchange–correlation functional on the results of the simulations. In addition we will be able to compare our results to the simulations performed on the RASPT2 level of theory. Furthermore, the impact of solute–solvent interactions on the computed RIXS cross-sections will be discussed.General applicability of the method
The results of our spectrum simulations for scattering through the N 1s → π* resonance for 2-TP in its different protonation states are presented in Fig. 3. The spectrum of the protonated 2-TP species contains spectral lines which have been attributed to RIXS final states characterised by a hole in molecular orbitals located dominantly on the pyridine ring. In contrast the deprotonated species 2-TP− exhibits an additional intense RIXS channel at 5 eV energy loss resulting from transitions between a nitrogen based lone pair orbital into the nitrogen 1s hole. All transitions are well captured in the TD-DFT based simulation of the spectra and the matching of transition intensities is improved compared to the RASPT2 simulations especially for the protonated 2-TP species. This improvement originates from the inclusion of more orbitals in the donor space, as well as from the explicit inclusion of solvent water molecules in the proximity of the 2-TP molecule (see next paragraph). Additionally, the transition energies between the ground and the valence excited states are very well described by the TD-DFT simulation. This results in accurate peak positions for the RIXS transitions on an energy loss scale, so they do not require an energy shift of the simulated RIXS spectra, which is required for other computational methods.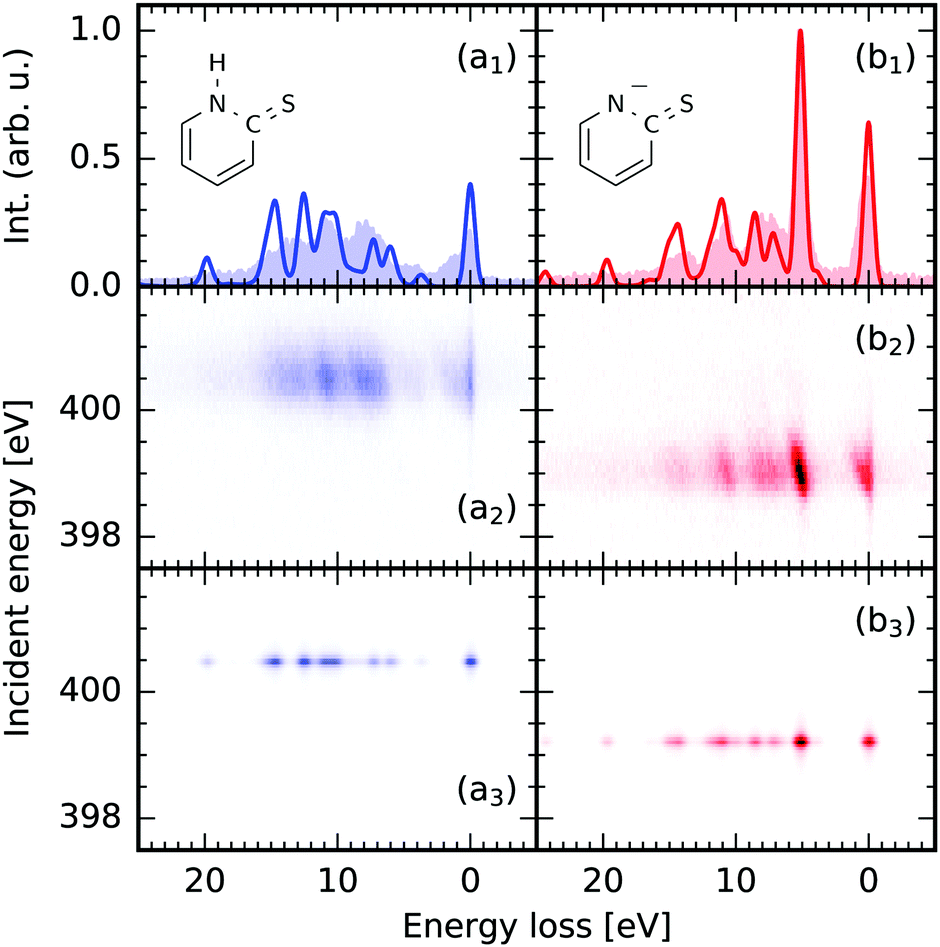 | ||
| Fig. 3 Comparison between the theoretical nitrogen 1s → π* RIXS simulations of 2-thiopyridone (2-TP, a) and the anion 2-TP− (b) and experimental data from ref. 14. (1) on resonance RIXS spectra extracted from the experimental (2) and theoretical (3) maps. The simulations use the (1,30,20) space and include three water molecules in the proximity of the molecules. | ||
Explicit solvation
For small to medium sized organic molecules, both geometry optimizations, as well as the TD-DFT simulations within the proposed orbital restriction scheme can be performed efficiently, enabling the impact of solute–solvent interactions on the RIXS transitions to be studied through the inclusion of explicit solvent molecules in the TD-DFT simulation. Both the occupied, as well as the unoccupied solvent valence orbitals are included in the donor and the acceptor space. In Fig. 4, we present a comparison for RIXS simulations of 2-TP and 2-TP− considering solvation implicitly through a polarizable continuum model (CPCM) and additionally with explicit solvation through three water molecules allowing for hydrogen bond formation at the nitrogen and the sulfur site, in agreement with molecular dynamics simulations by Norell et al.51 (see ESI†). We see that the inclusion of the solvent improves the agreement of the spectrum 2-TP− with the experimental data mainly for the strong lone-pair resonance at 5 eV energy loss. The slight energy underestimation in the exclusive CPCM simulation is compensated through the inclusion of hydrogen bonding solute–solvent interaction in the explicit solvation model. The energetically higher lying transitions are only mildly affected, as they correspond to transitions between π-orbitals in the pyridine ring and the nitrogen 1s core hole. Depending on the solvent, this approach leads to a drastic increase the number of roots necessary to account for all core to valence as well as valence to valence transitions. In our case, the number of roots increased from 360 to 600 to reach the core states for the spaces (1,18,20) and (1,30,20) for the exclusive CPCM and the explicit solvation model. Each of the explicitly considered solvent molecules thus increases the number of roots to be computed by k × Nu, where k is the number of occupied valence orbitals per solvent molecule.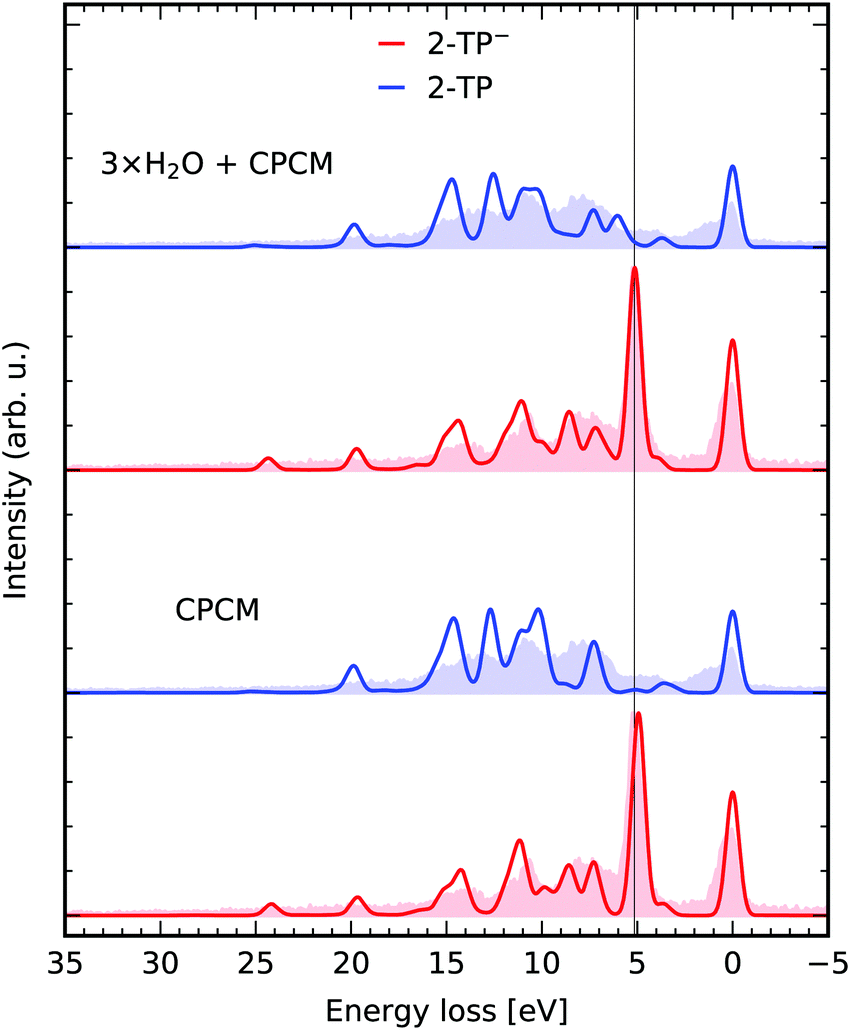 | ||
| Fig. 4 Explicit solvation. RIXS simulations for 2-TP and 2-TP− with a polarizable continuum model and explicit solvation with three water molecules. The theoretical spectra are compared to the experimental data from ref. 14. | ||
Functional dependence
Results obtained with TD-DFT are expected to display a dependence on the exchange–correlation functional employed. Here we briefly address this question where it pertains to the quality of the RIXS spectrum of our showcase systems, however a full benchmark is beyond the scope of the present article. In Fig. 5 we compare the simulated RIXS spectra for a group of selected functionals, namely the pure functional PBE, the global-hybrids PBE0,50 B3LYP, M06 and M06-2X,52 the range-corrected CAM-B3LYP and the range- and dispersion- corrected ωB97X-V.53 The peak positions for the most intense transition of 2-TP− are listed in Table 1 to enable a quantitative comparison. It can be seen that HF exchange is crucial for a correct description of the involved states, with PBE yielding the worst peak positions and comparatively worse intensities. Peak positions are also slightly worse for the two range-corrected functional considered. Overall, the agreement for the global-hybrids for the system considered is near-quantitative, but it can be argued to be marginally in favor of PBE0.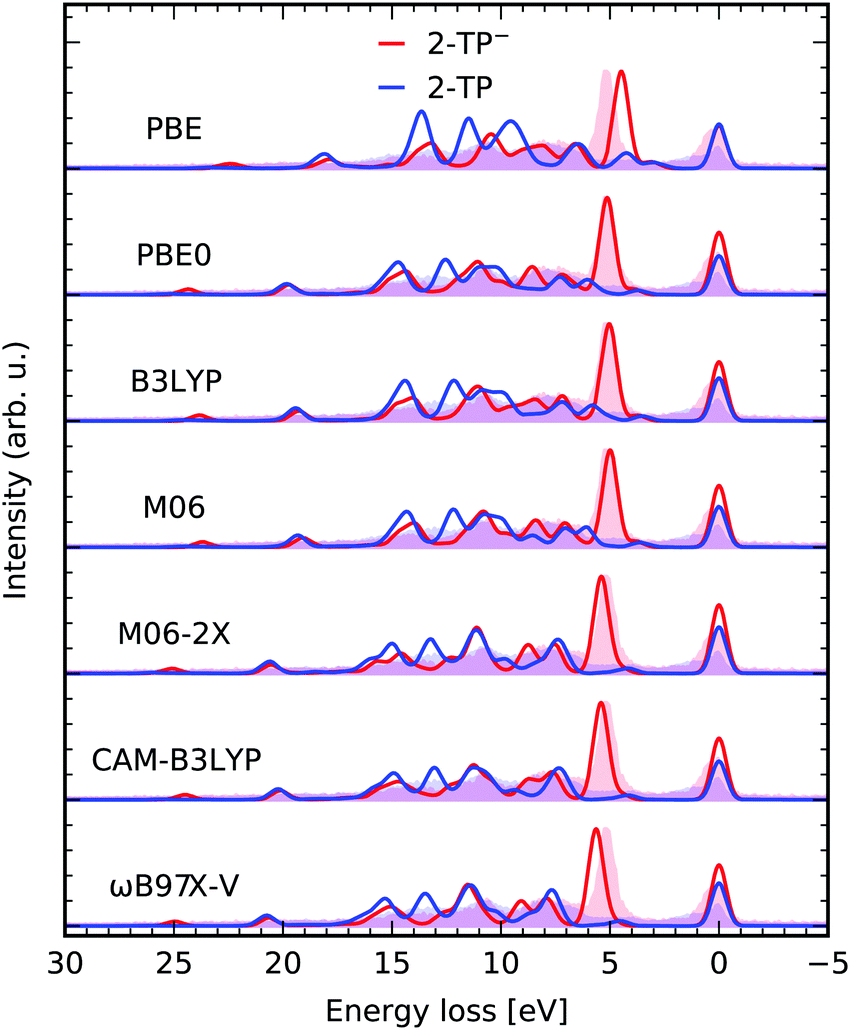 | ||
| Fig. 5 Comparison between different functionals used in the presented TD-DFT RIXS scheme in a constrained orbital space (1,30,20) with one N 1s core, 30 occupied valence and 20 virtual orbitals for the molecules 2-TP and 2-TP−. The theoretical spectra are compared to the experimental data from ref. 14. | ||
| xc-funct. | ε f − ε0 [eV] | xc-funct. | ε f − ε0 [eV] | xc-funct. | ε f − ε0 [eV] |
|---|---|---|---|---|---|
| PBE | 4.483 | M06-2X | 5.404 | PBE0 (CPCM) | 4.928 |
| PBE0 | 5.128 | CAM-B3LYP | 5.416 | ||
| B3LYP | 5.030 | ωB97X-V | 5.646 | PBE0 (frozen) | 6.809 |
| M06 | 4.988 | PBE (frozen) | 4.410 | ||
| Exp. | 5.150 | ||||
Impact of constrained orbital spaces on spectral intensities
Fig. 6a depicts the effect of our orbital constraint scheme on the description of the X-ray induced transitions. We compare the spectra of different (1,30,Nu) spaces to the one of the (1,0,−1) space. The latter contains the nitrogen 1s level as the only donating and all virtual orbitals as accepting orbitals, which represents the conventional way to simulate X-ray absorption spectra with TD-DFT employing the CVS. The extent of the absorption spectra in Fig. 5a is strongly dependent on the size of the acceptor space. The low energy transitions, here the nitrogen 1s → π* transitions, as well as the transitions at the onset of the continuum absorption up to 406 eV remain nearly unaffected even under strong orbital restriction down to Nu ≥ 20. RIXS through energetically higher lying transitions is usually superimposed with the non-resonant X-ray emission spectrum formed by valence to core transitions in the core-ionized molecule. Therefore, we focus here on simulations for energetically lower lying, well isolated core-excited states. Excluding the lower lying unoccupied valence orbitals might even be considered to model scattering through higher lying core-excited states. If the acceptor space is reduced even further to (1,30,5), only the nitrogen 1s → π* is described accurately.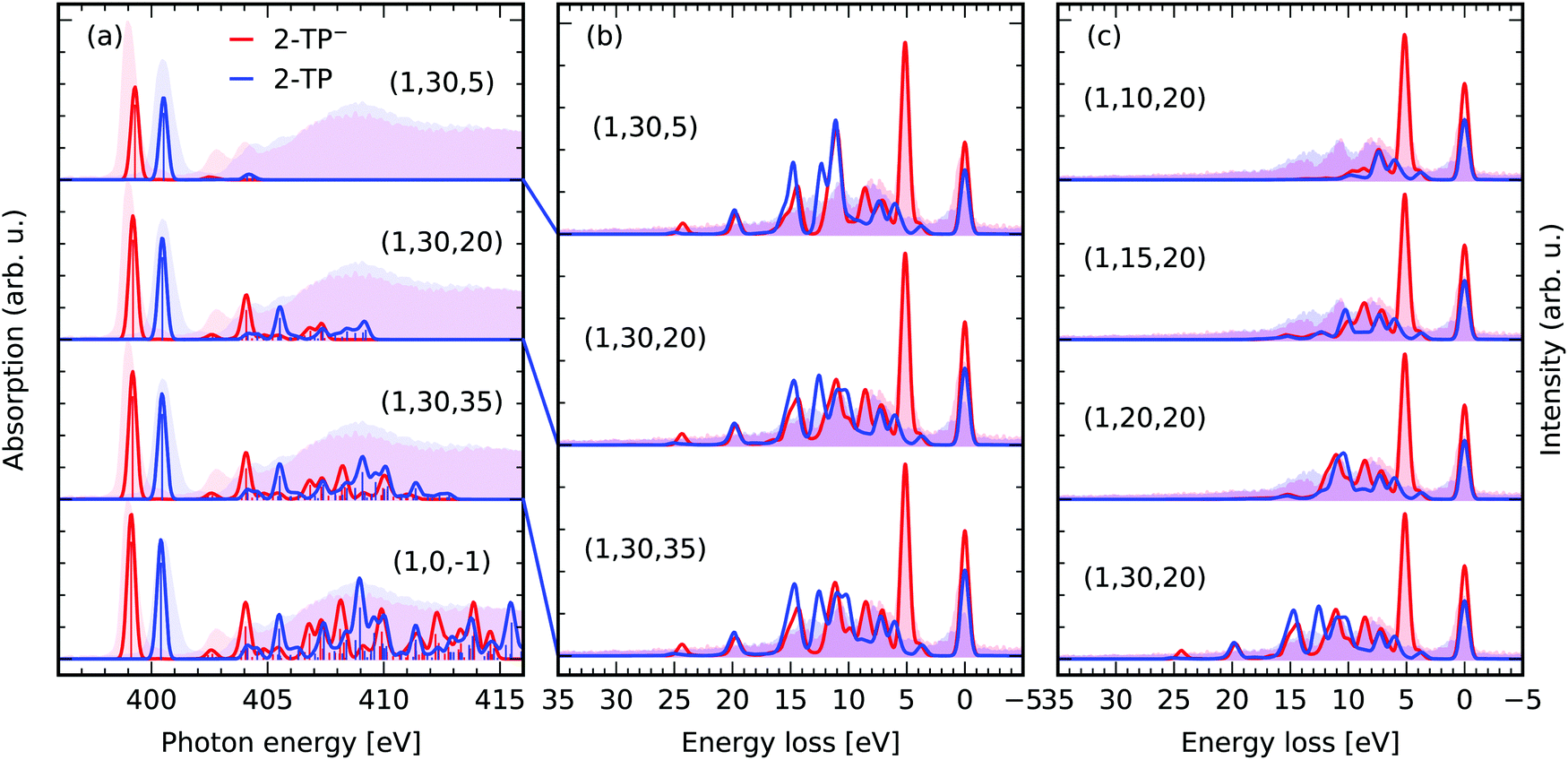 | ||
| Fig. 6 Impact of the orbital restriction on the simulated X-ray spectra in comparison the experimental data for 2-TP from ref. 14 and 54. (a) Comparison between the conventional TD-DFT XAS scheme (1,0,−1) including one N 1s core-orbital and all virtual orbitals to generate the roots and the proposed constrained orbital space (1,No,Nu) including in contrast No occupied valence and Nu virtual orbitals. The theoretical spectra were shifted by 11.25 eV to overlap the π*-resonances of 2-TP with experimental partial fluorescence yield XAS spectra. (b) RIXS simulations for the spaces with a restricted number of unoccupied acceptor orbitals Nu discussed in (a). (c) Impact of restrictions of the number of donating occupied orbitals No in the RIXS simulations. | ||
Let us focus on the RIXS simulations for excitation at the π*-resonances of the two systems in the different spaces in Fig. 6b. We compare the spectra for the largest space (1,30,35) to the ones of the reduced spaces (1,30,20) and (1,30,5). The impact of the space reduction on the RIXS spectrum via the lowest resonance is not as pronounced as the one on the X-ray absorption spectrum. This is because the core-excited state filters the accessible final states in the RIXS process. One can thus reduce the number of roots being computed, by simply contracting the subspace Nu to the core-excitations of interest. In the case of the investigated π*-resonance, states characterised by excitations into the LUMO orbital have the dominant contribution to the RIXS spectrum. As a result, even the (1,30,5) space yields reasonable qualitative agreement between the experimental and the theoretical RIXS spectra. The agreement is only qualitative, as the description of both the core and valence excited states is deteriorated by the restriction of the acceptor space. This lack of multi-configurational description of the states is not present for the (1,30,20) and (1,30,35) space. Additionally, the inclusion of higher lying core-excited state resonances has a negligible impact on the shape of the spectra for the two spaces. The amplitude for scattering through these states in the Kramers–Heisenberg formula (eqn (3)) for incident photon energies tuned to the π*-resonance is minimal.
Restricting the subspace of occupied valence orbitals, as illustrated in Fig. 6c, generally deteriorates the quality of the RIXS spectrum. Transitions between deep lying occupied valence orbitals and the lowest unoccupied molecular orbital are not considered as final states of the RIXS process. Therefore, the RIXS intensity at high energy losses is not modeled correctly. The authors recommend to include all occupied valence orbitals in the active space and rather reduce the acceptor space in computationally expensive cases, e.g. if heavy elements are present in the studied system or a large number of solvent molecules are treated explicitly. In general the number of roots needed to reach the core-excited states is given by the product No × Nv. Table 2 lists the number of roots for the approximations presented in Fig. 6.
| N c | N o | N v | # of roots |
|---|---|---|---|
| — | — | — | 10452 |
| 1 | 30 | 5 | 150 |
| 1 | 30 | 20 | 600 |
| 1 | 30 | 35 | 1050 |
| 1 | 10 | 20 | 200 |
| 1 | 15 | 20 | 300 |
| 1 | 20 | 20 | 400 |
| 1 | 30 | 20 | 600 |
Improvements regarding frozen orbital based RIXS schemes
In the proposed approach both the energy and the character of the valence and core-excited states is affected by the contribution of different excited state determinants to the roots resulting from the TD-DFT treatment of the photo-induced transitions. To estimate the effect of the accurate treatment of the excited states, we compare the results of our simulation to ones performed within the frozen orbital approximation.In the frozen orbital approximation, the photo-absorption and emission processes are expected to affect exclusively the orbitals changing occupation during the transitions, fully neglecting relaxation effects in the core-excited state. In this approximation the transition dipole matrix elements between the many-electron states in eqn (3) contract to the moments between the involved orbitals. This scheme to approximate the RIXS cross-section can be employed using Kohn–Sham orbitals from a ground state DFT simulation of the electronic structure of a system.
In Fig. 7 the RIXS simulations in the two frameworks are compared for scattering through the N 1s → π* state of 2-TP and 2-TP−. The frozen orbital scheme considers decay from all occupied valence orbitals, as well as from the π*-orbital, populated in the excitation step of the RIXS process. It yields qualitative agreement between the detected transitions for both considered functionals PBE and PBE0. Mainly the transition energies are not modeled correctly, i.e. the transition energies are underestimated for the PBE functional, which can also be seen from the values given in Table 1.
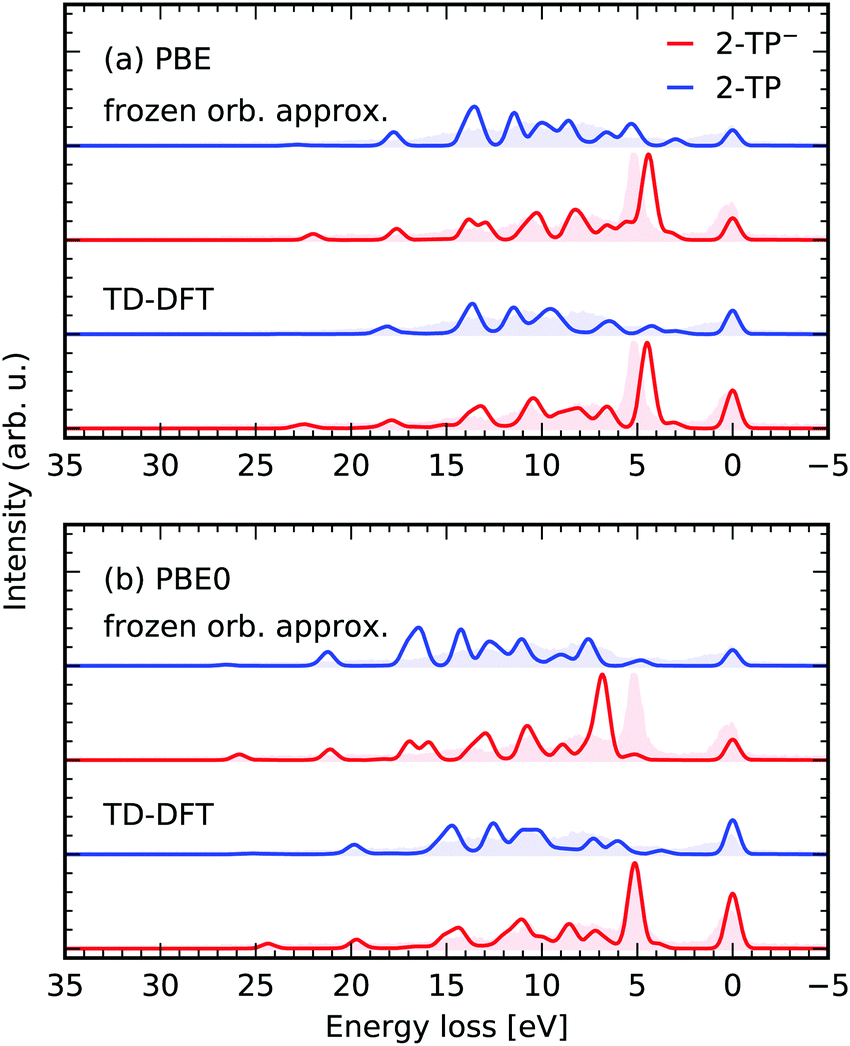 | ||
| Fig. 7 Performance of the presented TD-DFT RIXS scheme using the (1,30,20) space and in comparison to a RIXS scheme in the frozen orbital approximation for the functionals PBE (a) and PBE0 (b). The spectra are simulated for scattering through N 1s → π* state of 2-TP and 2-TP−. The experimental data is taken from ref. 14. | ||
The transfer from single electron excitations to excited states with mixed excitation character in the TD-DFT framework only mildly affects the spectra for the PBE functional. Naturally, the use of the hybrid PBE0 functional in the frozen orbital approximation yields an overestimation of the transition energies as in this case the orbital energies are no longer a good approximation for the excited state energies. In contrast, the RSA-TD-DFT RIXS simulations for the PBE0 functional yield an accurate description of both transition energies, as well as peak intensities.
Application to electronic structure analysis at soft X-ray K-edges
To present additional cases, in which our simulation scheme can be used to investigate molecular electronic structure, we present results of the RIXS simulations for different molecules in the gas and liquid phase, as well as in aqueous solutions in Fig. 8. The experimental spectra for the different systems were extracted from the figures in the publications specified in the figure-caption, if not declared differently below. Exclusively, RIXS spectra for dominant excitation at the energetically lowest lying core excited state are investigated. The experimental spectra are shifted by the incident energy stated in the different studies to compare to the simulated spectra on an energy loss scale. The polarization of the exciting radiation with respect to the detection direction is accounted for through the angle χ defined in eqn (7). The RSA-TD-DFT calculations were set up for each molecule including the relevant core-orbitals along with all occupied valence orbitals in the donor space, while the 20 lowest virtual orbitals were included in the acceptor space.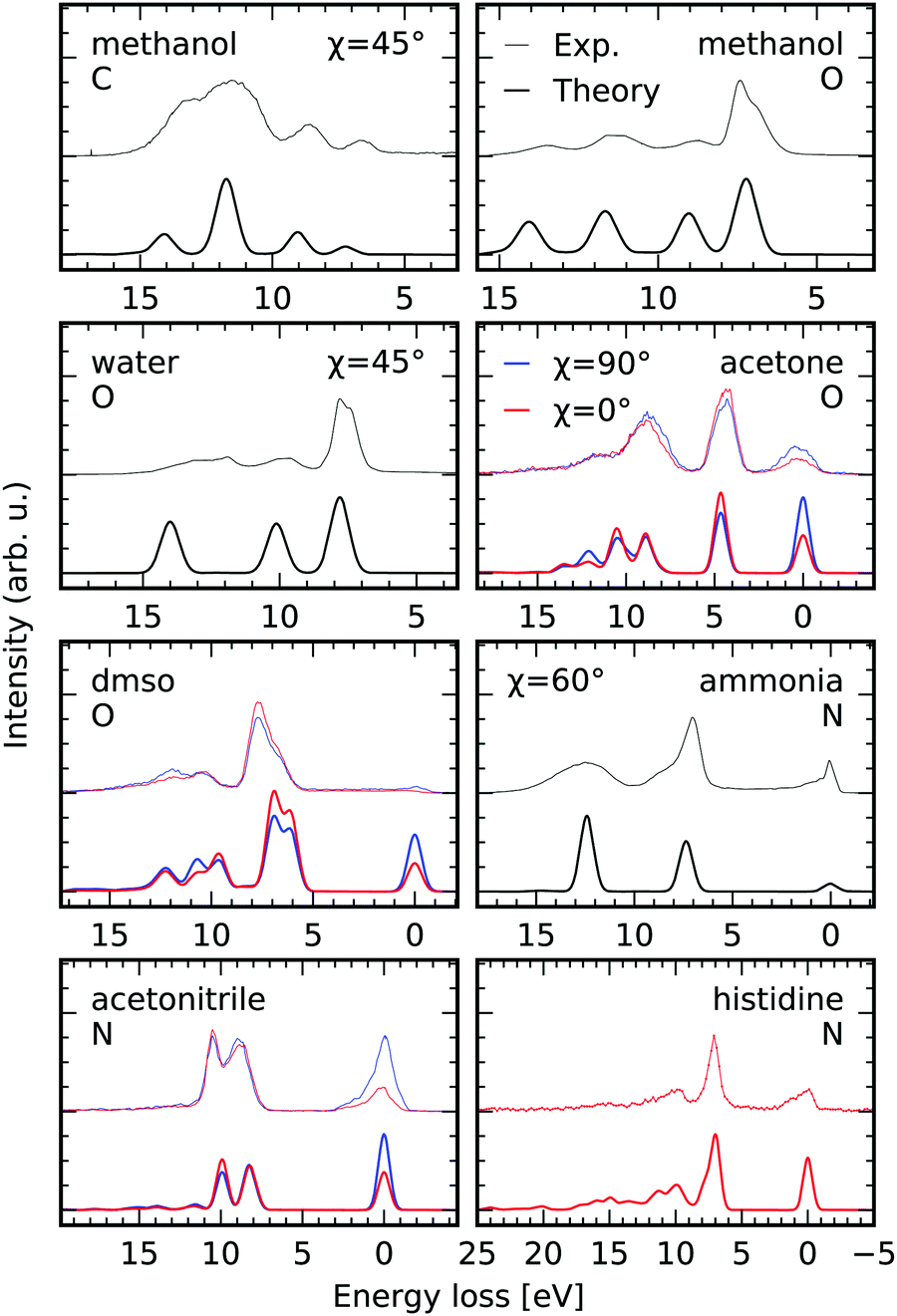 | ||
| Fig. 8 Comparison between experimental K-edge RIXS of gas phase methanol55 and water,56 liquid acetone, dimethylsulfoxide and acetonitrile,57 as well as aqueous ammonia58 and neutral histidine (lowest π* core excited state)6 and the results of the RSA-TD-DFT scheme to simulate electronic RIXS transitions. The experimental spectra are offset with respect to the simulations. The polarization of the incident X-ray radiation is considered through the angle χ defined in eqn (7). The edges i.e. carbon, oxygen and nitrogen are abbreviated by their initials. | ||
We consider scattering through core-excited states characterised by the 8a′ orbital of methanol in the gas phase, both at the carbon and the oxygen K-edge based on the experimental data by Benkert et al.55 The excellent agreement of both peak intensities and positions between the simulated and the experimental RIXS spectra allows for clear assignment of the detected transitions. As expected, the profile of the transition at an energy loss of 7 eV in the oxygen K-edge RIXS spectrum is not modeled correctly in our static RIXS model. As Benkert et al. concluded from their comparison to deuterated methanol, the RIXS transition is strongly affected by the dissociation process the |2a′′−18a′1〉 state. In a subsequent study by Vaz da Cruz et al. it could be shown that the splitting of the aforementioned transition is caused by a substantial spectral contribution of decay processes in the dissociated fragments, conserving the kinetic energy upon dissociation.59 Based on a fully quantum-dynamical treatment of the dissociation processes the spectral contribution of the so called molecular band on the low energy loss side of the peak, corresponding to scattering in the intact molecule, could be separated from the quasi-atomic peak at the high energy loss side, which results from scattering to the electronic ground state of the dissociated fragments.
Similar agreement is obtained for gas-phase water in the comparison between the experimental spectra upon scattering through the |1sO−14a11〉 state by Weinhardt et al.,56 who provided the experimental spectrum, and the theoretical simulations. Again the only qualitative agreement between the experimental and theoretical spectra results form the static treatment of the scattering process. In the comparison to deuterated water by Weinhardt et al.56 and the following ultra high-resolution RIXS investigations,2,19,60,61 it could be shown that ultrafast dissociation has a strong impact on RIXS intensity profiles at the |1sO−14a11〉 resonance and causes the same substructure of the lowest detected transition as in methanol through the contribution of the quasi-atomic peak.
For acetone, dimethylsulfoxide (DMSO) and acetonitrile presented by Dierker et al.57 RIXS spectra are available for horizontal and vertical polarization, allowing us to evaluate the capability of simulating the polarization anisotropy according to eqn (7). The simulations consider solvation implicitly through a polarizable continuum model for the different liquids. The spectral signatures, as well as the polarization anisotropy are modeled excellently. Differences in peak positions can probably be ascribed to the neglect of explicit solvation and core-excited state dynamics in the simulations.
For aqueous ammonia we achieve qualitative agreement in comparison to the data by Weinhardt et al.58 In their study both the peak shapes and relative peak intensities are strongly affected by deuteration of the system, reflecting the large impact of core-excited state dynamics on the spectrum. Also, hydrogen bonding effects can be expected to play an important role in the spectrum of ammonia, as already evidenced in the XAS,62 which further accounts for the discrepancy.
Finally, we compare our simulations for excitation at the π*-resonance of the deprotonated nitrogen site in the imidazole ring of aqueous histidine in neutral conditions. The spectrum is extracted from the data set presented by Eckert et al.6 The agreement between the experimental and the simulated spectrum is quantitative. Exclusively the vibrational progression at the elastic line at 0 eV energy loss is not modeled as it is the case for the systems discussed previously.
All presented cases reflect the need to consider explicit solvation and more importantly core-excited state dynamics to achieve quantitative agreement between the experimental spectra and the simulations. Ways to consider the dynamical aspect of the scattering process to model both the electronically elastic and inelastic transitions in the presented RSA-TD-DFT framework are discussed in the following section.
The role of core-excited nuclear dynamics
It is well established that RIXS signals are particularly sensitive to ultra-fast geometric distortions that can happen in the core-excited states,1 more strictly the RIXS profile is dependent on the difference between the potential energy surfaces between the states involved in the scattering process. Nuclear dynamics also often lead to violation of the electronic quadrupole selection rules in polyatomic systems.1,63,64 Furthermore, a growing effort is being devoted to the development of RIXS as a technique to study vibrational excitations and dynamics in molecules,2,59,65,66 liquids and solutions,10,67,68 as well as in solids.11,69 Therefore, it is important to discuss how nuclear dynamics effects can be incorporated into the framework discussed in the previous sections. This will be done by revisiting the high-resolution RIXS spectra of gas-phase H2O,2 molecular Oxygen66 and liquid acetone.67 The calculations will consider TD-DFT core-excited states computed with the PBE, PBE0 and M06-2X as a proxy systematic series on the increase of the fraction of HF exchange in the functional, from 0% to 25% and 56% respectively. Additionally, the equivalent core-hole approximation, henceforth called Z + 1, will be considered as a reference point, as it has been shown to be a robust and straightforward way of estimating core-hole induced relaxation.70,71 All vibrationally resolved spectra were computed using the time-dependent wave packet implementation of the Kramers–Heisenberg amplitudes as described by Vaz da Cruz et al.59Nuclear dynamics in gas-phase H2O
Water is a prime example of the effect of nuclear dynamics on X-ray spectra. The vibrationally resolved spectra of water have been carefully studied in previous investigations, specifically excitations to different intermediate states lead to an interesting mode selectivity,2,60 namely excitation from the O 1s orbital into the 4a1 valence orbital leads to an ultra-fast dissociation of the OH bond, while excitation to the 2b2 orbital drives the system along the symmetric stretching coordinate. Although mode-coupling is crucial for the correct description of the core-excited dynamics in water,2 it has been demonstrated by Eckert et al.61 that in water the RIXS cross-section can be described by an effective one-dimensional Hamiltonian along the driven mode. We will adopt this approximation here.In Fig. 9a the potential energy curve (PEC) along the OH bond of the water molecule, for the ground state (GS) and lowest core-excited state |O 1s−14a11〉 are shown for different functionals. A PEC computed within the Z + 1 approximation computed with the PBE0 functional and the high-quality RASPT2 potentials from ref. 2 and 19 are also shown. The curves were computed with a (1,4,20) orbital space using either a restricted or unrestricted Kohn–Sham ground state reference, denoted as RKS and UKS, respectively. The behaviour of the potential energy curves is qualitatively correct for the UKS calculations, up to the Coulson–Fisher (CF) point,72–74 beyond this point unphysical kinks are observed due to spin-contamination in the ground-state reference determinant.
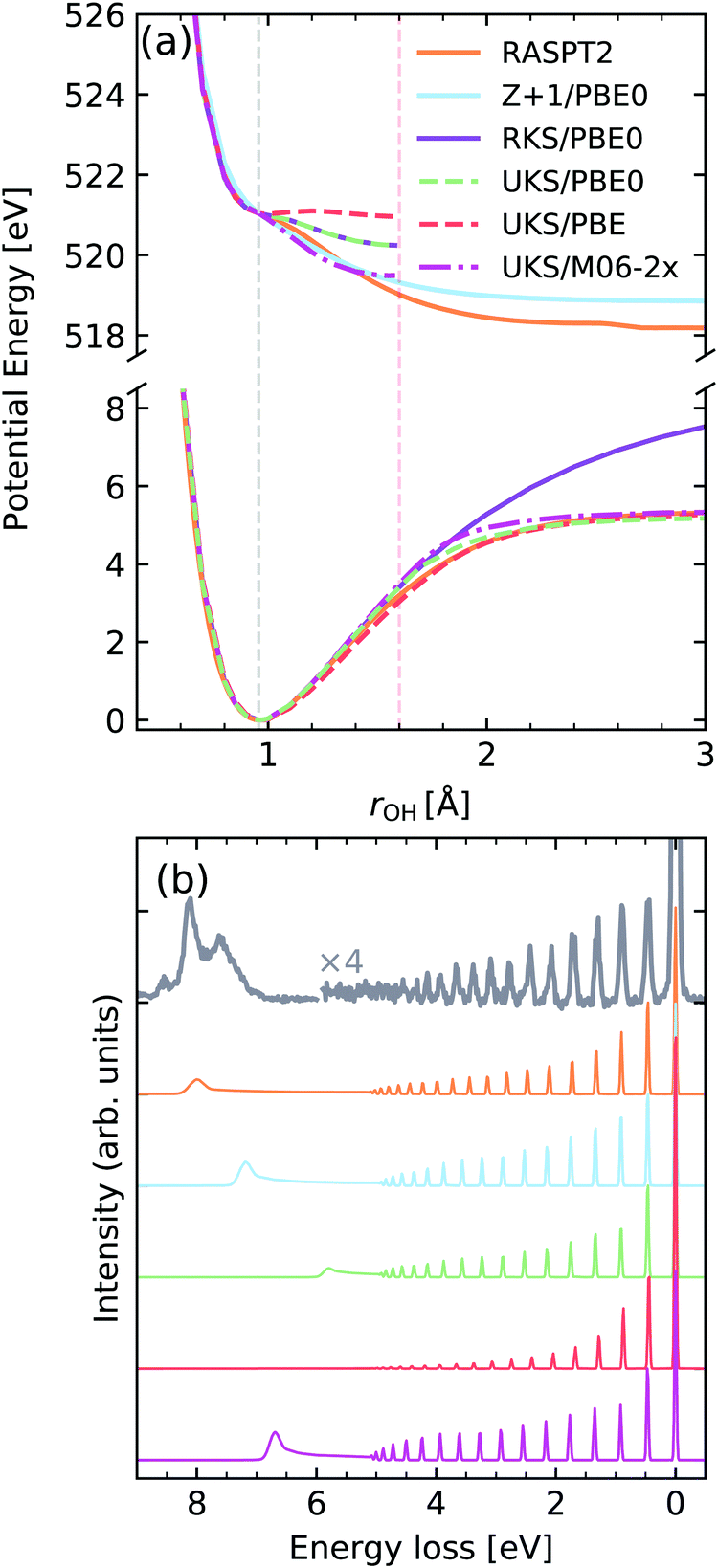 | ||
| Fig. 9 (a) Potential energy curves along the OH bond of water for the ground state (bottom) and |O 1s−14a11〉 core-excited state computed employing different functionals and methods. For comparability, the vertical transition point of all curves were matched to the one of the RKS/PBE0 PEC. The vertical transition point is marked by a dashed gray line, while the CF point is marked by a red dashed line. (b) Experimental vibrationally resolved RIXS spectrum of water2 along with the theoretical spectra computed with the respective potential curves displayed in panel (a). | ||
We observe a very strong sensitivity of the quality of the core-excited potential energy curve on the choice of functional, with the behaviour ranging from a quasi-bound state up to the correct dissociative behaviour. In this context, the fraction of Hartree–Fock exchange included in the functional seems to be the main attribute responsible for the observed changes. The Z + 1 approximation, on the other hand, leads to a more robust behaviour of the PEC, with the caveat that it leads to an incorrect dissociation limit, when compared to the RASPT2 reference, due to spurious interactions with the additional electron in the core shell.
In Fig. 9b The computed vibrationally resolved RIXS spectra are compared to high-resolution experimental data from ref. 2 and 61. Although all functionals lead to a qualitatively correct spectrum two main factors seem to be the most disparaging, namely the intensity distribution of the vibrational progression and the position of the atomic-peak. The intensity distribution of the vibrational peaks is determined by the evolution of the nuclear wave packet in the core-excited state, being very sensitive to the gradient near the vertical transition point. Hence we see that the shallower potential computed by PBE leads to much lower intensity for higher energy vibrational losses and an absent atomic peak. On the other hand the steeper curve computed with M06-2X shows much higher intensity near the >4 eV losses, being actually over-estimated with respect to the RASPT2 reference. The spectrum computed with the Z + 1 approximation shows a better intensity distribution, however, as mentioned has an incorrect atomic-peak2,19 position. The vibronic XAS spectra for each functional are shown in the ESI.†
We turn our attention now to the second core-excited state of water, namely the one arising from excitation of the O 1s electron into the 2b2 orbital, henceforth denoted by |O 1s−12b12〉. The computed potentials are shown in Fig. 10. In this case, we have a bound core-excited state which is driven along the symmetric stretching coordinate qs, again we notice a large sensitivity on the functional choice, with the core-excited state minimum displacement being heavily affected by the HF exchange fraction, with a higher fraction of HF exchange leading to a larger core-excited state displacement. In this figure the curve labelled as Z + 1 refers to the lowest TD-DFT root computed for the ground state of the FH2 equivalent core system. From the spectra shown in Fig. 10b we see that the resulting RIXS spectra are highly sensitive to the quality of the intermediate state potential. It should be noted that even though the core-excited PEC computed with M06-2X displays a significant shift, it leads to a qualitatively incorrect intensity distribution since the force constant of the potential is too high. This is because the RIXS spectrum is highly sensitive to lifetime vibrational interference (LVI) effects.1,75 The spectrum computed with the Z + 1 approximation in this case is still superior to the TD-DFT ones, however it is still too steep and shows a larger displacement than the RASPT2 reference. It should be added that for simplicity of comparison, the bending mode was neglected in the simulations, which accounts for the missing resonances seen between the stretching peaks in the experimental spectrum. The vibronic XAS spectra for each functional are shown in the ESI.†
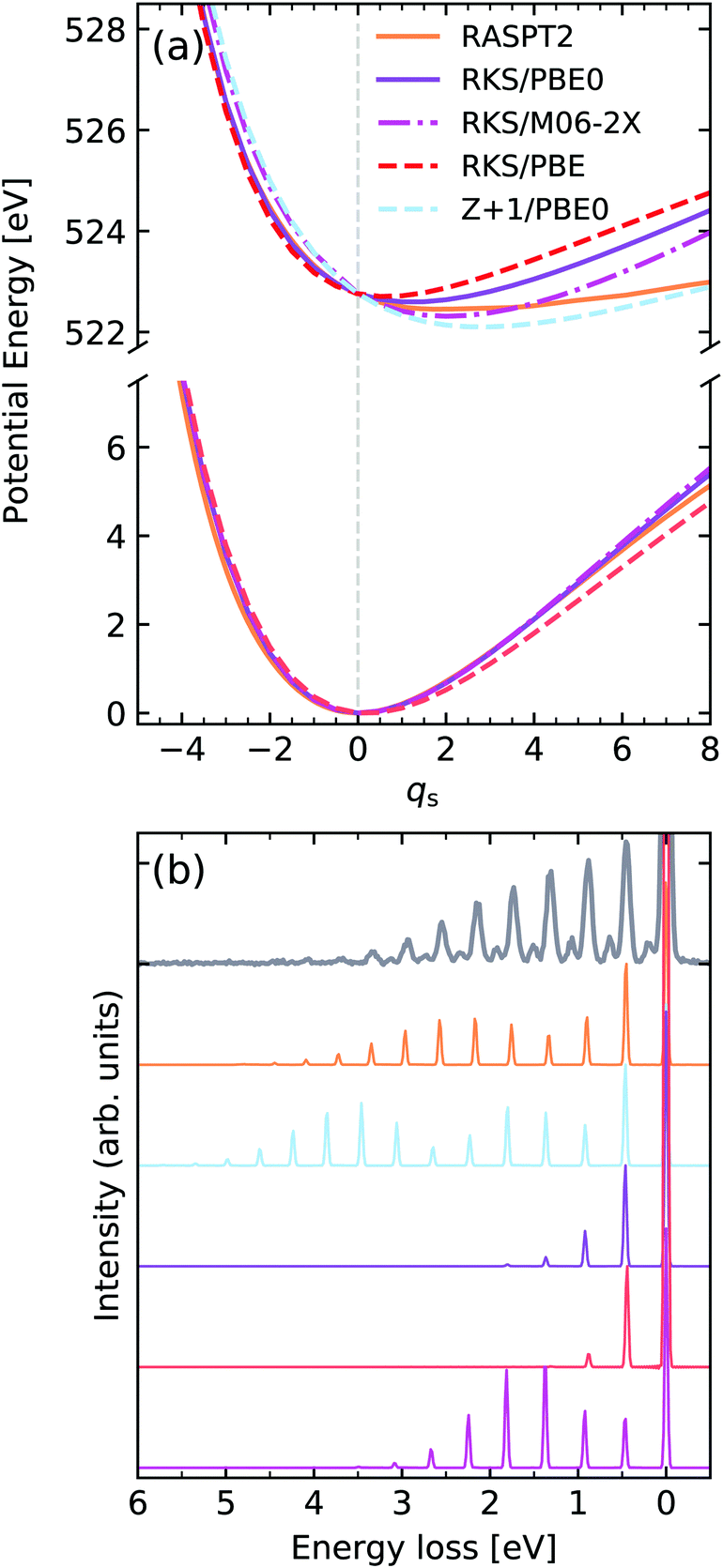 | ||
| Fig. 10 (a) Potential energy curves along the dimensionless symmetric stretching coordinate of water for the ground state (bottom) and |O 1s−12b12〉 core-excited state computed employing different functionals and methods. For comparability, the vertical transition point of all curves were matched to the one of the RKS/PBE0 PEC. The vertical transition point is marked by a dashed gray line. (b) Experimental vibrationally resolved RIXS spectrum of water2 along with the theoretical spectra computed with the respective potential curves displayed in panel (a). | ||
Molecular oxygen
We now focus on molecular oxygen, which was the first molecule to be measured with enough resolution to identify individual vibrational peaks in the RIXS spectrum.66Fig. 11a shows the potentials computed for the GS and the core-excited state arising from excitation from the 1σu to the half-filled 1πg orbital, denoted here by |1σu−11πg+1〉. We observe a reverse trend from the calculations for gas-phase water, where the pure functional PBE leads to the largest core-excited state displacement, while the M06-2X functional leads to the smallest. The Z + 1 leads to the most reasonable result in this case, showing a displacement very close to the one determined experimentally from the vibrationally resolved XAS.76 The behaviour of the PECs is clearly reflected in the RIXS spectrum. We see that all the TD-DFT potentials computed under-estimate the length of the vibrational progression. In contrast, the spectrum computed using the Z + 1 PEC leads to the most robust result, with respect to progression length and intensity distribution. The vibronic XAS spectra for each functional are shown in the ESI.†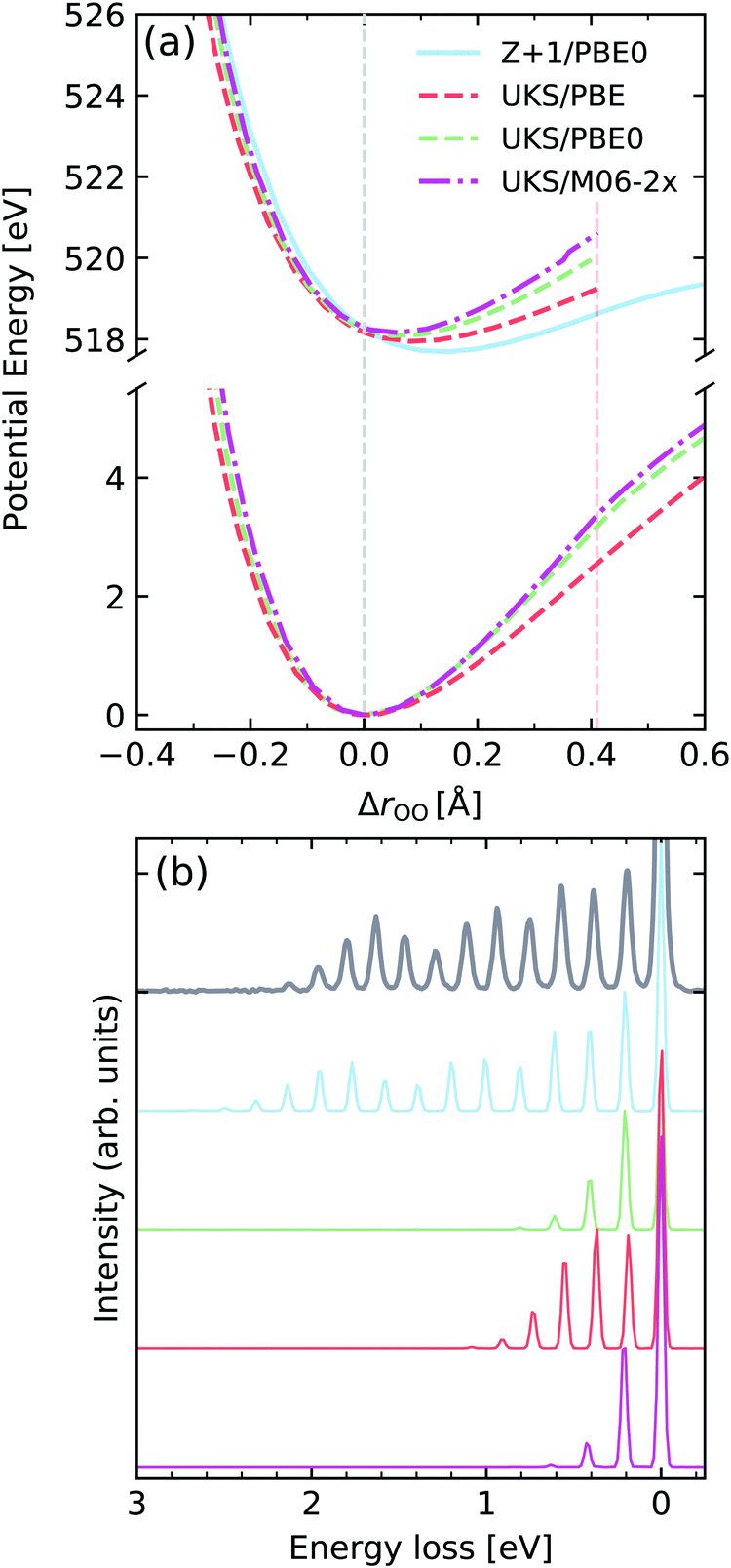 | ||
| Fig. 11 (a) Potential energy curves along change of internuclear distance in O2 for the ground state (bottom) and |1σu−11πg+1〉 core-excited state computed employing different functionals and methods. For comparability, the vertical transition point of all curves were matched to the one of the RKS/PBE0 PEC. The vertical transition point is marked by a dashed gray line. (b) Experimental vibrationally resolved RIXS spectrum of oxygen66 along with the theoretical spectra computed with the respective potential curves displayed in panel (a). | ||
Excitation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O stretching in acetone
O stretching in acetone
As a last example we examine the high-resolution RIXS spectrum of liquid acetone, which was first investigated by Sun et al.67 Here we will consider the decays to the ground state and to the first excited state. Fig. 12a shows the potentials computed for the GS, lowest valence excited state and the |O 1s−1π*1〉 core-excited state along the CO stretching coordinate qCO. In terms of the core-excited displacement along the CO stretching coordinate, we see a similar trend to O2, where the magnitude of the displacement decreases with the fraction of HF exchange in the functional, with PBE yielding the largest geometric distortion. Once again, the Z + 1/PBE0 approximation leads to a reasonable result, and is a significant improvement over the RKS/PBE0 curve. The valence excited state for the hybrid functionals PBE0 and M06-2X was found to be rather similar, in contrast to the PBE functional, which leads to a shallower PEC. Similar behaviour is seen for the GS PEC.
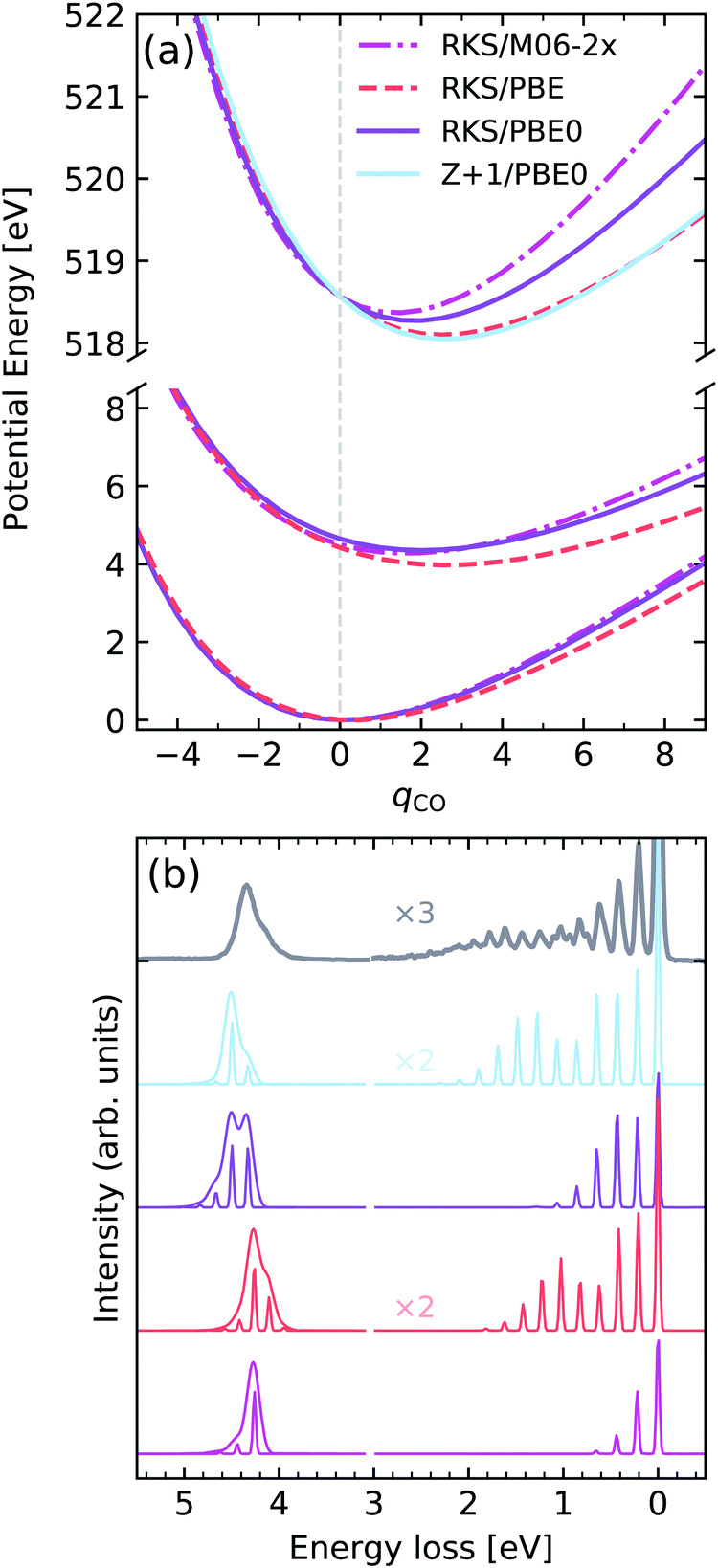 | ||
| Fig. 12 (a) Potential energy curves along the dimensionless CO stretching coordinate of acetone for the ground state (bottom) and |O 1s−1π*1〉 core-excited state computed employing different functionals and methods. For comparability, the vertical transition point of all curves were matched to the one of the RKS/PBE0 PEC. The vertical transition point is marked by a dashed gray line. (b) Experimental vibrationally resolved RIXS spectrum of acetone67 along with the theoretical spectra computed with the respective potential curves displayed in panel (a). | ||
Analysing the experimental and theoretical RIXS spectra in Fig. 12, we see that good agreement is reached for the spectra computed with Z + 1/PBE0 and PBE, both in terms of vibrational progression length and intensities, as well as the asymmetry of the lowest electronic band. Here it should be noted that all soft-modes were neglected in the calculations, however a 0.075 eV HWHM broadening was added to the inelastic band, as deduced in ref. 67. The calculations using PBE0 and M06-2X lead to unsatisfactory agreement, with progressions that are too short, as well as an incorrect profile asymmetry for the inelastic band. It is also noteworthy to mention that the inclusion of the vibrational motion greatly improves the peak ratios between elastic and inelastic bands, initially computed for the low-res spectrum in Fig. 8. The vibronic XAS spectra for each functional are shown in the ESI.†
Conclusions
We have explored the application of RSA-TD-DFT to the simulation of RIXS spectra at the K-edge of light elements. The quality of the obtained spectra is near quantitative on the energy loss scale, in spite of the notoriously large errors associated with TD-DFT X-ray absorption transition energies. The convergence of the RIXS spectrum with respect to the truncation used in the RSA is found to be rapid, as also observed for UV-vis transitions.29 Moreover, the RSA enables a consistent description of both core- and valence-excited states. This set of states can be used, in future applications, to simulate full RIXS planes and intermediate-state interference can be treated intrinsically.The simulations of purely electronic RIXS transitions in the GS molecular geometry, within the described framework, are computationally inexpensive. This allows the simulation of molecules of medium size including important effects, such as hydrogen bonding interactions and quantum nuclear dynamics, which are more challenging to tackle within wave function based theories. The RSA can be set up in an automatized way without the necessity of expert-designed orbital spaces. Furthermore, the low computational cost also enables investigations in solution phase where sampling over fluctuating solvated structures might be important.
From the analysis of the functional dependence of the results it was found that global-hybrids lead to very good peak positions, on the energy loss scale, and we found PBE0 to be reasonably applicable across a wide range of systems. The functional choice becomes more nuanced when it comes to studying nuclear dynamics and high-resolution RIXS spectra. The results seem to indicate that 1s excitations to unoccupied orbitals of σ character benefit from a higher fraction of Hartree–Fock exchange, while the opposite is observed for excitations into unoccupied π orbitals, which improve with a reduction of the amount of HF exchange. Nevertheless, the use of the Z + 1 approximation was found to be a more robust method of estimating core-induced nuclear dynamics at the DFT level, and can be efficiently combined with the transition dipole moments and final state potential energy surfaces computed at the RSA-TD-DFT level. In the present work we restricted the analysis to widely available functionals, but we expect that the application of short-range corrected functionals77 aimed specifically at X-ray spectroscopy could prove a worthwhile direction of investigation.
We envisage that the method described can become an additional tool in the fast-growing field of solution phase RIXS experiments. It can be easily implemented in most computational packages on the already existing TD-DFT routines, since it relies only on the ground state Kohn–Sham orbitals and a subspace restriction of the standard linear response equations. The low computational cost and ease of set-up allow non-expert users to simulate RIXS for both interpretative and predictive purposes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. F. acknowledges funding from the ERC-ADG-2014 Advanced Investigator grant no. 669531 EDAX under the Horizon 2020 EU Framework, Programme for Research and Innovation.References
- F. Gelmukhanov and H. Ågren, Phys. Rep., 1999, 244 Search PubMed.
- V. Vaz da Cruz, E. Ertan, R. C. Couto, S. Eckert, M. Fondell, M. Dantz, B. Kennedy, T. Schmitt, A. Pietzsch and F. F. Guimarães, et al. , Phys. Chem. Chem. Phys., 2017, 19, 19573, 10.1039/C7CP01215B.
- V. Vaz da Cruz, E. Ertan, N. Ignatova, R. C. Couto, S. Polyutov, M. Odelius, V. Kimberg and F. Gel'mukhanov, Phys. Rev. A, 2018, 98, 012507, DOI:10.1103/PhysRevA.98.012507.
- A. E. A. Fouda, L. C. Seitz, D. Hauschild, M. Blum, W. Yang, C. Heske, L. Weinhardt and N. A. Besley, J. Phys. Chem. Lett., 2020, 7476, DOI:10.1021/acs.jpclett.0c01981.
- V. Ekholm, G. S. Chiuzbian, C. Såthe, A. Nicolaou, M. Guarise, M. Simon, N. Jaouen, J. Lüning, C. F. Hague and F. Gel'mukhanov, et al. , J. Phys. B: At., Mol. Opt. Phys., 2020, 53, 185101 CrossRef CAS.
- S. Eckert, J. Niskanen, R. M. Jay, P. S. Miedema, M. Fondell, B. Kennedy, W. Quevedo, M. Iannuzzi and A. Föhlisch, Phys. Chem. Chem. Phys., 2017, 19, 32091, 10.1039/C7CP05713J.
- K. Kunnus, W. Zhang, M. G. Delcey, R. V. Pinjari, P. S. Miedema, S. Schreck, W. Quevedo, H. Schröder, A. Föhlisch and K. J. Gaffney, et al. , J. Phys. Chem. B, 2016, 120, 7182, DOI:10.1021/acs.jpcb.6b04751.
- R. M. Jay, S. Eckert, M. Fondell, P. S. Miedema, J. Norell, A. Pietzsch, W. Quevedo, J. Niskanen, K. Kunnus and A. Föhlisch, Phys. Chem. Chem. Phys., 2018, 20, 27745, 10.1039/C8CP04341H.
- L. Weinhardt, A. Benkert, F. Meyer, M. Blum, D. Hauschild, R. G. Wilks, M. Bär, W. Yang, M. Zharnikov and F. Reinert, et al. , Phys. Chem. Chem. Phys., 2019, 21, 13207, 10.1039/C9CP02481F.
- V. Vaz da Cruz, F. Gel'mukhanov, S. Eckert, M. Iannuzzi, E. Ertan, A. Pietzsch, R. C. Couto, J. Niskanen, M. Fondell and M. Dantz, et al. , Nat. Commun., 2019, 10, 1013 CrossRef.
- M. Rossi, R. Arpaia, R. Fumagalli, M. Moretti Sala, D. Betto, K. Kummer, G. M. De Luca, J. van den Brink, M. Salluzzo and N. B. Brookes, et al. , Phys. Rev. Lett., 2019, 123, 027001, DOI:10.1103/PhysRevLett.123.027001.
- A. S. M. Ismail, Y. Uemura, S. H. Park, S. Kwon, M. Kim, H. Elnaggar, F. Frati, Y. Niwa, H. Wadati and Y. Hirata, et al. , Phys. Chem. Chem. Phys., 2020, 22, 2685, 10.1039/C9CP03374B.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grübel, M. Scholz and D. Nordlund, et al. , Nature, 2015, 520, 78 CrossRef CAS.
- S. Eckert, J. Norell, P. S. Miedema, M. Beye, M. Fondell, W. Quevedo, B. Kennedy, M. Hantschmann, A. Pietzsch and B. E. Van Kuiken, et al. , Angew. Chem., Int. Ed., 2017, 56, 6088, DOI:10.1002/anie.201700239.
- N. Ignatova, V. Vaz da Cruz, R. C. Couto, E. Ertan, M. Odelius, H. Ågren, F. F. Guimarães, A. Zimin, S. P. Polyutov and F. Gel'mukhanov, et al. , Phys. Rev. A, 2017, 95, 042502, DOI:10.1103/PhysRevA.95.042502.
- R. M. Jay, J. Norell, S. Eckert, M. Hantschmann, M. Beye, B. Kennedy, W. Quevedo, W. F. Schlotter, G. L. Dakovski and M. P. Minitti, et al. , J. Phys. Chem. Lett., 2018, 9, 3538, DOI:10.1021/acs.jpclett.8b01429.
- L. Kjellsson, K. D. Nanda, J.-E. Rubensson, G. Doumy, S. H. Southworth, P. J. Ho, A. M. March, A. Al Haddad, Y. Kumagai and M.-F. Tu, et al. , Phys. Rev. Lett., 2020, 124, 236001, DOI:10.1103/PhysRevLett.124.236001.
- P. Norman and A. Dreuw, Chem. Rev., 2018, 118, 7208 CrossRef CAS.
- E. Ertan, V. Savchenko, N. Ignatova, V. Vaz da Cruz, R. C. Couto, S. Eckert, M. Fondell, M. Dantz, B. Kennedy and T. Schmitt, et al. , Phys. Chem. Chem. Phys., 2018, 20, 14384 RSC.
- J. Norell, R. M. Jay, M. Hantschmann, S. Eckert, M. Guo, K. J. Gaffney, P. Wernet, M. Lundberg, A. Föhlisch and M. Odelius, Phys. Chem. Chem. Phys., 2018, 20, 7243, 10.1039/C7CP08326B.
- D. Maganas, P. Kristiansen, L.-C. Duda, A. Knop-Gericke, S. DeBeer, R. Schlögl and F. Neese, J. Phys. Chem. C, 2014, 118, 20163, DOI:10.1021/jp505628y.
- D. R. Rehn, A. Dreuw and P. Norman, J. Chem. Theory Comput., 2017, 13, 5552, DOI:10.1021/acs.jctc.7b00636.
- R. Faber and S. Coriani, Phys. Chem. Chem. Phys., 2020, 22, 2642, 10.1039/C9CP03696B.
- L. Triguero, L. G. M. Pettersson and H. Ågren, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8097, DOI:10.1103/PhysRevB.58.8097.
- M. W. D. Hanson-Heine, M. W. George and N. A. Besley, J. Chem. Phys., 2017, 146, 094106 CrossRef.
- M. Roemelt, D. Maganas, S. DeBeer and F. Neese, J. Chem. Phys., 2013, 138, 204101, DOI:10.1063/1.4804607.
- D. Maganas, S. DeBeer and F. Neese, Inorg. Chem., 2017, 56, 11819, DOI:10.1021/acs.inorgchem.7b01810.
- Y. Zhang, J. D. Biggs, D. Healion, N. Govind and S. Mukamel, J. Chem. Phys., 2012, 137, 194306, DOI:10.1063/1.4766356.
- M. W. D. Hanson-Heine, M. W. George and N. A. Besley, Mol. Phys., 2018, 116, 1452 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291 CrossRef CAS.
- C. Davidson, J. Comput. Phys., 1975, 17, 87 CrossRef.
- M. Stener, G. Fronzoni and M. D. de Simone, Chem. Phys. Lett., 2003, 373, 115 CrossRef CAS.
- S. DeBeer George, T. Petrenko and F. Neese, J. Phys. Chem. A, 2008, 112, 12936 CrossRef CAS.
- N. A. Besley and F. A. Asmuruf, Phys. Chem. Chem. Phys., 2010, 12, 12024, 10.1039/C002207A.
- B. N. C. Tenorio, T. Moitra, M. A. C. Nascimento, A. B. Rocha and S. Coriani, J. Chem. Phys., 2019, 150, 224104 CrossRef.
- G. Tu, V. Carravetta, O. Vahtras and H. Ågren, J. Chem. Phys., 2007, 127, 174110 CrossRef.
- G. Tu, Z. Rinkevicius, O. Vahtras, H. Ågren, U. Ekström, P. Norman and V. Carravetta, 76 , Phys. Rev. A: At., Mol., Opt. Phys., 2007, 022506, DOI:10.1103/PhysRevA.76.022506.
- N. A. Besley, A. T. B. Gilbert and P. M. W. Gill, J. Chem. Phys., 2009, 130, 124308 CrossRef.
- D. Hait and M. Head-Gordon, J. Phys. Chem. Lett., 2020, 11, 775 CrossRef CAS.
- Y. Mochizuki, K. Tanaka, K. Ohno and H. Tatewaki, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 11907, DOI:10.1103/PhysRevB.39.11907.
- N. A. Besley, Chem. Phys. Lett., 2004, 390, 124 CrossRef CAS.
- N. A. Besley, M. T. Oakley, A. J. Cowan and J. D. Hirst, J. Am. Chem. Soc., 2004, 126, 13502, DOI:10.1021/ja047603l.
- D. Robinson, J. Chem. Theory Comput., 2014, 10, 5346, DOI:10.1021/ct500687j.
- M. W. D. Hanson-Heine, J. Phys. Chem. A, 2019, 123, 2949, DOI:10.1021/acs.jpca.8b09642.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73, DOI:10.1002/wcms.81.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 054107, DOI:10.1063/1.4927476.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158, DOI:10.1063/1.478522.
- J. Norell, S. Eckert, B. E. Van Kuiken, A. Föhlisch and M. Odelius, J. Chem. Phys., 2019, 151, 114117, DOI:10.1063/1.5109840.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215, DOI:10.1007/s00214-007-0310-x.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904 RSC.
- S. Eckert, P. Miedema, W. Quevedo, B. OCinneide, M. Fondell, M. Beye, A. Pietzsch, M. Ross, M. Khalil and A. Föhlisch, Chem. Phys. Lett., 2016, 647, 103 CrossRef CAS.
- A. Benkert, F. Meyer, D. Hauschild, M. Blum, W. Yang, R. G. Wilks, M. Bär, F. Reinert, C. Heske and L. Weinhardt, J. Phys. Chem. A, 2016, 120, 2260, DOI:10.1021/acs.jpca.6b02636.
- L. Weinhardt, A. Benkert, F. Meyer, M. Blum, R. G. Wilks, W. Yang, M. Bär, F. Reinert and C. Heske, J. Chem. Phys., 2012, 136, 144311, DOI:10.1063/1.3702644.
- B. Dierker, E. Suljoti, K. Atak, K. M. Lange, N. Engel, R. Golnak, M. Dantz, K. Hodeck, M. Khan and N. Kosugi, et al. , New J. Phys., 2013, 15, 093025, DOI:10.1088/1367-2630/15/9/093025.
- L. Weinhardt, M. Weigand, O. Fuchs, M. Bär, M. Blum, J. D. Denlinger, W. Yang, E. Umbach and C. Heske, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104202, DOI:10.1103/PhysRevB.84.104202.
- V. Vaz da Cruz, N. Ignatova, R. C. Couto, D. A. Fedotov, D. R. Rehn, V. Savchenko, P. Norman, H. Ågren, S. Polyutov and J. Niskanen, et al. , J. Chem. Phys., 2019, 150, 234301, DOI:10.1063/1.5092174.
- R. C. Couto, V. Vaz da Cruz, E. Ertan, S. Eckert, M. Fondell, M. Dantz, B. Kennedy, T. Schmitt, A. Pietzsch and F. F. Guimarães, et al. , Nat. Commun., 2017, 8, 12725 Search PubMed.
- S. Eckert, V. Vaz da Cruz, F. Gel’mukhanov, E. Ertan, N. Ignatova, S. Polyutov, R. C. Couto, M. Fondell, M. Dantz and B. Kennedy, et al. , Phys. Rev. A, 2018, 97, 053410, DOI:10.1103/PhysRevA.97.053410.
- M. Ekimova, W. Quevedo, L. Szyc, M. Iannuzzi, P. Wernet, M. Odelius and E. T. J. Nibbering, J. Am. Chem. Soc., 2017, 139, 12773, DOI:10.1021/jacs.7b07207.
- A. Cesar, F. Gelmukhanov, Y. Luo, H. Ågren, P. Skytt, P. Glans, J. Guo, K. Gunnelin and J. Nordgren, J. Chem. Phys., 1997, 106, 3439, DOI:10.1063/1.474111.
- P. Skytt, P. Glans, J.-H. Guo, K. Gunnelin, C. Såthe, J. Nordgren, F. K. Gel’mukhanov, A. Cesar and H. Ågren, Phys. Rev. Lett., 1996, 77, 5035, DOI:10.1103/PhysRevLett.77.5035.
- J. Söderström, R. Stefanuik, F. Hennies, T. Schmitt, V. N. Strocov, J. Andersson, B. Kennedy, J. Schlappa, A. Föhlisch and A. Pietzsch, et al. , Phys. Rev. A, 2020, 101, 062501, DOI:10.1103/PhysRevA.101.062501.
- F. Hennies, A. Pietzsch, M. Berglund, A. Föhlisch, T. Schmitt, V. Strocov, H. O. Karlsson, J. Andersson and J.-E. Rubensson, Phys. Rev. Lett., 2010, 104, 193002, DOI:10.1103/PhysRevLett.104.193002.
- Y.-P. Sun, F. Hennies, A. Pietzsch, B. Kennedy, T. Schmitt, V. N. Strocov, J. Andersson, M. Berglund, J.-E. Rubensson and K. Aidas, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 132202, DOI:10.1103/PhysRevB.84.132202.
- S. Schreck, A. Pietzsch, B. Kennedy, C. SÅthe, P. S. Miedema, S. Techert, V. N. Strocov, T. Schmitt, F. Hennies and J.-E. Rubensson, et al. , Sci. Rep., 2016, 7, 20054 CrossRef.
- A. Geondzhian and K. Gilmore, Phys. Rev. B: Condens. Matter Mater. Phys., 2020, 101, 214307, DOI:10.1103/PhysRevB.101.214307.
- O. Plashkevych, T. Privalov, H. Ågren, V. Carravetta and K. Ruud, Chem. Phys., 2000, 260, 11 CrossRef CAS.
- W. Hua, G. Tian and Y. Luo, Phys. Chem. Chem. Phys., 2020, 22, 20014–20026, 10.1039/D0CP02970J.
- C. A. Coulson and I. Fischer, London, Edinburgh Dublin Philos. Mag. J. Sci., 1949, 40, 386 CrossRef CAS.
- H. Myneni and M. E. Casida, Comput. Phys. Commun., 2017, 213, 72 CrossRef CAS.
- D. Hait, A. Rettig and M. Head-Gordon, Phys. Chem. Chem. Phys., 2019, 21, 21761, 10.1039/C9CP04452C.
- F. Gel'Mukhanov, L. Mazalov and A. Kondratenko, Chem. Phys. Lett., 1977, 46, 133 CrossRef.
- M. Coreno, M. de Simone, K. Prince, R. Richter, M. Vondráček, L. Avaldi and R. Camilloni, Chem. Phys. Lett., 1999, 306, 269 CrossRef CAS.
- N. A. Besley, M. J. G. Peach and D. J. Tozer, Phys. Chem. Chem. Phys., 2009, 11, 10350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04726k |
| This journal is © the Owner Societies 2021 |