
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic deflection of neutral sodium-doped ammonia clusters†
J. V.
Barnes
a,
M.
Beck
 a,
S.
Hartweg
a,
A.
Luski
b,
B. L.
Yoder
a,
J.
Narevicius
b,
E.
Narevicius
b and
R.
Signorell
*a
a,
S.
Hartweg
a,
A.
Luski
b,
B. L.
Yoder
a,
J.
Narevicius
b,
E.
Narevicius
b and
R.
Signorell
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Zürich, 8093, Switzerland. E-mail: rsignorell@ethz.ch
bDepartment of Chemical Physics, Weizmann Institute of Science, Rehovot, 76100, Israel
First published on 8th December 2020
Abstract
We describe the setup and the performance of a new pulsed Stern–Gerlach deflector and present results for small sodium-doped ammonia clusters Na(NH3)n (n = 1–4) in a molecular beam. NaNH3 shows the expected deflection of a spin ½ system, while all lager clusters show much smaller deflections. Experimental deflection ratios are compared with the values calculated from molecular dynamics simulations. The comparison reveals that intracluster spin relaxation in NaNH3 takes place on a time scale significantly longer than 200 μs. Assuming that intracluster relaxation is the cause of the reduced deflection, relaxation times seem to be on the order of 200 μs for all larger clusters Na(NH3)n (n = 2–4). Our work is a first attempt to understand the magnetic properties of isolated, weakly-bound clusters with relevance to the variety of diamagnetic and paramagnetic species expected in solvated electron systems.
1. Introduction
The discovery of concentration-dependent colours of alkali metal–ammonia solutions in the early 19th century by Sir Humphry Davy and later reported by W. Weyl1 sparked a large series of experimental and theoretical works on excess electrons in alkali metal ammonia solutions2 (and references therein). In dilute blue coloured solutions, a variety of diamagnetic and paramagnetic species may exist (solvated electrons, electron–cation pairs, solvated metal atoms and bipolarons).2–5 In bulk solutions the paramagnetic species have been investigated via electron spin resonance (ESR)6 and nuclear magnetic resonance (NMR) spectroscopy7 (and references therein). Neutral sodium doped ammonia clusters Na(NH3)n have previously served as model systems to study the electronic properties of the ‘ammoniated’ electron via photoion spectroscopy, photoelectron spectroscopy and ab initio methods8–20 (and references therein). Photoion spectroscopy has revealed that the ion appearance energy of Na(NH3)n decreases systematically with increasing number of solvent molecules15 (and references therein). Various effects such as increasing delocalization of the 3s electron with increasing cluster size, surface and bulk localization of the electron, closing of solvation shells and cluster symmetry can contribute to the decrease in the ion appearance energy and the ionization (binding) energy. From n = 1 to 4, for example, a substantial decrease of ∼1.4 eV was observed. A similar trend for the ionization (binding) energy was later found in photoelectron studies and in high-level ab initio calculations.17–19 Experiments and calculations for the smaller clusters are consistent with surface electrons (compared to internally solvated electrons), which essentially can be described as an unpaired electron of Na that is perturbed by the solvent molecules.17–19 These studies also reveal many structural isomers for a given cluster size. An intriguing result of the high-level ab initio study by Gunina and Krylov19 concerns the electronic properties. The study found that the hydrogen-bond network of the NH3 solvent molecules contributes significantly to the overall electronic properties, such as the dipole moment. Angle-resolved photoelectron spectroscopy provides experimental access to the photoelectron anisotropy (photoelectron anisotropy parameter β) and thus to the orbital character of the unpaired electron. Experimental and calculation results17,18 show that the lowest electronic states of small clusters have high s-character and thus large β values. The β values decrease with increasing cluster size, except for highly symmetric clusters (e.g. n = 4; magic number for the photoelectron anisotropy). Generally, the decrease in β is accompanied by an increase in the electric dipole moment. Effective polarizabilities of Na(NH3)n were determined from electric deflection studies.21 Yet, detailed knowledge of the magnetic properties of the solvated electron in free clusters is still not established. To the best of our knowledge, we present the first study on the magnetic properties of small (n = 1–4) sodium doped ammonia clusters Na(NH3)n investigated via Stern–Gerlach deflection.The original Stern–Gerlach experiment was designed to determine the magnetic moment of isolated silver atoms;22 later various other isolated atoms,23 several molecules with a nonzero spin S24–34 and metallic and bi-metallic clusters35–47 were investigated with Stern–Gerlach setups (except for ref. 46). For molecular systems and metal clusters, additional degrees of freedom like vibrations and rotations are accessible compared with atomic systems. It has been shown that the overall molecular rotations can induce Zeeman-like sublevels.27,28 Several groups48–50 gave theoretical explanations of why transitions between Zeeman-like sublevels can occur and how Stern–Gerlach deflection experiments could be influenced by so-called intramolecular spin relaxation (ISR) and its intracluster analog. Amirav and Navon provided experimental evidence of ISR effects in isolated molecules.25,26 In their deflection studies on the molecular beams of paramagnetic molecules and stable organic radicals, they observed smaller-than predicted deflection magnitudes and incomplete spin refocusing involving two Stern–Gerlach magnets. These two experimental results were interpreted as ISR processes occurring on faster time scales than the interaction time with the magnetic field. In one case, however, this interpretation was disputed.28 Later, various groups investigated the magnetic properties of free Fem, Com, and Nim clusters (m = 10–1000) via Stern–Gerlach deflection.36,38,41,44 Hereby, one-sided deflection toward high field instead of symmetric magnetic deflection was observed. This asymmetric deflection behaviour was interpreted as being due to rapid ISR49 occurring among the perturbed, spin-rotation induced Zeeman sublevels. Studies on one-dimensional metal–organic sandwich clusters51–54 show similar one-sided deflection in the high-field direction, indicating that intracluster spin relaxation occurs within the complexes as they traverse the magnetic field. In recent studies, Schäfer and co-workers investigated spin relaxation processes via spin refocusing of paramagnetic superatoms with two Stern–Gerlach magnets.55,56
Here, we present a new setup for a pulsed Stern–Gerlach experiment, characterize its performance and study the magnetic properties of neutral sodium doped ammonia clusters, which can be seen as solvated electron precursors. An overview of previous Stern–Gerlach setups is provided in ref. 57 along with a description of a new strong permanent magnet gradient deflector and its comparison with commonly employed electromagnets. Similar to the experiments of various groups highlighted above, we investigate the deflection magnitude in relation to a predicted (from a molecular dynamics (MD) approach) deflection magnitude. With this we aim to probe the cluster size dependent magnetic properties of neutral Na(NH3)n (n = 1–4) clusters. Studying the size dependent magnetic properties of sodium doped ammonia clusters will lead to a better understanding of the involved paramagnetic species in bulk sodium ammonia solutions. In the present study, we focus on the interpretation of cluster size dependent intracluster relaxation times.
2. Methods
2.1. Experimental setup
Fig. 1 shows the experimental setup for the current study of the size-dependent magnetic properties of neutral sodium doped ammonia clusters Na(NH3)n (n = 1–4). It consists of four chambers (A–D) which are separated by skimmers, in order to produce a well-defined molecular beam along with differential pumping (Fig. 1a). For the detection of ion kinetic energies, we use velocity map imaging (VMI).58 Mass spectra were produced by collecting time-of-flight (TOF) signals using the same ion optics as that in the case of VMI. | ||
| Fig. 1 (a) Sketch of the experimental setup consisting of the source chamber (A), the Na-oven chamber (B), the deflection chamber (C) and the ionization/detection chamber (D). See text for details. (b) Side view with dimensions along the molecular beam propagation axis. (c) Side view of the deflector consisting of three electromagnets with relevant dimensions and the axis system (right). Shaded areas correspond to epoxy parts. (d) Cross section of the deflector flight channel and calculated z-component of the magnetic field gradient. A detailed description of the mechanical and electronic design is given in Sections S1 and S2 of the ESI.† | ||
Upon ionization, the ions are accelerated by a Wiley-McLaren type extractor optimized for either TOF conditions or VMI conditions (0 to ±15 kV). The ionization region is located between the repeller and the extractor plate. The extraction region and a 20 cm field-free drift region are shielded by two concentric mu-metal cylinders. The position sensitive detector is composed of a pair of 40 mm diameter microchannel plates (MCPs) in a chevron stack which are coupled to a phosphor screen (Photonis USA, Inc.; APD), followed by a charge coupled device (CCD) camera. The front of the MCP assembly is kept at ground potential when the voltage on the back plate is used to ‘gate’ the detector. A high voltage switch (Behlke) is used to vary the MCP bias voltage by 500 V so that the gain is only high when the ions of interest are impinging on the detector. The luminescence of the screen is recorded by the CCD camera. Individual frames (12 bit grayscale image, 1/3 inch sensor, 1024 × 768 pixel) are taken for each laser shot, summed up in real time using NuAcq software63 and saved to a computer connected to the CCD camera.
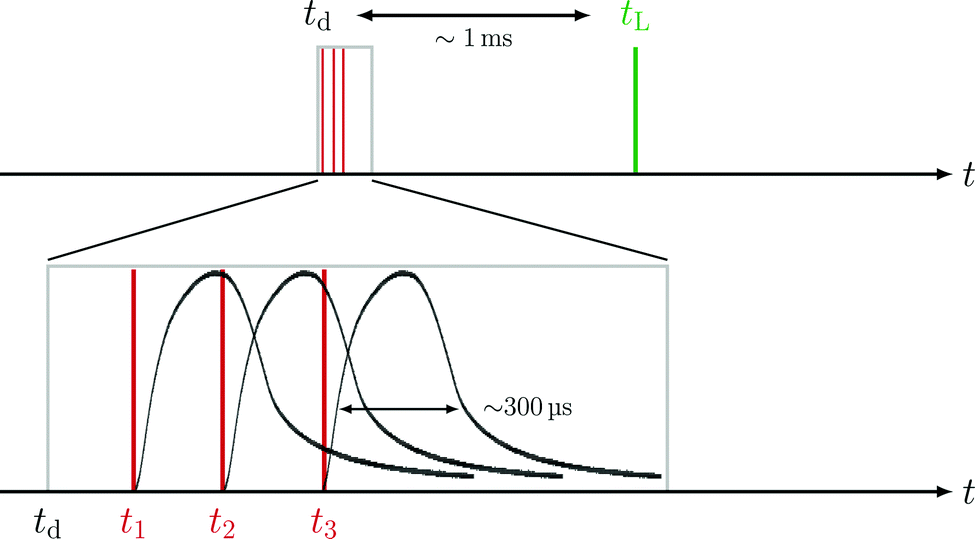 | ||
| Fig. 2 Sketch of the timing sequence of a deflection measurement. The relative timing between deflection (td) and ionization (tL) is varied for various TOF measurements (upper sketch). The deflector consists of three individual coils, which can be operated with different offset timings (t1, t2, t3) relative to td. The lower sketch shows the experimentally measured magnetic field pulses (Id = 300 A) for each coil timing, t1, t2, and t3. | ||
The lower sketch of Fig. 2 shows that each individual coil can be operated with timings t1, t2, and t3. Throughout one deflection measurement, these coil timings are constant relative to the absolute deflector timing td. The relative integrated TOF mass signal θrel is defined as the ratio of the integrated TOF mass signal with the deflector switched on and the integrated TOF mass signal with the deflector switched off.
 | (1) |
 | (2) |
2.2. Measured velocity distributions of Na(NH3)n
Since the deflector is operated in a pulsed manner, the coil timings ti (i = 1, 2, 3) have to be synchronized with tL. The time delay between deflection and ionization (Fig. 2) depends on the cluster size specific velocity distributions. The knowledge of individual cluster beam velocity distributions is thus crucial to optimize the deflector timings for high deflection ratios. To determine cluster velocity distributions, photoions were extracted perpendicular to the molecular beam propagation axis (Fig. 1a). The extraction field was set to optimized VMI conditions. Time of flight based mass-gating was used to record the size-selected photoion images of Na(NH3)n (n = 1–4) shown in Fig. 3a. The velocities of the neutral clusters in the molecular beam correspond to a displacement of the photoions in the detector plane. The cluster-resolved velocities can be determined by the displacement of the image centre (white cross in Fig. 3a). The velocity v is calculated using the equation:| v = (2·C·VR·r2/m)1/2 | (3) |
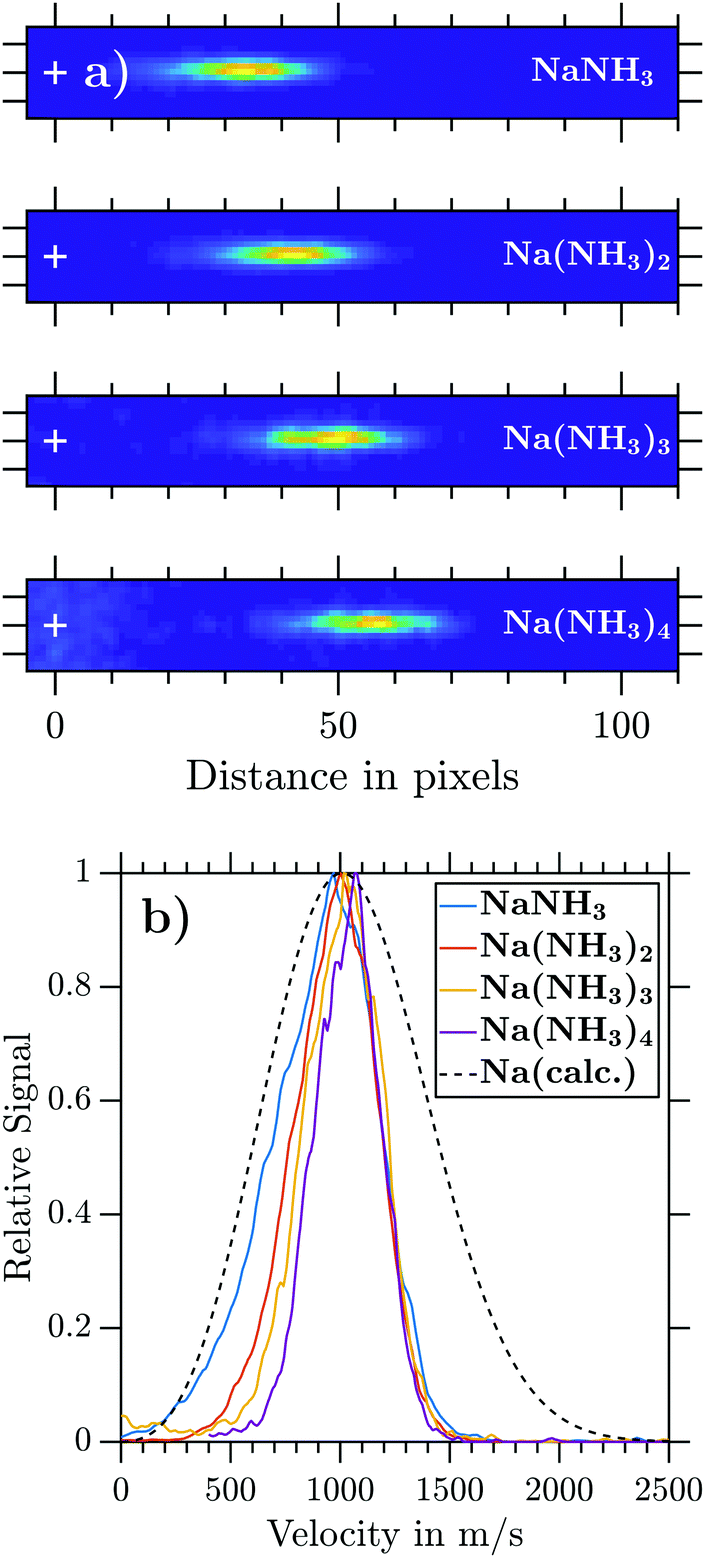 | ||
| Fig. 3 (a) Na(NH3)n (n = 1–4) ion images measured in perpendicular extraction at VR = 7 kV. (b) Experimental Na(NH3)n velocity distributions retrieved from ion images. The calculated velocity distribution for an effusive Na-beam at 265 °C is shown as a dashed line (see the ESI,† Section S3). Its distribution function is given by the convolution of a 1D Maxwell–Boltzmann distribution with an exponential transmission function. | ||
Comparing the velocity distributions of doped ammonia clusters, two effects can be seen with increasing cluster size. Firstly, the centre velocity vc shifts to slightly higher velocities, and secondly the FWHM decreases (Table 1). These differences can be attributed to collisions occurring between the bare clusters and sodium atoms inside the oven during the doping process. Due to momentum conservation, lighter particles deviate further from their initial molecular beam velocity than heavier particles. This explains why the velocity profiles are essentially cluster size independent on the high velocity side of the distributions (Fig. 3b). This part of the velocity distributions reflects collisions between Na and clusters with little momentum transfer, thus largely retaining the initial neat NH3 supersonic velocity distribution.
| Cluster | v c/ms−1 | FWHM/ms−1 |
|---|---|---|
| NaNH3 | 970 | 540 |
| Na(NH3)2 | 1000 | 445 |
| Na(NH3)3 | 1020 | 420 |
| Na(NH3)4 | 1070 | 350 |
2.3. Modelling of the deflection
To verify our experimental results, we chose a MD approach to simulate the cluster trajectories from the entry of the deflection chamber until ionization. For this approach, firstly, a detailed model of the cluster beam is essential. Secondly, a detailed description of the inhomogeneous magnetic field is needed. In our model we describe the cluster beam by the initial velocity and mass of each particle. The initial velocities in the y-direction are randomly sampled from the cluster size specific molecular beam velocity distribution, which is discussed in detail in Section 2.2. The initial velocities in the x- and z-directions are calculated using their beam divergence angle and the velocity in the y-direction. The initial particle coordinates are defined by the position and diameter of the entrance skimmer (see Fig. 1b). Hereby the skimmer position defines the y-coordinate and the diameter defines the possible x, z-coordinates of the Gaussian beam profile. By sampling enough particles (>105) a nearly continuous distribution of initial properties is achieved. The experimental dimensions described in Section 2.1 are accounted for by our MD approach.The force caused by the inhomogeneous potential in the flight channel is defined as the negative gradient of the Zeeman potential (neglecting the contribution of the electron orbital angular momentum L = 0).
 | (4) |
 | (5) |
![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) ,
, ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) the magnetic field and
the magnetic field and ![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) is the force. The force field for the MD simulations is defined by the magnetic field gradients, obtained from a COMSOL simulation assuming a 2D model of the deflector cross section (see Fig. 1d) and a DC current of 1000 A. As in the experiment the force field is only applied during the on-time of the magnetic field pulses. For an accurate description of the magnetic pulse profiles, we used a Hall probe for the time-resolved measurement of the pulses inside the deflector flight channel. These magnetic pulse profiles (see Fig. 2) were measured for various peak currents and used as input for our MD simulations. Current specific scaling factors are determined in relation to the integrated magnetic field pulse profile at 1000 A. An additional magnetic field measurement showed that the induction at the end of each metal bridge is approximately 20% of the peak pulse inside the deflector flight channel. We therefore define the magnetic field magnitude in the epoxy layers to be 20% of the current specific scaling factor of the electromagnetic coils. The forces applied during the pulse are calculated using eqn (5). As in the experiment, the three electromagnets can be pulsed with individual timings t1, t2, and t3. Due to the experimental dimensions, not all particle trajectories will reach the ionization region, and consequently we introduce the following conditions to simulate the particle trajectories. Particles that collide with the deflector walls are considered ‘lost’. Only particles that are transmitted through the detection skimmer can be detected. Only particles that are in the ionization region at time tL can be detected.
is the force. The force field for the MD simulations is defined by the magnetic field gradients, obtained from a COMSOL simulation assuming a 2D model of the deflector cross section (see Fig. 1d) and a DC current of 1000 A. As in the experiment the force field is only applied during the on-time of the magnetic field pulses. For an accurate description of the magnetic pulse profiles, we used a Hall probe for the time-resolved measurement of the pulses inside the deflector flight channel. These magnetic pulse profiles (see Fig. 2) were measured for various peak currents and used as input for our MD simulations. Current specific scaling factors are determined in relation to the integrated magnetic field pulse profile at 1000 A. An additional magnetic field measurement showed that the induction at the end of each metal bridge is approximately 20% of the peak pulse inside the deflector flight channel. We therefore define the magnetic field magnitude in the epoxy layers to be 20% of the current specific scaling factor of the electromagnetic coils. The forces applied during the pulse are calculated using eqn (5). As in the experiment, the three electromagnets can be pulsed with individual timings t1, t2, and t3. Due to the experimental dimensions, not all particle trajectories will reach the ionization region, and consequently we introduce the following conditions to simulate the particle trajectories. Particles that collide with the deflector walls are considered ‘lost’. Only particles that are transmitted through the detection skimmer can be detected. Only particles that are in the ionization region at time tL can be detected.
As an output, the MD simulation creates x,z-coordinates for each trajectory in the detection skimmer plane. These coordinates resemble the molecular beam cross section. In Fig. 4, the visualization of such an output (106 trajectories) is shown for three spin states −1/2, 0 and +1/2 for particles with the same mass as NaNH3. The magnetic field gradient in the z-direction deflects paramagnetic particles in either the positive or the negative z-direction depending on mS. In the centre of the flight channel (x = 0), the field gradient along the z-axis is close to constant. The gradient in the x-direction causes one spin component to diverge (see Fig. 4a), while it focuses the other spin component (see Fig. 4c). The relative signal passing through the detection skimmer, shown as a white circle in Fig. 4, and reaching the ionization region at the same time, is calculated using the following equation:
 | (6) |
 | ||
| Fig. 4 MD simulations of a particle with the same mass as NaNH3 for spin states (a) mS = –1/2, (b) S = 0 and (c) mS = +1/2. 106 trajectories were calculated for all simulations. The white lines indicate the cross section of the flight channel, while the white dotted lines represent the borders of the epoxy layers. The white circle represents the detection skimmer. | ||
In a further step, we simulated the trajectories reaching the ionization region. Trajectories that pass through the detection skimmer and overlap with the Gaussian laser beam profile of 4.8 mm FWHM are treated as ionized. For those ionized particles, an output file is created. It contains the spin state, ionization coordinates and velocity vectors. In addition, the output files are translated into input files for SIMION. SIMION® 8.064 is used to simulate the trajectories of the generated photoions in the electrostatic extraction field and at which positions they impinge on the 2D plane of the ion detector. With this combination of simulations, we are able to simulate the full trajectories from neutral clusters entering the deflector to photoions colliding with the position-sensitive detector (see Section 3.1).
3. Results
3.1. Coil contributions to the deflection of NaNH3
Fig. 5 visualizes the influence of the individual coils on the deflection for the example of NaNH3. For that purpose, mass-gated NaNH3 photoions were extracted perpendicular to the molecular beam propagation axis and detected via VMI while operating the deflector at Id = 300 A (Fig. 5a). For the experiment where all coils (‘all coils’) were used, the electromagnets were fired in an optimized t2 < t1 < t3 sequence. Hereby, td was set to a timing where the highest possible deflection ratio is achievable. By operating each electromagnet separately (1st, 2nd, and 3rd coils), we aim to understand the individual deflection contribution. A further ion image was recorded when the magnetic field was turned off. These recorded ion images are shown in Fig. 5a on the same colour code intensity scale. Operating the deflector clearly reduces the ions impinging the 2D detector plane. Each coil acts on a subset of the cluster distribution and all coils acting together cause a decreased ion signal of θrel = 0.19.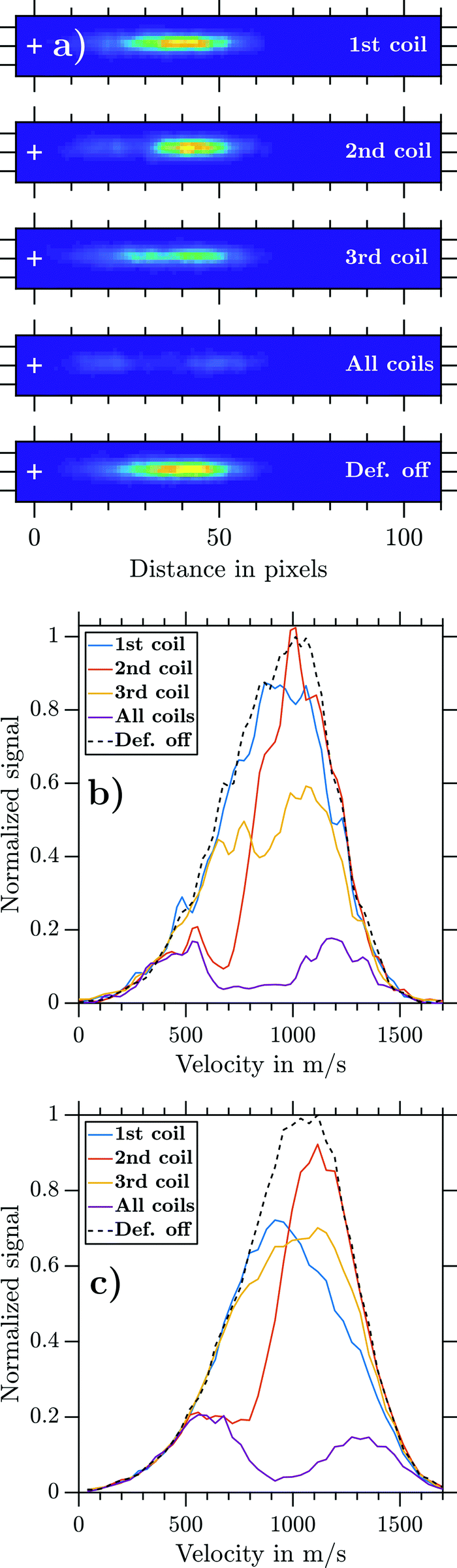 | ||
| Fig. 5 (a) NaNH3 ion images measured with VR = 5 kV for different coil settings. The coils of the deflector were pulsed individually (1st, 2nd, or 3rd coil), together (all coils) with a t2 < t1 < t3 coil sequence and Id = 300 A, or not at all (Def. off). (b) Experimental NaNH3 velocity distributions retrieved from the photoion VMIs in panel a. (c) Corresponding simulated NaNH3 velocity distributions obtained by combining MD and SIMION simulations. The displacement coordinates on the detector were calculated and then transformed into velocity coordinates. | ||
The retrieved experimental NaNH3 velocity distributions for each ion image are shown in Fig. 5b. The 2nd coil (orange line), being switched on first, mainly acts on particles with slow velocities of about 500 ms−1. The 1st coil (blue line) acts on particles traveling with about 1000 ms−1 and the 3rd coil (yellow line) acts on almost the full width of the distribution. The violet line shows all coils being fired, with residual signal remaining from particles at the fast and slow ends of the velocity distribution. These results visualize the functioning of the pulsed deflector and clearly show how critical it is to properly set the coil timings for a given cluster velocity distribution. To achieve reasonable deflection ratios, the magnetic field pulses have to act on the full velocity distribution. If the coil timings are not set well, a certain part of the velocity distribution will not be deflected. Finding such optimal coil timings in the experiment can be challenging. Due to the repetition rate (1–5 Hz) of the deflector, tracking of experimental deflection signals would be prohibitively time consuming. Therefore, we decided to rely on our simplified model for global optimization of the coil timings (Section 2.1.4).
Fig. 5c displays the corresponding simulated NaNH3 velocity distributions obtained from a combination of MD simulations with SIMION simulations (Section 2.3). The experimental and simulated data show good general agreement if all coils are operated simultaneously. Yet the experimental results for the 1st and 3rd coils are only partially verified. While the simulation overestimates the deflection ratio of the 1st coil, deflection by the 3rd coil is underestimated. The experimental and simulated deflection ratios γd (eqn (2)) of the individual coils are summarized in Table 2.
| 1st coil | 2nd coil | 3rd coil | All coils | |
|---|---|---|---|---|
| γ d (experiment) | 0.09 | 0.25 | 0.33 | 0.81 |
| γ d (simulation) | 0.23 | 0.25 | 0.19 | 0.77 |
3.2. Deflection ratios for Na(NH3)n: experiment vs. MD simulations
 and the inhomogeneous magnetic field
and the inhomogeneous magnetic field  . The same holds for larger currents (not shown). NaNH3 TOF signals of four representative td (0, 4.6, 4.8 and 10.0 ms) are marked as open black circles in Fig. 6a and shown as representative TOF traces in Fig. 6b. The TOF traces are baseline subtracted, background corrected and smoothed before the signal peaks are integrated. The dashed lines indicate the integration window applied to retrieve integrated NaNH3 TOF signals for each td.
. The same holds for larger currents (not shown). NaNH3 TOF signals of four representative td (0, 4.6, 4.8 and 10.0 ms) are marked as open black circles in Fig. 6a and shown as representative TOF traces in Fig. 6b. The TOF traces are baseline subtracted, background corrected and smoothed before the signal peaks are integrated. The dashed lines indicate the integration window applied to retrieve integrated NaNH3 TOF signals for each td.
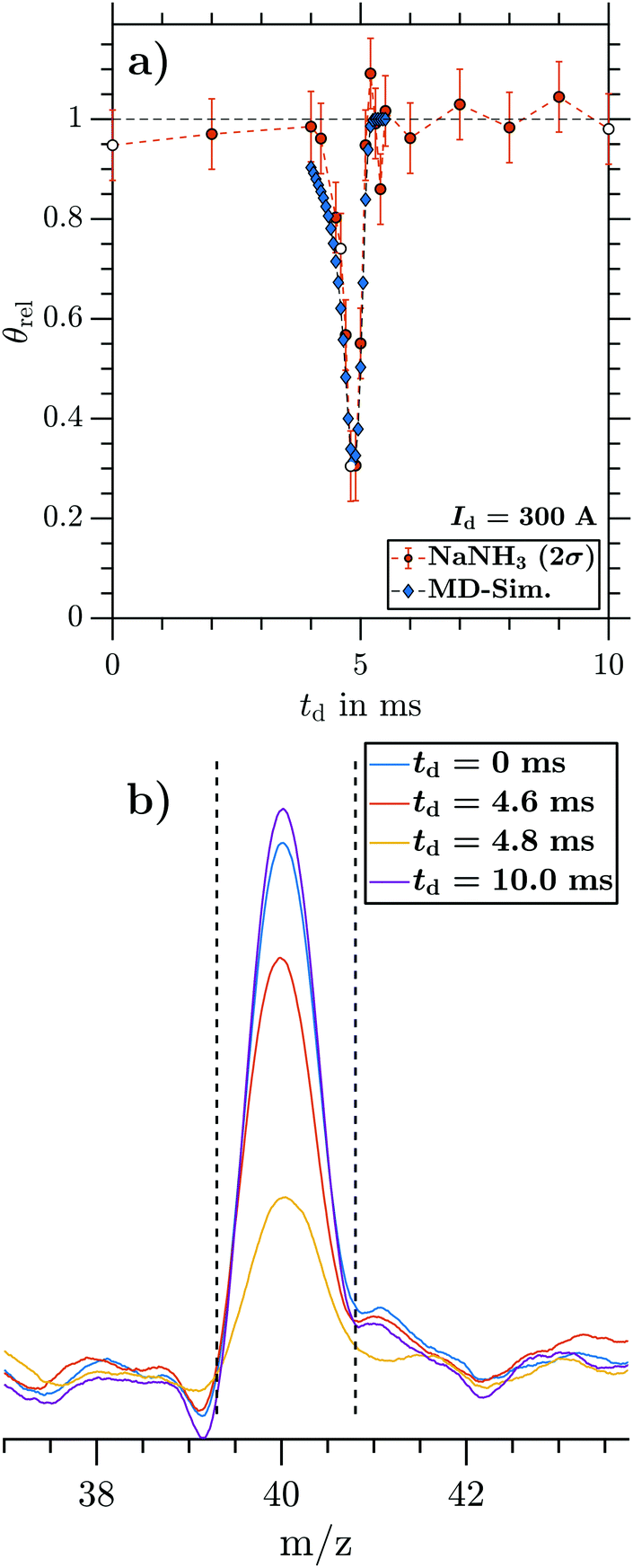 | ||
| Fig. 6 (a) Relative integrated NaNH3 TOF signal θrel (2000 shots per data point) as a function of td (circles with 2σ error), compared with MD simulations (diamonds). (b) NaNH3 TOF signals for four representative td corresponding to the open black circles at 0, 4.6, 4.8 and 10.0 ms in panel a. The deflector was operated at Id = 300 A. | ||
By changing the integration window to larger cluster sizes, we are able to obtain equivalent data as in Fig. 6 for all cluster sizes Na(NH3)n (n = 1–4) from the same TOF traces (see Sections 3.2.2 and 3.2.3 for results on larger clusters). Repeating measurements and MD simulations for several magnetic field strengths by varying Id provides the maximum γd (corresponding to the minima as in Fig. 6a) as a function of Id. Fig. 7 shows the maximal γd as a function of Id for NaNH3, obtained from the experimental data (circles) and from the simulations (diamonds). With increasing magnetic field strength, the experimental γd increases reaching a maximum of 0.78(7) at Id = 700 A. For all different Id, the experimental and simulated γd agree very well within error bars (2σ). Due to the fact that the applied MD approach does not take any intracluster spin relaxation (ISR) times30,48,49 into account, the good agreement between experiment and simulation reveals that for NaNH3 the intracluster spin relaxation time has to be significantly longer than the interaction time with the magnetic field gradient25,26 (see discussion in Section 4).
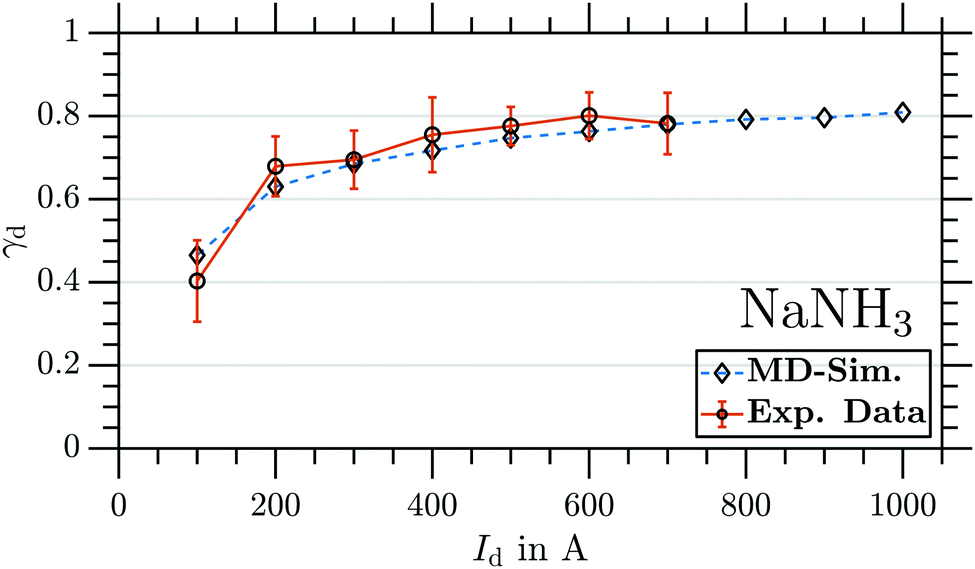 | ||
| Fig. 7 Maximal γd as a function of Id for NaNH3. Circles: experiment. The error indicates 2σ. Diamonds: MD simulation. The deflector was operated in a t1 < t2 < t3 coil timing sequence. | ||
 | ||
| Fig. 8 Maximal γd as a function of Id for Na(NH3)2. Circles: experiment. The error indicates 2σ. Diamonds: MD simulation. The deflector was operated in a t1 < t2 < t3 coil timing sequence. | ||
 | ||
| Fig. 9 θ rel as a function of td of Na(NH3)3 recorded at (a) Id = 300 A and (b) Id = 700 A. (c) Maximal γd as a function of Id for Na(NH3)3. Circles: experiment. The error indicates 2σ. Diamonds: MD simulation. | ||
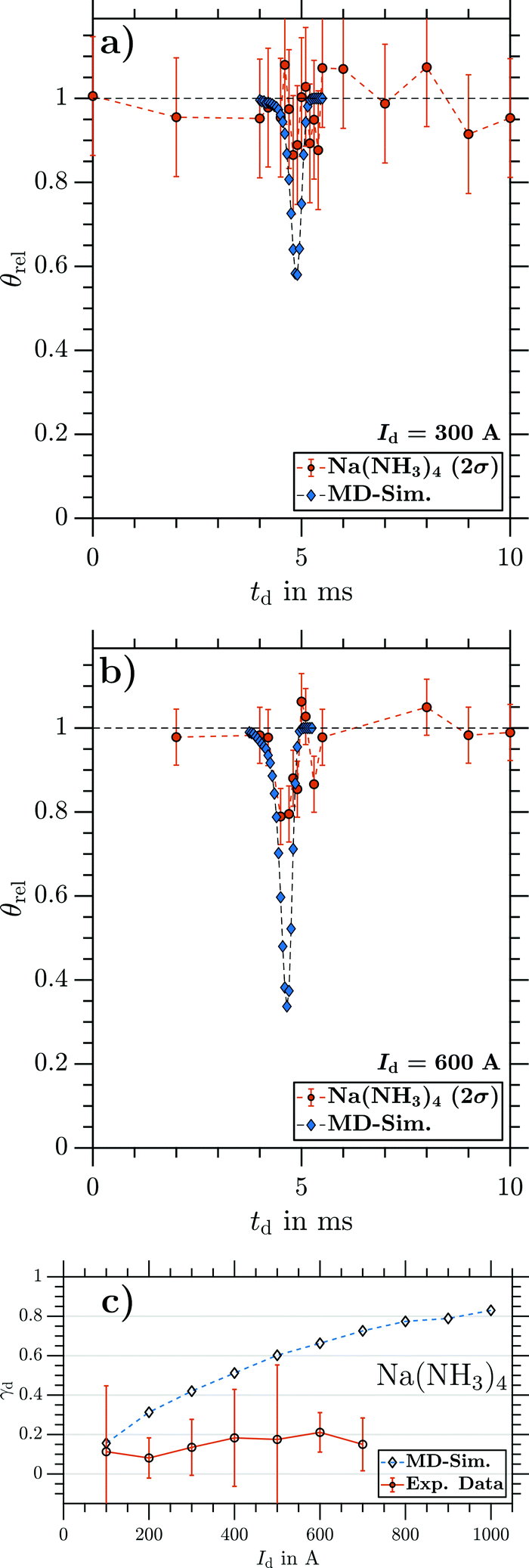 | ||
| Fig. 10 θ rel as a function of td of Na(NH3)4 recorded at (a) Id = 300 A and (b) Id = 600 A. (c) Maximal γd as a function of Id for Na(NH3)4. Circles: experiment. The error indicates 2σ. Diamonds: MD simulation. | ||
In summary, we observe the same deflection as expected from MD simulations for the smallest cluster NaNH3 at all currents (Fig. 7), while the larger clusters Na(NH3)n (n = 2–4; Fig. 8–10) show strongly reduced deflection compared with the simulations. Na(NH3)2 experiences small but clear deflection at all currents. For Na(NH3)3 and Na(NH3)4, in contrast, clear deflection within error bars could be seen only at higher currents. For comparison, at a current of Id ∼ 700 A, the simulations predict γd values around 0.8 for all cluster sizes, while the experimentally observed deflection ratios systematically decrease from γd ∼ 0.8 for NaNH3 to γd ∼ 0.4 for Na(NH3)2 to γd ∼ 0.2 for Na(NH3)3 and Na(NH3)4. As discussed in the following section this hints that spin relaxation times decrease with increasing cluster size.
4. Discussion
Since the effect of increasing cluster mass is already accounted for by our MD-simulations, the reduced deflection we observe for larger clusters might at first sight be interpreted phenomenologically as an indication of a reduction in the effective magnetic moment with increasing cluster size. Such dependence of the magnetic moment on cluster size and structure was found to explain the magnetic deflection experiments of isolated bi-metal clusters. Thus, the different magnetic moments of a single magnetic impurity (Co) in non-magnetic metallic hosts of discrete size (Nbn) have been explained within the framework of the Anderson impurity model, in terms of the increase of the local moment with increasing host band gap (HOMO–LUMO gap).47 Such arguments, however, are not applicable to our system of single spin 1/2 doped weakly interacting molecular clusters. The host molecules (NH3) are closed shell with a very large HOMO–LUMO gap (liquid ammonia band gap: 7 eV).65 The intermolecular interaction (a few tenths of an eV) is too weak to perturb the electronic structure of the host significantly. The effect of clustering on the electronic structure of the dopant is more pronounced. According to ab initio calculations the HOMO–LUMO (s–p) gap of about 2 eV in bare Na is reduced to 1.5 eV, 0.9 eV and 0.8 eV in Na(NH3), Na(NH3)2 and Na(NH3)3, respectively.19 But the unpaired electron still largely retains its s-character. Within an earlier study of the photoelectron spectrum of the solvated electron in Na-doped ammonia clusters, we performed extensive DFT calculations on Na(NH3)n≤30.17,18 The s-character of the unpaired electron of 96% in Na(NH3) decreases only slightly to about 93% in Na(NH3)4. In all these cases the unpaired electron mainly resides on the surface of the cluster (see Fig. 11). There is some delocalization of the spin density over the NH3 host molecules, which tends to increase with cluster size, but with 16% in Na(NH3)4 it still remains rather modest for the cluster sizes considered in the present work. It appears unlikely that any of these effects could change the magnetic moment significantly in these pure spin 1/2 systems. This leaves intracluster spin relaxation as a more plausible reason for the reduced deflection observed in our experiments. | ||
| Fig. 11 Isosurfaces of the HOMOs of Na(NH3)n clusters: (a) NaNH3, (b) Na(NH3)2, (c) Na(NH3)3, (d) Na(NH3)4 isomer IA with all eclipsed arrangement of NH3 and (e) asymmetric isomer IC. (d) and (e) are adapted with permission from ref. 17 Copyright © 2015 American Chemical Society. | ||
In order to discuss our results in terms of possible intracluster spin relaxation processes, a characteristic spin relaxation time τ is compared with the cluster specific interaction time tm with the magnetic field for two different cases. If τ > tm, the electronic spin state does not ‘flip’ when the cluster traverses the deflector setup (i.e. when the cluster interacts with the magnetic field) and free spin (atom-like) deflection behaviour would be observed. This matches the assumptions of our MD simulation, which, therefore, should reproduce the experimentally observed deflection ratios. This was successfully verified using a beam of Na atoms, which exhibit no spin relaxation (Fig. S1, ESI†) during tm. Very good agreement between the MD simulation and experimental data was observed for NaNH3 (Fig. 7), suggesting negligible intracluster spin relaxation during tm. In our experiment, tm is ∼200 μs considering the centre velocity of NaNH3 (∼1000 ms−1) and the deflector length. Therefore, the intracluster spin relaxation time for NaNH3 must be longer than 200 μs.
If τ ≤ tm, the electron spin state stochastically ‘flips’ while travelling through the magnetic field. These statistical fluctuations of the spin reduce deflection and cause a broadening of the molecular beam cross section.25,26 In our setup, this would result in partial deflection. Since these effects are not included in our MD model, the simulations for cases with τ ≤ tm no longer reproduce the measured deflection ratios. If τ ≪ tm, γd values of zero are expected, while τ values on the order of tm would result in reduced, but non-zero deflection ratios. The latter would explain our observations for Na(NH3)2 at all currents and for Na(NH3)3 and Na(NH3)4 at higher currents. With this explanation, typical intracluster relaxation times would be on the order of 200 μs.
If the decrease of intracluster relaxation times in larger clusters is the reason for the observed reduced deflection, how does this size dependence come about? The electronic structure might play a role here, as both the spatial extension of the unpaired electron and its average separation from the Na core increase with cluster size.19 Given that these effects are not dramatic we expect other contributions to be more important. A detailed theoretical description on spin relaxation processes in isolated molecules and clusters was previously given by Knickelbein.49 In his interpretation of Stern–Gerlach experiments, intracluster/intramolecular spin relaxation (ISR) becomes more likely the higher the density of the rovibrational states. Overall rotations of molecular clusters induce Zeeman-like eigenstates in the presence of an external magnetic field. Transitions between such Zeeman sublevels may occur within the conservation laws of internal energy and total angular momentum. The free molecule or cluster can undergo intramolecular or intracluster spin relaxation processes. In such processes, the molecule or cluster serves as a ‘thermal bath’ for its own spin, especially when the system is large and the temperature is high enough to populate many intramolecular states.
In our study of sodium doped ammonia clusters, we expect analogous Zeeman-like sublevels with possible transitions occurring between the sublevels. Yet, the weak non-covalent bonds between the sodium atom and ammonia molecules and between ammonia molecules cause distinct differences to previously investigated free molecules25–29,31–34 and cluster systems.30,35–45,47,51–56 They are rather floppy systems undergoing large amplitude motions with low characteristic frequencies. Many of these represent hindered internal rotations whose vibrational angular momenta provide an effective coupling mechanism to the spin of the unpaired electron. While the rovibrational dynamics of these systems are evidently highly anharmonic, a harmonic analysis of the DFT calculations for the structures shown in Fig. 11 already provides a qualitative picture. The Na–N stretching and bending harmonic wavenumbers of NaNH3 are calculated to be 231 cm−1 and 323 cm−1, respectively. Considering the level of theory and experimental uncertainties, this is consistent with experimental results with values for the fundamental wavenumbers ranging from 195 cm−1 to 215 cm−1 for the stretching and 278 cm−1 for the bending fundamental.66,67 At estimated cluster temperatures in the range of 110–145 K,21 excited states of these low-frequency vibrations (vibrational temperature ∼300 K) are barely populated. This situation changes drastically for the larger clusters Na(NH3)n≥2.68 Here new types of low frequency modes arise, i.e. internal rotation of NH3 around the Na–N axis and N–Na–N bending, with calculated harmonic wavenumbers of 20 cm−1 and 31 cm−1 for the NH3 rotation and 41 cm−1 for the N–Na–N bending. This is consistent with a value of 25 cm−1 derived for the latter via ZEKE-PFI spectroscopy.67 The corresponding vibrational temperature falls in the range between 30 K and 60 K. Compared with NaNH3, this leads to an abrupt increase in thermally accessible states. With each NH3 molecule added, the number of internal modes with vibrational temperatures well below the estimated cluster temperature increases further with a corresponding exponential increase in the thermally accessible density of states.
The excited states of the NH3 internal rotation and N–Na–N bending (hindered orbiting of NH3 around Na) are associated with large vibrational angular momenta, which couple to the cluster's rotational states and subsequently induce a highly perturbed Zeeman-like splitting pattern. Thus the increase of the rovibrational density of states to which the spin of the unpaired electron can couple would explain the decrease of the magnetic deflection upon the addition of NH3 molecules to the Na-doped clusters. This density of coupling states is low for NaNH3 translating into slow ISR. Adding a second NH3 molecule produces a sudden increase in the density of coupling states as new types of low frequency internal motions arise. ISR accelerates accordingly and abruptly reduces the observed magnetic deflection. Adding more NH3 molecules further enhances the effect, but does so more gradually.
While we believe that this is the dominant cause for the cluster size dependence of the magnetic deflection we observe, other effects can also contribute. For example, hyperfine coupling might have to be taken into account. Furthermore, the molecular beam can contain multiple structural isomers for a given cluster size at the estimated cluster temperature. Each structural isomer would exhibit a particular Zeeman splitting pattern with possible transitions occurring between the Zeeman-like sublevels. Thereby spin relaxation would depend on the current geometry of the clusters, which might vary on the timescale of the experiment. The above phenomena would imply a marked temperature dependence of the spin relaxation process, which would be interesting to investigate in future work.
5. Conclusions
We have implemented a pulsed Stern–Gerlach deflection experiment to study the magnetic properties of sodium-doped ammonia clusters. The testing of the experimental setup for an effusive Na atom beam and the comparison with molecular dynamics simulations confirm the expected performance of the design.The present experimental study for Na(NH3)n (n = 1–4) is a first attempt to understand the magnetic properties of sodium doped ammonia clusters, which can be seen as a model system for solvated electron precursors. The combination of deflection experiments with molecular dynamics simulations provides, for the first time, insights into time-scales of intracluster spin relaxation in these small weakly-bound clusters. For NaNH3, we expect intracluster spin relaxation to take place on a timescale substantially longer than 200 μs. The partial deflection that is observed for the larger clusters of Na(NH3)n (n = 2–4) compared with molecular dynamics simulations, that do not take intracluster relaxation effects into account, suggests the relaxation times for these clusters to be in the order of 200 μs. We propose an acceleration of intra-cluster spin relaxation in n > 1 clusters as a result of Zeeman-like spin-rotation coupling. The emergence of very low frequency internal rotation modes for n > 1 leads to a drastic increase in the thermally accessible density of rovibrational states to which the spin can couple, which could explain the abrupt decrease of the deflection for clusters with n > 1. However, to obtain a more definite explanation of the observed cluster size dependence, further magnetic deflection experiments (e.g. spin refocusing with two Stern–Gerlach magnets) and modelling of intracluster relaxation processes should be carried out. Detailed studies of the magnetic and electronic properties of solvated electrons in clusters will lead to a better understanding of the paramagnetic and diamagnetic species in bulk alkali metal ammonia solutions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very grateful to Prof. Gunnar Jeschke for carrying out initial calculations to estimate approximate spin relaxation times for small sodium doped clusters and to Dr David Luckhaus for helping us to modify his molecular dynamics program. We would like to thank David Stapfer and Markus Steger from the mechanical and electronic LPC workshops of ETH Zurich and Haim Sade from the Weizmann CNC unit for their technical contributions to the experimental setup and their continuing support during the improvement of the setup. We also thank Nina Bersenkowitsch for her contribution as an exchange student at the early stage of this project. This project was financially supported by the Swiss National Science Foundation (project nr. 200020_172472), ETH Zurich and the Israel Science Foundation.References
- W. Weyl, Ann. Phys., 1864, 199, 350–367 CrossRef.
- E. Zurek, P. P. Edwards and R. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 8198–8232 CrossRef CAS PubMed.
- J. M. Herbert and L. D. Jacobson, J. Chem. Phys. A, 2011, 115, 14470–14483 CrossRef CAS PubMed.
- P. Voehringer, Annu. Rev. Phys. Chem., 2015, 66, 97–118 CrossRef CAS PubMed.
- D. Holton and P. Edwards, Chem. Br., 1985, 21, 1007 CAS.
- A. Meyer and M. van Gastel, J. Chem. Phys. A, 2011, 115, 1939–1945 CrossRef CAS PubMed.
- P. P. Edwards, J. Phys. Chem., 1984, 88, 3772–3780 CrossRef CAS.
- I. Hertel, C. Hüglin, C. Nitsch and C. Schulz, Phys. Rev. Lett., 1991, 67, 1767 CrossRef CAS PubMed.
- C. Nitsch, C. Schulz, A. Gerber, W. Zimmermann-Edling and I. Hertel, ZPhys-e.D: At., Mol. Clusters, 1992, 22, 651–658 CAS.
- C. Steinbach and U. Buck, J. Chem. Phys., 2005, 122, 134301 CrossRef CAS PubMed.
- I. Dauster, M. A. Suhm, U. Buck and T. Zeuch, Phys. Chem. Chem. Phys., 2008, 10, 83–95 RSC.
- K. Hashimoto and K. Daigoku, Chem. Phys. Lett., 2009, 469, 62–67 CrossRef CAS.
- T. Almeida and B. C. Cabral, J. Chem. Phys., 2010, 132, 094307 CrossRef CAS PubMed.
- B. L. Yoder, J. H. Litman, P. W. Forysinski, J. L. Corbett and R. Signorell, J. Phys. Chem. Lett., 2011, 2, 2623–2628 CrossRef CAS.
- T. Zeuch and U. Buck, Chem. Phys. Lett., 2013, 579, 1–10 CrossRef CAS.
- R. Signorell, B. L. Yoder, A. H. C. West, J. J. Ferreiro and C. M. Saak, Chem. Sci., 2014, 5, 1283–1295 RSC.
- A. H. West, B. L. Yoder, D. Luckhaus and R. Signorell, J. Chem. Phys. A, 2015, 119, 12376–12382 CrossRef CAS PubMed.
- A. H. C. West, B. L. Yoder, D. Luckhaus, C. M. Saak, M. Doppelbauer and R. Signorell, J. Phys. Chem. Lett., 2015, 6, 1487–1492 CrossRef CAS PubMed.
- A. O. Gunina and A. I. Krylov, J. Chem. Phys. A, 2016, 120, 9841–9856 CrossRef CAS PubMed.
- J. V. Barnes, B. L. Yoder and R. Signorell, J. Phys. Chem. A, 2019, 123, 2379–2386 CrossRef CAS PubMed.
- A. l. Carrera and E. Marceca, J. Chem. Phys. A, 2015, 119, 4207–4213 CrossRef CAS.
- W. Gerlach and O. Stern, Z. Phys., 1922, 9, 349–352 CrossRef CAS.
- N. Ramsey, Molecular Beams, OUP, Oxford, 1985 Search PubMed.
- D. J. Meschi and A. W. Searcy, J. Chem. Phys., 1969, 51, 5134–5138 CrossRef CAS.
- A. Amirav and G. Navon, Phys. Rev. Lett., 1981, 47, 906 CrossRef CAS.
- A. Amirav and G. Navon, Chem. Phys., 1983, 82, 253–267 CrossRef CAS.
- N. Kuebler, M. Robin, J. Yang, A. Gedanken and D. Herrick, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 737 CrossRef CAS PubMed.
- A. Gedanken, N. Kuebler, M. Robin and D. Herrick, J. Chem. Phys., 1989, 90, 3981–3993 CrossRef CAS.
- A. Gedanken, N. A. Kuebler, M. B. Robin and D. R. Herrick, J. Am. Chem. Soc., 1989, 111, 5568–5572 CrossRef CAS.
- D. Herrick, M. Robin and A. Gedanken, J. Mol. Spectrosc., 1989, 133, 61–81 CrossRef CAS.
- A. Gedanken, V. Kelner and E. Sominksa, Chem. Phys. Lett., 1994, 221, 274–278 CrossRef CAS.
- A. Malakhovskii and A. Gedanken, J. Chem. Soc., Faraday Trans., 1996, 92, 329–332 RSC.
- A. Malakhovskii, E. Sominska and A. Gedanken, J. Chem. Soc., Faraday Trans., 1996, 92, 1319–1322 RSC.
- M. Alagia, V. Aquilanti, D. Ascenzi, N. Balucani, D. Cappelletti, L. Cartechini, P. Casavecchia, F. Pirani, G. Sanchini and G. G. Volpi, Isr. J. Chem., 1997, 37, 329–342 CrossRef CAS.
- D. Cox, D. Trevor, R. Whetten, E. Rohlfing and A. Kaldor, J. Chem. Phys., 1986, 84, 4651–4656 CrossRef CAS.
- W. A. De Heer, P. Milani and A. Chtelain, Phys. Rev. Lett., 1990, 65, 488 CrossRef CAS PubMed.
- P. Ballone, P. Milani and W. de Heer, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 10350 CrossRef PubMed.
- J. Bucher, D. Douglass and L. Bloomfield, Phys. Rev. Lett., 1991, 66, 3052 CrossRef CAS PubMed.
- W. De Heer, P. Milani and A. Châtelain, ZPhys-e.D: At., Mol. Clusters, 1991, 19, 241–245 CAS.
- I. M. Billas, A. Chatelain and W. A. de Heer, Science, 1994, 265, 1682–1684 CrossRef CAS PubMed.
- T. Hihara, S. Pokrant and J. Becker, Chem. Phys. Lett., 1998, 294, 357–362 CrossRef CAS.
- A. Châtelain, Philos. Mag. B, 1999, 79, 1367–1378 CrossRef.
- N. Hamamoto, N. Onishi and G. Bertsch, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 1336 CrossRef CAS.
- M. B. Knickelbein, J. Chem. Phys., 2002, 116, 9703–9711 CrossRef CAS.
- X. Xu, S. Yin, R. Moro and W. A. de Heer, Phys. Rev. Lett., 2005, 95, 237209 CrossRef PubMed.
- D. Dieleman, M. Tombers, L. Peters, J. Meyer, S. Peredkov, J. Jalink, M. Neeb, W. Eberhardt, T. Rasing and G. Niedner-Schatteburg, Phys. Chem. Chem. Phys., 2015, 17, 28372–28378 RSC.
- A. Diaz-Bachs, L. Peters, R. Logemann, V. Chernyy, J. Bakker, M. Katsnelson and A. Kirilyuk, Phys. Rev. B, 2018, 97, 134427 CrossRef CAS.
- D. Herrick, M. Robin and A. Gedanken, Chem. Phys., 1989, 130, 201–209 CrossRef.
- M. B. Knickelbein, J. Chem. Phys., 2004, 121, 5281–5283 CrossRef CAS PubMed.
- H. Wennerstrom and P. O. Westlund, Phys. Chem. Chem. Phys., 2012, 14, 1677–1684 RSC.
- K. Miyajima, A. Nakajima, S. Yabushita, M. B. Knickelbein and K. Kaya, J. Am. Chem. Soc., 2004, 126, 13202–13203 CrossRef CAS PubMed.
- K. Miyajima, M. B. Knickelbein and A. Nakajima, Polyhedron, 2005, 24, 2341–2345 CrossRef CAS.
- K. Miyajima, M. B. Knickelbein and A. Nakajima, Eur. Phys. J. D, 2005, 34, 177–182 CrossRef CAS.
- K. Miyajima, S. Yabushita, M. B. Knickelbein and A. Nakajima, J. Am. Chem. Soc., 2007, 129, 8473–8480 CrossRef CAS PubMed.
- U. Rohrmann and R. Schäfer, J. Chem. Phys. C, 2015, 119, 10958–10961 CrossRef CAS.
- T. M. Fuchs and R. Schafer, Phys. Rev. A, 2018, 98, 063411 CrossRef CAS.
- J. Liang, T. M. Fuchs, R. Schäfer and V. V. Kresin, Rev. Sci. Instrum., 2020, 91, 053202 CrossRef CAS PubMed.
- A. T. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- B. L. Yoder, J. H. Litman, P. W. Forysinski, J. L. Corbett and R. Signorell, J. Phys. Chem. Lett., 2011, 2, 2623–2628 CrossRef CAS.
- S. Schütte and U. Buck, Int, J. Mass Spectrom., 2002, 220, 183–192 Search PubMed.
- C. Bobbert, S. Schütte, C. Steinbach and U. Buck, Eur. Phys. J. D, 2002, 19, 183–192 CAS.
- R. Signorell, M. Goldmann, B. L. Yoder, A. Bodi, E. Chasovskikh, L. Lang and D. Luckhaus, Chem. Phys. Lett., 2016, 658, 1–6 CrossRef CAS.
- W. Li, S. D. Chambreau, S. A. Lahankar and A. G. Suits, Rev. Sci. Instrum., 2005, 76, 063106 CrossRef.
- D. A. Dahl, Int, J. Mass Spectrom., 2000, 200, 3–25 CAS.
- T. Vogler and P. Vöhringer, Phys. Chem. Chem. Phys., 2018, 20, 25657–25665 RSC.
- D. A. Rodham and G. A. Blake, Chem. Phys. Lett., 1997, 264, 522–530 CrossRef CAS.
- X. Peng and W. Kong, J. Chem. Phys., 2002, 117, 9306–9315 CrossRef CAS.
- C. P. Schulz, A. Scholz and I. V. Hertel, Isr. J. Chem., 2004, 44, 19–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04647g |
| This journal is © the Owner Societies 2021 |