
Effects of ion migration and improvement strategies for the operational stability of perovskite solar cells
Yao
Zhao
a,
WenKe
Zhou
ab,
Zhengyuan
Han
a,
Dapeng
Yu
ac and
Qing
Zhao
 *ade
*ade
aState Key Lab for Mesoscopic Physics and Frontiers Science Center for Nano-optoelectronics, School of Physics, Peking University, Beijing, 100871, China. E-mail: zhaoqing@pku.edu.cn
bArmy Engineering University of PLA, State Key Laboratory for Disaster Prevention & Mitigation of Explosion & Impact, Nanjing, 210007, China
cInstitute for Quantum Science and Technology and Department of Physics, South University of Science and Technology of China (SUSTech), Shenzhen, 518055, China
dPeking University Yangtze Delta Institute of Optoelectronics, Nantong, 226010, Jiangsu, China
eCollaborative Innovation Center of Quantum Matter, Beijing, 100084, China
First published on 16th December 2020
Abstract
The fundamental factor affecting the stability of perovskite solar cells, ion migration, has been reviewed, which is found to be closely related to the degradation of perovskite solar cells. Characterization methods like impedance spectroscopy and galvanostatic measurement to identify ion migration in perovskite films have been reviewed. The influence of light on ion migration was further discussed, which could largely explain the photo-stability decay in most perovskite solar cells. Finally, several solutions to inhibit ion migration for better operational stability of perovskite solar cells were summarized, including bulk passivation, interface passivation and grain boundary passivation. Several strategies have also been proposed to further improve the stablity of perovskite solar cells.
 Yao Zhao | Yao Zhao is a graduate student in Prof. Dapeng Yu and Prof. Qing Zhao's group at the School of Physics, Peking University. He has been doing perovskite solar cell research, mainly on interfacial passivation and stability improvement of perovskite solar cells. |
 Qing Zhao | Qing Zhao is a professor at the School of Physics, Peking University. She obtained her PhD degree from Peking University and did her postdoctoral research at the University of Washington. She is interested in research on nanostructures and low-dimensional physics. Her group has recently focused on the study of perovskite solar cells, mainly on the mechanistic study of stability and developing general strategies to improve the stability of perovskite solar cells. |
Introduction
Solar energy is so abundant, which makes it one of the best clean energy sources. Conversion of solar energy to electricity is likely to be the most cost-effective energy conversion solution.1 Many photovoltaic technologies, such as passivated emitters and rear cells, heterojunction technology and tunnel oxide passivated carrier-selective contact silicon based solar cells have been commercialized.2–4 Perovskite materials have many superior properties for photovoltaic technology, such as an adjustable direct band gap,5 charge carrier diffusion lengths in the micrometre range,6 defect tolerance,7 low manufacturing cost, etc. Perovskite materials for the state-of-art perovskite solar cells (PSCs) commonly have a 3D ABX3 crystal structure, where A is a monovalent cation of methylammonium CH3NH3 (denoted as MA), formamidinium (denoted as FA), cesium (Cs), etc. B is a divalent metal cation (Pb or Sn), and X is a halide anion (chlorine, bromide, or iodine). There is a tolerance factor t to describe the stability of the perovskite crystal structure: when t is located in the range of 0.81 < t < 1, halide perovskite easily forms a 3D-crystal structure.8,9 The power conversion efficiency (PCE) of PSCs increased from 3.8% to 25.2% in the last ten years.10–15 However, the stability of PSCs is not as good as its Si-based predecessor, which restricts their commercialization.16 On one hand, PSCs are sensitive to moisture, heat and oxygen.17–19 On the other hand, PSCs are easily destroyed by intrinsic factors: ion migration and the related grain boundary defect.20–24 Ion migration means ions, like I− and MA+, would migrate across the perovskite layer under light illumination and electric field, and the migrated ions would form vacancies or other defects, which would lead to the formation of recombination centers.25,26 The disordered grain boundary lattice has many fractured chemical bonds, which act as recombination centers, which could also lead to the performance degradation of solar cells.27–29 The performance of solar cells would be decreased as the number of recombination centers increased. Intensive efforts have been made to protect perovskites from environmental factors, but intrinsic factors seem to play a more important role in PSCs while working for a long time.30The perovskite layer can be synthesized by various techniques, such as spraying,31 ultrasonic spraying,32 chemical vapor deposition,33 blade coating,34 screen printing,35etc. Among them, the spin coating method is the most widely used due to its convenience. According to the device structure, PSCs can be divided into many types such as mesoporous PSCs,36 inverted planar PSCs37 and planar PSCs.38 Massive strategies, such as solvent engineering, interfacial engineering and bandgap engineering, have been developed to improve the photovoltaic performance of PSCs.39 In terms of stability, moisture and oxygen induced perovskite decomposition would be trivial after careful encapsulation, therefore, improving the intrinsic stability of PSCs is more important to expand its future applications.34 Ion migration is thought to be the origin of intrinsic instability of PSCs due to their ionic crystal nature, therefore, investigation of ion migration is of vital importance to the development of PSCs. In this review, we survey the discovery and characterization methods of ion migration in perovskite materials, the influence of light on ion migration in perovskites, the effect of ion migration on the photovoltaic performance of PSCs, and effective strategies to mitigate the ion migration in order to improve the operational stability of PSCs.
1. Detection method of ion migration in perovskites
Ion migration is an intrinsic effect of perovskite materials because of their ionic conductor nature, loose crystal structures and the existence of vacancies and other defects. Ion migration occurs within perovskite materials under an electric field and would cause hysteresis and instability of PSCs.40–45 Therefore, the determination of ion migration in perovskite materials is vitally important.1.1 Impedance spectroscopy
In general, impedance spectroscopy (IS) is thought to be one of the most efficient and convenient techniques to analyze perovskite photovoltaic devices. Sanchez et al. found a slow dynamic process in PSCs, corresponding to hysteresis, and they identified the process by IS measurement and ascribed the slow dynamic process to the polarizability of MA cations in perovskites.46 Dualeh et al. suggested that the perovskite materials would exhibit ionic charge transport mixed with electronic conduction from impedance spectroscopy results, indicating the existence of the ion migration effect.47 In the low frequency region at high forward bias (Fig. 1a), the PSCs show a so-called “negative capacitance” effect, which stems from the imbalanced ion distribution at contacts originated from ion migration. From then on, researchers started to observe the evidence of the existence of ion migration in PSCs.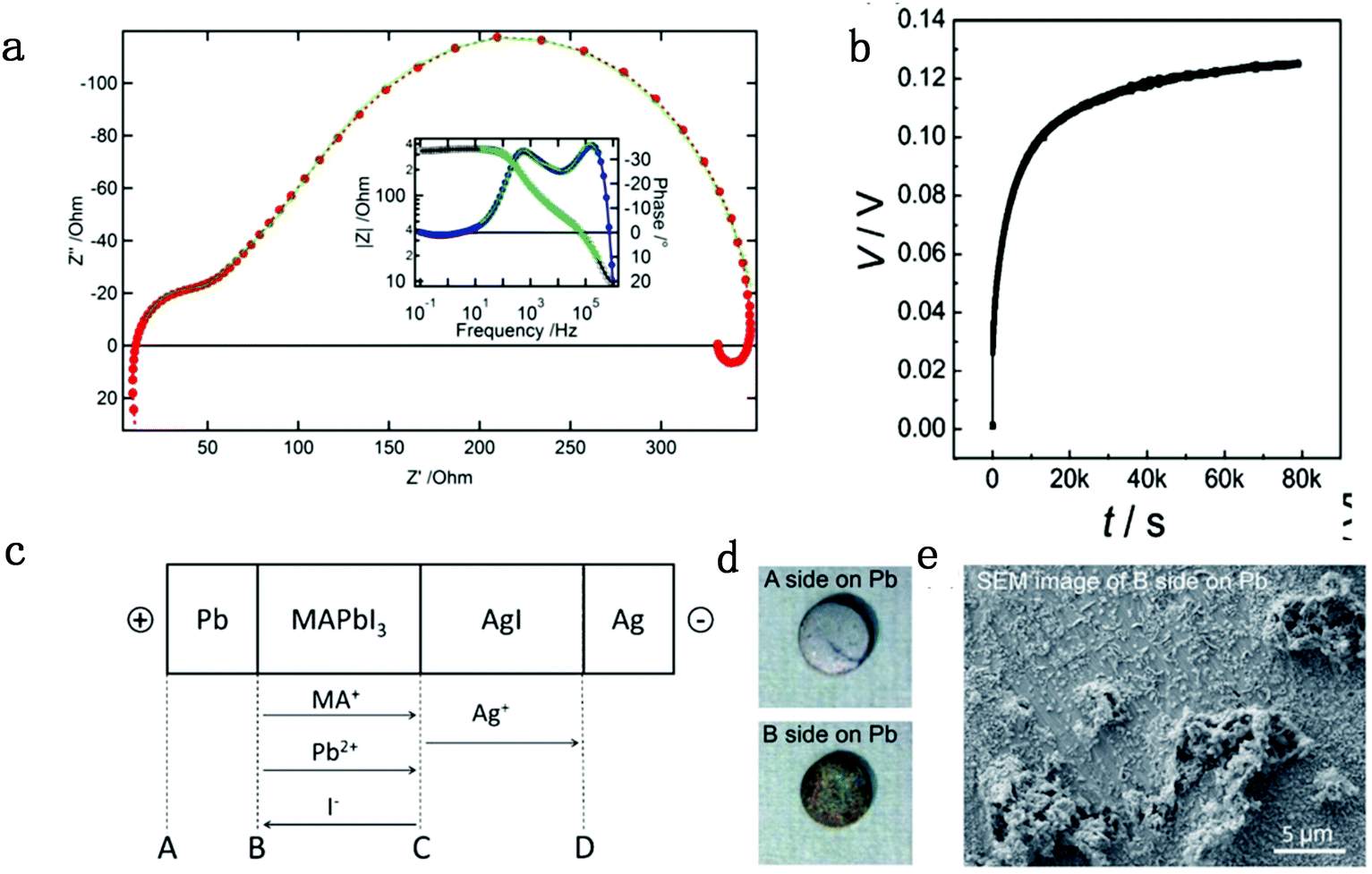 | ||
| Fig. 1 (a) Nyquist and Bode plot (inset) of a standard solid-state perovskite-based device at 550 mV forward bias shows negative capacitance. Reproduced with permission from ref. 47. Copyright 2014 American Chemical Society. (b) DC polarization curve for a carbon|MAPbI3|carbon device measured at room temperature by applying a current of 2 nA, consisting of ion mixed electron and pure electron contribution parts. (c) Flow directions of the charged ion species in a Pb|MAPbI3|AgI|Ag cell under electrical bias. (d) Images for surfaces A and B. (e) SEM image of surface B on the Pb pellet. Reproduced with permission from ref. 48. Copyright 2015 John Wiley & Sons, Inc. | ||
1.2 Galvanostatic measurement
The galvanostatic method consists of placing a constant current pulse upon the measured sample between two electrodes and measuring the variation of voltage along with time. Yang et al. used galvanostatic measurements to clarify the properties of the ion and electron conduction of perovskite materials. They designed a carbon–perovskite–carbon sandwich structure to carry out galvanostatic measurements.48 As illustrated in Fig. 1b, under the tiny set current (i = 2 nA), the voltage instantaneously reaches 25 mV and then slowly increases to a saturation Vs. When applying constant current to a perovskite film, the ions and electrons would migrate simultaneously at the first stage, but the accumulated ions would reach a saturation state in a few minutes, therefore, there is only electron conductance existing in the saturation state. The increasing part of the voltage corresponds to the electrical and ionic resistance whereas the steady part of the voltage corresponds to pure electrical resistance. A relationship between ion/electron resistance and voltage could be obtained: V = i(ReonRion/(Reon + Rion)), and only electron flow contributes to the saturation voltage (i = ieon, iion = 0), that is, Vs = iReon. The electronic conductivity could be deduced from Vs, which is calculated to be 1.9 × 10−9 S cm−1 and hence the ionic conductivity can be obtained as 7.7 × 10−9 S cm−1 (stot = sion + seon) (s refers to conductance).They also indirectly validated the 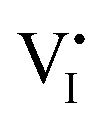 (iodine vacancies) contribution to the ion migration in DC galvanostatic measurement by altering the partial pressure of I2 in the atmosphere. They found that the conduction of ions would decrease along with the increasing partial pressure of I2 because the high partial pressure of I2 will lead to the decrease of
(iodine vacancies) contribution to the ion migration in DC galvanostatic measurement by altering the partial pressure of I2 in the atmosphere. They found that the conduction of ions would decrease along with the increasing partial pressure of I2 because the high partial pressure of I2 will lead to the decrease of 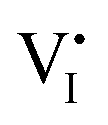 concentration and thus result in the reduced ion conduction. To further investigate which kinds of ions contribute to the migration, they designed a Pb|MAPbI3|AgI|Ag structure device and applied current on Pb and Ag electrodes to study the chemical effect at the various interfaces A, B, C and D (Fig. 1c). As displayed in Fig. 1d, the A side maintained the original morphology and Pb appearance, while the interface B exhibited a yellow and rough appearance. In the scanning electron microscopy (SEM) micrograph (Fig. 1e), the formation of grains with irregular shape at the B interface could be observed whereas MAPbI3 and AgI did not reveal any change during the test. These experiments illustrated that I− ions contribute to the ion migration in perovskite films.
concentration and thus result in the reduced ion conduction. To further investigate which kinds of ions contribute to the migration, they designed a Pb|MAPbI3|AgI|Ag structure device and applied current on Pb and Ag electrodes to study the chemical effect at the various interfaces A, B, C and D (Fig. 1c). As displayed in Fig. 1d, the A side maintained the original morphology and Pb appearance, while the interface B exhibited a yellow and rough appearance. In the scanning electron microscopy (SEM) micrograph (Fig. 1e), the formation of grains with irregular shape at the B interface could be observed whereas MAPbI3 and AgI did not reveal any change during the test. These experiments illustrated that I− ions contribute to the ion migration in perovskite films.
Later on, Zhao's group25 further identified ion migration in perovskite materials in a lateral structure by combining galvanostatic measurements with photoluminescence (PL) and microscope apparatus (Fig. 2a). The migrated ions accumulate at the anode and cathode after the lateral structure was applied with a tiny constant current, therefore, a compositional gradient was formed to prevent the voltage from continuously increasing. The decrease of the PL peak intensity and the shift in the peak position at the cathode and anode after electric poling for several seconds indicated that ion migration induced more defects near the electrode (Fig. 2b and c).
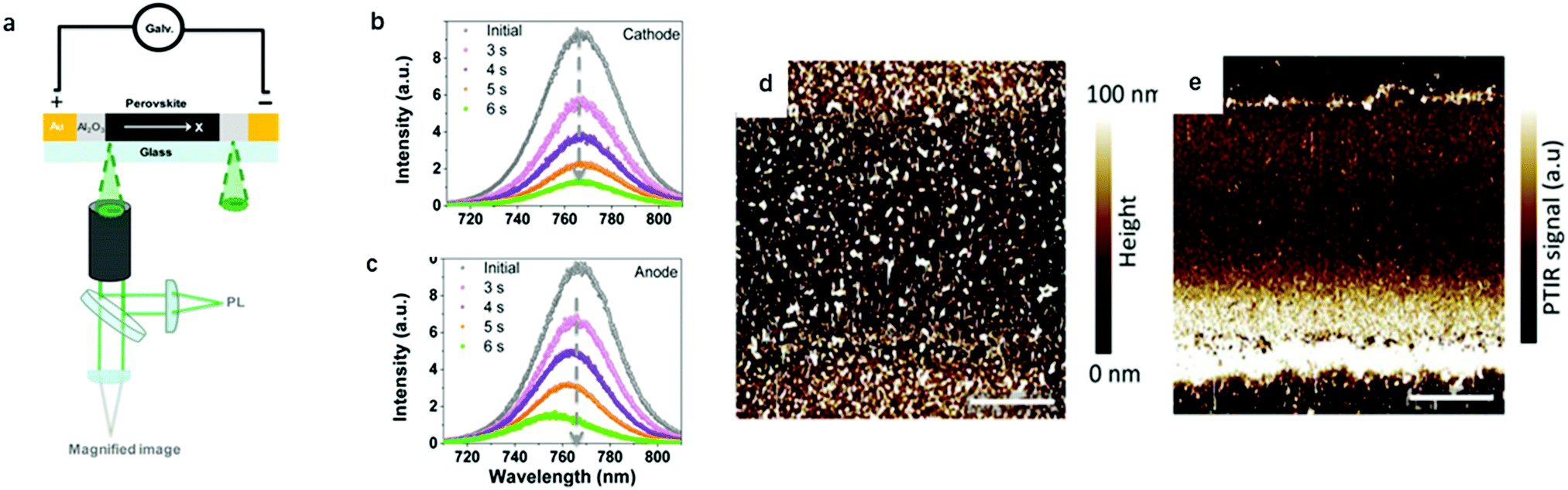 | ||
| Fig. 2 (a) Schematic illustration of the PL and galvanostatic measurement of a perovskite lateral structure. (b and c) PL spectra in the cathode and anode after electric poling at 1 V μm−1 for 6 s. Adapted with permission from ref. 25. Copyright 2016 American Chemical Society. (d and e) PTIR microscopy image of the lateral device before and after electric poling. Adapted with permission from ref. 49. Copyright 2015 John Wiley & Sons, Inc. | ||
1.3 Photothermal induced resonance (PTIR) microscopy
In PTIR microscopy, a pulsed IR laser for excitation and an atomic force microscope (AFM) tip as a detector of IR spectra are used. Because of the specificity of IR spectroscopy to the chemical composition, PTIR allows mapping the distribution of given chemical species, such as MA+. Yuan et al. used PTIR microscopy to further study the ion migration effect in a lateral structure of perovskite films. The function of the AFM tip is to convert the sample thermal expansion into large cantilever oscillations, which are detected in the far field by reflecting a diode laser into the AFM four-quadrant detector.49Fig. 2d and e show the PTIR image before and after electric poling for 200 s, respectively. The bright line along the Au electrode edge (upper and bottom in Fig. 2d and e) in the PTIR map is caused by the plasmonic enhancement of the PTIR signal induced by MA+ ion migration. This is the first time that the electric field-driven MA+ ion migration is directly observed in a MAPbI3 film. It has been demonstrated that MA+ would rotate within the inorganic framework with a relaxation time of few ps at room temperature, indicating a weak interaction between MA+ and the inorganic framework. These characteristics make MA+ ion migration likely to occur under an electric field.1.4 Kelvin probe force microscopy (KPFM) and conducting atomic force microscopy (c-AFM)
While I− and MA+ ion migrations have been identified by DC galvanostatic and PTIR measurements, respectively, the detailed migration mechanism in perovskites has not been yet directly observed. Yuan et al.50 designed a lateral device with perovskite films deposited on glass substrates to identify the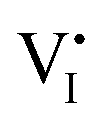 ion migration process by observation under a microscope and electron dispersive spectra (EDS) measurement. Fig. 3a and b show the optical image and schematic illustration diagram of their experiment device. After applying a constant electrical field of ∼3 V μm−1 for 20–60 s, a thread formed near the anode region, and then gradually moved toward the cathode along the applied electric field direction (Fig. 3c and d). The thread region is composed of PbI2, which could be identified from EDS mapping and XRD result; the thread has a smaller I− concentration than MAPbI3 and the diffraction angle is 15°.
ion migration process by observation under a microscope and electron dispersive spectra (EDS) measurement. Fig. 3a and b show the optical image and schematic illustration diagram of their experiment device. After applying a constant electrical field of ∼3 V μm−1 for 20–60 s, a thread formed near the anode region, and then gradually moved toward the cathode along the applied electric field direction (Fig. 3c and d). The thread region is composed of PbI2, which could be identified from EDS mapping and XRD result; the thread has a smaller I− concentration than MAPbI3 and the diffraction angle is 15°.
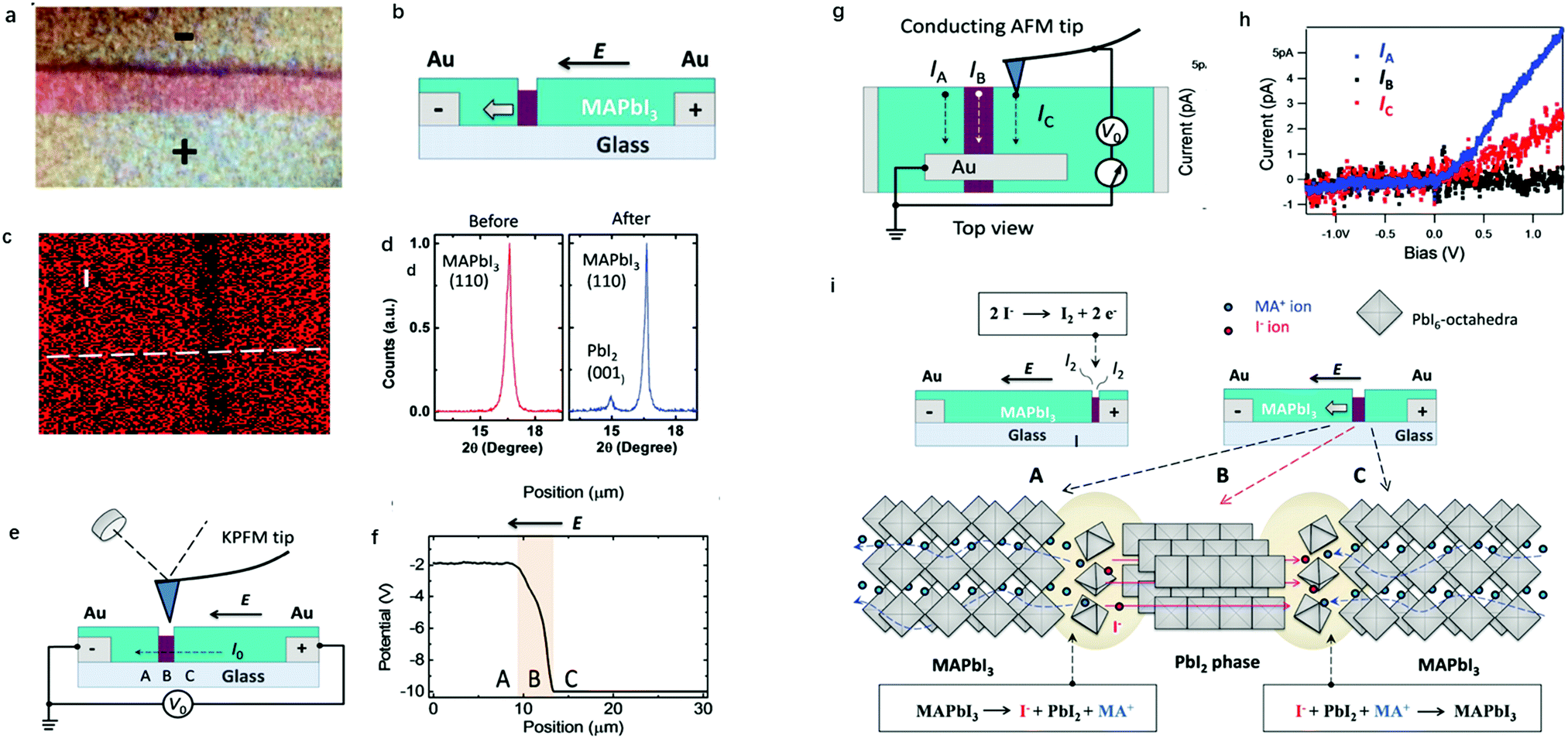 | ||
| Fig. 3 (a) Optical images of the lateral MAPbI3 device. (b) Schematic illustration of the lateral MAPbI3 perovskite devices with the PbI2 thread. (c) EDS mapping of I in lateral perovskite devices. (d) XRD for fresh and reformed MAPbI3. (e) Schematic illustration of the KPFM measurement around the PbI2 thread. (f) Potential information obtained from KPFM measurements. (g) Schematic illustration of the c-AFM measurement around the PbI2 thread. (h) Current information obtained from c-AFM. (i) The mechanism of ion migration in the lateral MAPbI3 device. Adapted with permission from ref. 49. Copyright 2016 John Wiley & Sons, Inc. | ||
To identify the property differences of the A, B and C areas shown in Fig. 3e, corresponding to pristine perovskites, thread and newly formed perovskites, KPFM and c-AFM measurements were used. KPFM could detect the potential information of given materials, and the conductivity of the materials can be estimated from the slope of the potential-position curve. Fig. 3e shows the schematic illumination of the KPFM measurement upon a lateral perovskite device. It can be observed from Fig. 3f that the potential drop across the PbI2 thread is 50 times faster than that across the fresh MAPbI3 film, indicating that almost all the potential applied to the device drops at the PbI2 thread.
C-AFM could identify the conductance information of given materials through current information, which could provide more detailed information for KPFM measurements. An Au electrode was prepared using the PbI2 thread as the ground electrode in the c-AFM measurements to identify the current of perovskite devices at different places (Fig. 3g). The conductivity of the thread and MAPbI3 can be obtained by applying a bias between the tip and the ground electrode in this way. From Fig. 3h, it can be observed that the resulting currents in fresh, reformed MAPbI3 and PbI2 thread are 6 pA, 2 pA and 0.1 pA, respectively, which are 60 and 20 times higher than the lateral current obtained in PbI2. Both KPFM and c-AFM results suggested that PbI2 has a relatively poor conductance than the MAPbI3 film and the fresh MAPbI3 has better conductance than the reformed one. Two reactions, as shown in Fig. 3i, happened at the front and back edges of PbI2 simultaneously. A conversion between MAPbI3 and PbI2 illustrates that both I− anions and MA+ cations contribute to the ion migration, which provides a qualitative explanation of the migration mechanism in perovskite materials.
2. Light influence on ion migration in perovskites
Apart from electrical factors, ion migration could also be affected by light illumination, which is thought to be an intrinsic factor that influences the PSC performance. Xing et al.44 illustrated the impact of light illumination on ion migration through a poling experiment on a lateral structure device (Fig. 4a). The activation energy of ionic conduction directly represents how easily ions move, and thus was applied here to quantitatively characterize the influence of light illumination on ion migration.51,52 The ion migration rate in perovskite films could be determined by the activation energy (Ea) by the Nernst–Einstein relationship: , where k is the Boltzmann constant, σ0 is a constant, and activation energy Ea can be derived from the slope of the ln(σT) − 1/kT relationship. The light illumination could obviously accelerate the moving speed of the PbI2 line when the light intensity increases from 2 to 25 mW cm−2, which could be ascribed to the reduced activation energy of ion migration (Fig. 4b).
, where k is the Boltzmann constant, σ0 is a constant, and activation energy Ea can be derived from the slope of the ln(σT) − 1/kT relationship. The light illumination could obviously accelerate the moving speed of the PbI2 line when the light intensity increases from 2 to 25 mW cm−2, which could be ascribed to the reduced activation energy of ion migration (Fig. 4b).
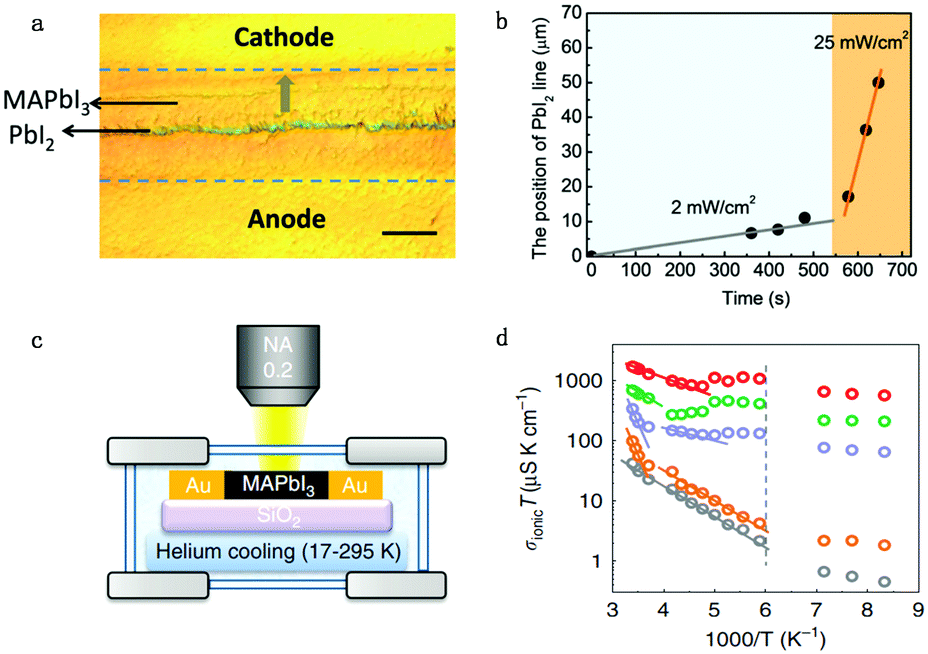 | ||
| Fig. 4 (a) The optical image showing the ion migration effect under light and electrical poling for the perovskite lateral device. (b) The moving speed of the PbI2 thread reflecting the light enhanced ion migration effect: the moving speed of the line is proportional to light illumination. Adapted with permission from ref. 44. Copyright 2016 Royal Society of Chemistry. (c) Apparatus used to measure the ion conductivity in a lateral structure under different temperatures, with a helium cooling system. (d) Ionic conductivity multiplied by temperature (from 17 to 295 K), σionT, as a function of 1000/T under various illumination intensities (grey: 0 mW cm−2; orange: 0.05 mW cm−2; purple: 1 mW cm−2; green: 5 mW cm−2; red: 20 mW cm−2). The dashed line indicates the phase transition temperature. Adapted with permission from ref. 26. Copyright 2017 Springer Nature. | ||
However, activation energy was deduced from the mixed conductance of ions and electrons, which cannot thoroughly explain the ion behavior under light illumination. Zhao et al.26 proposed a light-enhanced ionic transportation mechanism in MAPbI3 and measured the ion migration activation energy of perovskite materials by means of combined cryogenic galvanostatic and voltage–current measurements over the temperature range of 17–295 K (Fig. 4c). As the illumination intensity increased, the Au/MAPbI3/Au device under an electrical field would degrade faster because of the light-enhanced ion migration effect. The ion activation energy relationship is: 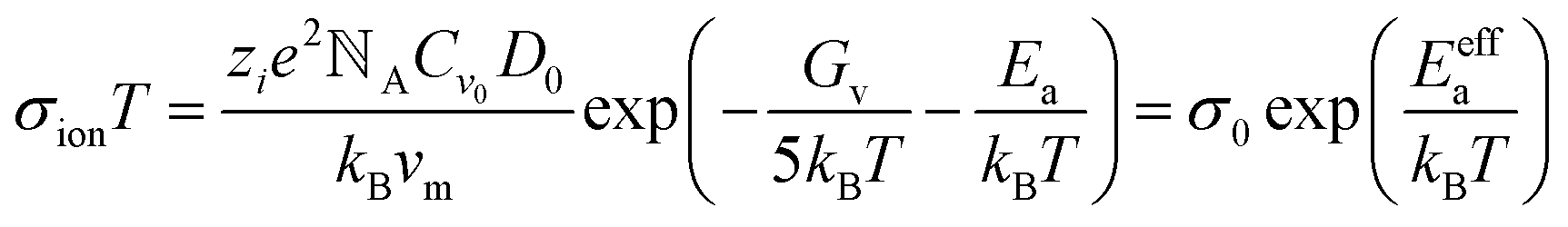 , where Zi is the charge of ions, NA is Avogadro's constant, Cv is the concentration of intrinsic defects, kB is the Boltzmann constant, Vm is the molar volume of the perovskite, D is the diffusion coefficient, Gv is the formation energy for vacancy defects, Ea is the activation energy for ionic diffusion, and Eeffa is defined for considering excess vacancy formation energy in the vacancy-mediated mechanism.
, where Zi is the charge of ions, NA is Avogadro's constant, Cv is the concentration of intrinsic defects, kB is the Boltzmann constant, Vm is the molar volume of the perovskite, D is the diffusion coefficient, Gv is the formation energy for vacancy defects, Ea is the activation energy for ionic diffusion, and Eeffa is defined for considering excess vacancy formation energy in the vacancy-mediated mechanism.
To obtain ion migration activation energy, one needs to segregate the ion conduction from the mixed electronic and ionic conduction. They carried out a fast scan I–V curve to obtain mixed conductance and deduct electron conductance, which was obtained by galvanostatic measurements, to finally obtain the ion conductance. By doing such experiments at a series of temperatures, the relationship of σionT and activation energy Ea could be obtained (Fig. 4d). The ion migration activation energy of the MAPbI3 perovskite material is found to decrease from 824 to 144 meV when the light illumination intensity increases from 0.05 to 20 mW cm−2 under electric field poling, which tells us that ions would migrate when the PSCs are under operation, and more severe ion migration would occur under stronger light intensity. The migrated ions accumulating at the interface would cause the formation of deep energy level defects and thus lead to the degradation of the PSCs.
Kim et al.53 also validated the light-enhanced ion migration effect by the experiment illustrated in Fig. 5a and c. They compared the iodine flux induced by applying a high iodine partial pressure on one side and Cu as the target on the other side of a MAPbI3 film. The iodine was identified by the formation of CuI, which only occurred for the light illumination side (Fig. 5d). They also measured the rate of iodine desorption obtained by contacting MAPbI3 with toluene, the iodine content of which is followed by comparing the dark and light conditions. The result suggested that the iodine content is much higher under light illumination conditions (Fig. 5b). They also observed a strong enhancement of σion when the immersion was done under illumination, corresponding to an increased generation of mobile iodine vacancies. They also provide insight into this enhanced ion migration effect: interaction between holes and iodine would lead to the formation of iodine vacancies. Light illumination leads to the formation of extra holes, which would interact with the I ions.54,55 The neutral iodine has a smaller radius than the iodide ions (ionic radius 1.5 Å instead of 2.1 Å) and would easily fit into interstitial sites. Furthermore, the interstitial particle, Ii (i stands for neural), can be stabilized by structural relaxation owing to polarizability and covalency (I2−, I32−), for which relaxation effects are commonly observed in alkaline compounds, mentioned as self-trapped holes.56–60 The increased concentration of I iodine vacancies leads to the enhancement of ion conductance.
 | ||
Fig. 5 (a) The toluene treatment is performed in the dark and under light to dissolve migrated I ions. (b) Conductivity variation as a function of immersion time in toluene measured in the dark and light. (c) Permeation cell in the dark and under light: an iodine flux through a MAPbI3 film is induced by applying a defined iodine partial pressure (3.6 × 10−6 bar) on one side and Cu as the iodine sink on the other side. (d) X-ray diffraction patterns of MAPbI3 films deposited on Cu foil, after exposure to 3.6 × 10−6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) barP(I2) at 50 °C for 2days with and without 1 mW cm−2 light illumination. Adapted with permission from ref. 53. Copyright 2018 Springer Nature. barP(I2) at 50 °C for 2days with and without 1 mW cm−2 light illumination. Adapted with permission from ref. 53. Copyright 2018 Springer Nature. | ||
3. Effect of ion migration on PSCs
3.1 Hysteresis
Hysteresis is a unique characteristic of PSCs: the forward scan PCE is usually lower than the reverse one in I–V measurements (Fig. 6a).61 Although there are several proposed mechanisms, like ferroelectric polarization effect,62 used in attempts to explain the hysteresis effect, more and more attention is focused on ion migration theory due to the consideration that decay time of hysteresis is on a second timescale.21,63,64 The ferroelectric polarization/depolarization process is much faster (in the picosecond range) than the hysteresis process. In addition, O’Regan et al.65 illustrated that the J–V curve hysteresis is not associated with the change of recombination rate and charge separation efficiency by transient photovoltage and capacitance measurements. Moreover, traditional trapping and detrapping of carriers in the bulk or at the interface defect is unlikely to be the primary cause for hysteresis due to the long duration and current decay magnitude.21 The trapping/detrapping process is associated with migrating induced defects, such as iodine interstitials and vacancies, which can be driven by an electrical field, accounting for the hysteresis of PSCs. In summary, perovskite hysteresis is generally believed to be caused by charge accumulation between interfaces; charge accumulation is mainly caused by ion migration, excess defect concentration and unbalanced charge transport caused by ion migration, so the hysteresis is a quality index of the interface of PSCs.66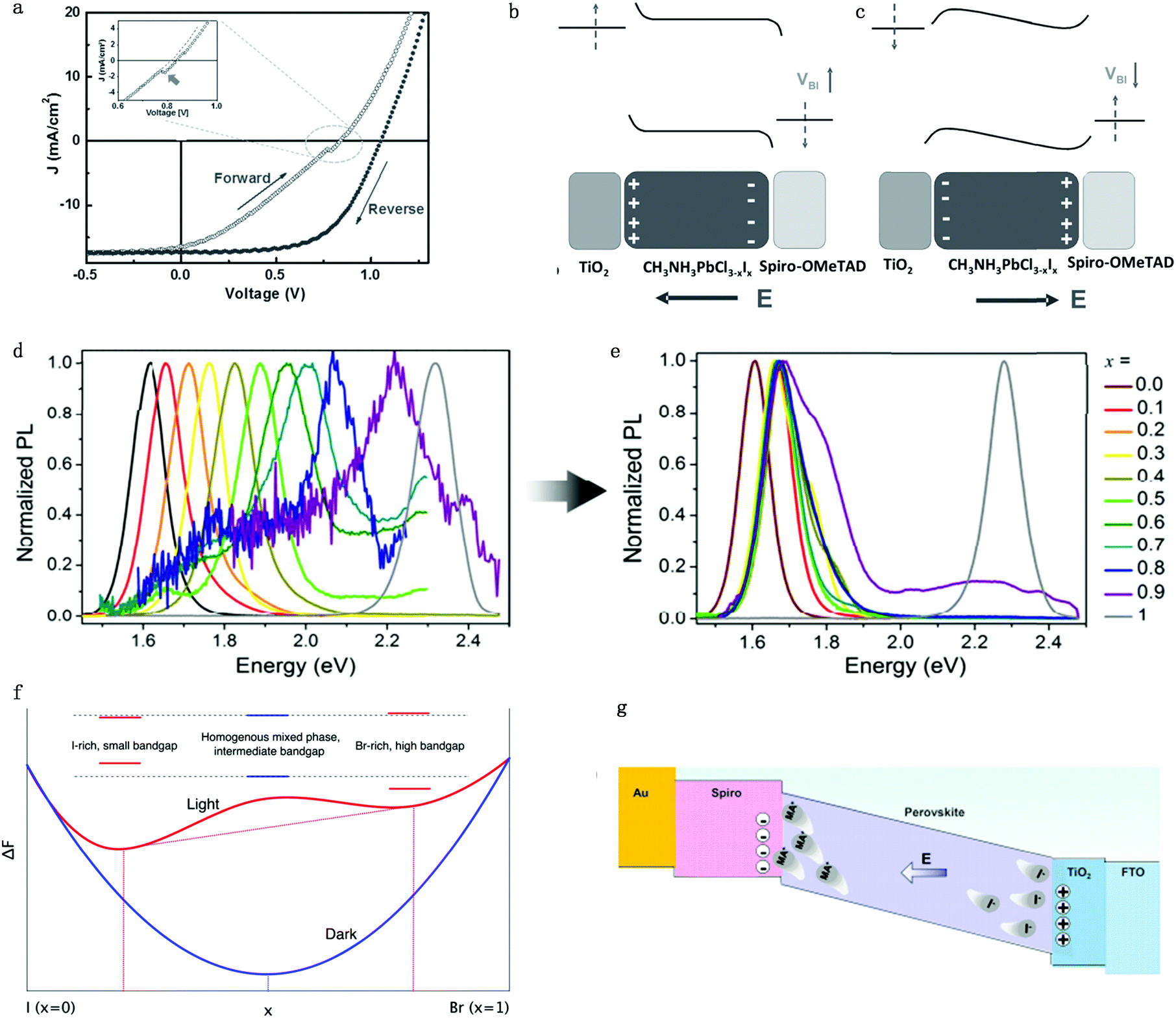 | ||
| Fig. 6 (a) Illumination of typical hysteresis I–V curve. Schematic of the electric energy band bending conditions under (b) position voltage bias and (c) negative voltage bias. Adapted with permission from ref. 61. Copyright 2016 John Wiley & Sons, Inc. (d and e) PL intensity for MAPb(BrxI1−x)3 with different x number before illumination (d) and after illumination (e). Adapted with permission from ref. 70. Copyright 2015 Royal Society of Chemistry. (f) Helmholtz free-energy for MAPb(BrxI1−x)3 with different x numbers under dark and light conditions. The red dashed lines show that the lowest attainable free energy in the light when the material segregates into I-rich and Br-rich phases. Adapted with permission from ref. 71. Copyright 2016 American Chemical Society. (g) Schematic illumination of the MA ion induced Spiro degradation mechanism under the electric field. Adapted with permission from ref. 72. Copyright 2017 American Chemical Society. | ||
For the n–i–p (TiO2–perovskite–Spiro-OMeTAD) structure planar devices, when applying a positive bias to PSCs, I− would move towards the perovskite/Spiro-OMeTAD side while leaving iodine vacancies at the TiO2 side, which leads to band bending within the perovskite layer itself and the two interfaces (Fig. 6b). The carrier injection and extraction efficiency would be improved by tunnelling through these narrow interfacial barriers and a positive charge accumulation effect would occur at the TiO2/perovskite interface. This would result in a compensating negative charge to accumulate at the TiO2 surface, which would further cause effective work functions of TiO2 and Spiro-OMeTAD shift upwards and downwards, respectively. Consequently, the built-in potential would increase whereas the local injection barriers at these interfaces would reduce due to local band bending. The open circuit voltage Voc and PCE of PSCs would be enhanced due to the improved charge transportation when applying reverse direction scan voltage on PSCs. However, when applying a negative bias to the n–i–p devices, an adverse effect would result in the formation of injection barriers at the two perovskite interfaces, which hinders charge injection/extraction efficiencies, which would lead to a decrease of Voc and PCE of PSCs (Fig. 6c). In summary, the variation of energy barrier under different voltage bias originating from migrating induced defects results in the hysteresis effect in PSCs.67
3.2 Phase segregation
Ion migration can cause stoichiometric polarization, which would further lead to phase segregation due to the Helmholtz free-energy equilibrium. Increasing the Br content in the MAPb(BrxI1−x)3 perovskite will commonly cause the increase of bandgap for perovskites, which leads to a corresponding increase in the open-circuit voltage.68,69 However, Hoke et al. showed that PCE degrades in mixed MAPb(BrxI1−x)3 perovskites solar cells because of a light induced halide phase segregation, that is, ion migration leads to the formation of smaller-bandgap and bigger-bandgap perovskite compositions and thus leads to the formation of trap states.70 As a matter of fact, for solar cells made from MAPb(BrxI1−x)3 (x > 0.2), they actually showed a reduced Voc along with the increasing bromide content, which originated from the inherent instability in the material introduced by interchanging X site halides. Initial photoluminescence (PL) measurements of MAPb(BrxI1−x)3 showed the expected simple increase in the bandgap from 1.6 to 2.3 eV with an increasing bromide content (Fig. 6d). However, the PL peak and intensity change with time: for MAPb(BrxI1−x)3 with x > 0.2, the pristine PL intensity slowly decreases while a lower-energy peak with much higher intensity appears after a few minutes of measurement (Fig. 6e). This newly appeared peak has almost the same energy for compositions in the range of 0.2 ≤ x < 1, corresponding to the initial PL peak energy of the x = 0.2 member, which is suggested in the x = 0.2 region; there exists a more stable phase and the segregated phase would fall into the composition of x = 0.2. However, the pure MAPbI3 or MAPbBr3 shows no obvious changes after illumination. The experimental results suggested that bromide and iodide ion migration leads to the phase segregation into higher bandgap Br-rich and lower-bandgap I-rich domains (high intensity) upon illumination.Since perovskites have relatively long carrier lifetimes and long carrier diffusion lengths, carriers would go across different crystallographic domains during the diffusion process and would be trapped upon encountering any I− rich low-bandgap region.6 The change in the band structure between a I− rich domain and the pristine composition perovskite could generate an electric field, helping carrier injection into the I− rich domain, which indicates that photoluminescence should come from the radiative relaxation of carriers trapped in I− rich regions, consistent with the results of photoluminescence measurement results. Moreover, Fig. 6f shows a schematic Helmholtz free-energy curves under the light and dark conditions as a function of energy band gaps of each phase.67 Light illumination will change the equilibrium of the Helmholtz free-energy state and thus induce two separate phases with stable quasi-equilibrium energy states.
3.3 Operational stability
On one hand, ion migration throughout the bulk perovskite layer would result in the stoichiometric polarization or even phase segregation, which would lead to the formation of iodine vacancies and iodine interstitials defects, which would cause the perovskite performance degradation. Moreover, light-enhanced ion migration can further lead to the photo-induced giant dielectric constant in the low frequency regime and the light-induced halide segregation in mixed perovskites as well as the photo-instability in perovskite-based devices. More ions tend to migrate toward the two sides of the device under illumination because of the reduced energy barrier and that the charge transfer is consequently suppressed by these excess defects.21,71 Therefore, the operational stability of PSCs is largely affected by ion migration under electric field and light illumination.On the other hand, ions would accumulate at the interface and even result in transmission of MA+ ions into the hole transportation layer (HTL) and further lead to the degradation of the HTL.72 Zhao et al.73 discovered that MA+ ions would come across the HTL (Spiro-OMeTAD) in planar n–i–p configuration during operation and cause the degradation of Spiro-OMeTAD (Fig. 6g). It is found from the mass spectra that MA+ signals could be detected in the HTL layer after the PSC is under operation for 20 h. The incorporation of MA+ ions into the HTL causes energy disorder and reduced conductivity in the Spiro-OMeTAD layer, resulting in rapid performance degradation of perovskite solar cells. Therefore, additional concerns regarding the operational stability of perovskite solar cells under illumination should be raised by the light-dependent ionic transport in MAPbI3.
4. Strategies to improve operational stability of PSCs
4.1 Importance of inorganic ions
Based on previous studies, we know that organic–inorganic hybrid perovskite materials exhibit the intrinsic light-enhanced ion migration effect under light and electric fields, for organic A site ions. However, this enhanced ion migration would be mitigated because Cs+ is not likely to protonize as MA+ does. This protonizing effect would cause the MA+ ion migration activation energy decrease by an order of magnitude while that of Cs+ remains invariant.26 Zhou et al.74 observed that Cs-based perovskite materials could suppress ion migration since Cs+ ions are not likely to exhibit the light-induced ion activation decrease effect from combined cryogenic galvanostatic and voltage–current measurements in the temperature range of 100–295 K. It is to be noted that the activation energy for CsPbI2Br is almost unchanged in the dark and under 20 mW cm−2 illumination from their experiments. However, for MAPbI3, the activation energy value is 0.824 and 0.144 eV, respectively. CsPbI2Br perovskite solar cells were fabricated and demonstrated more than 1500 h of operational stability under maximum power point (MPP) and light illumination due to light-independent ionic transport in inorganic perovskites. This work highlights the importance of inorganic ions in suppressing ion migration and improving the operational stability of PSCs.Based on above findings and understanding, a passivation strategy of using inorganic perovskites was put forward to increase the stability of PSCs by modifying the bulk organic-based perovskite by Cs-based perovskite materials. Zhou et al.75 managed to introduce CsPbBr3 into the Cs0.05FA0.80MA0.15Pb(I0.85Br0.15)3 (CsFAMA) precursor to form a quasi-core–shell perovskite structure to inhibit ion migration in PSCs (Fig. 7a). CsFAMA is the core and main material to absorb light while CsPbBr3 clusters act as the quasi shell located at the grain boundary of the CsFAMA grains. They successfully gained a synergetic effect of the two different perovskite materials. It is found that CsPbBr3 passivation greatly suppresses the ion migration-induced phase segregation in CsFAMA perovskites. The modified PSCs exhibit 20.6% PCE and 1.195 V open-circuit voltage, and could retain 90% of their initial performance at MPP under one sun illumination at 25 °C (65 °C) exceeding 500 h (100 h) of continuous operation. All in all, the Cs ion migration suppression effect was effectively utilized to improve the operational stability of PSCs without sacrificing the PCE of devices.
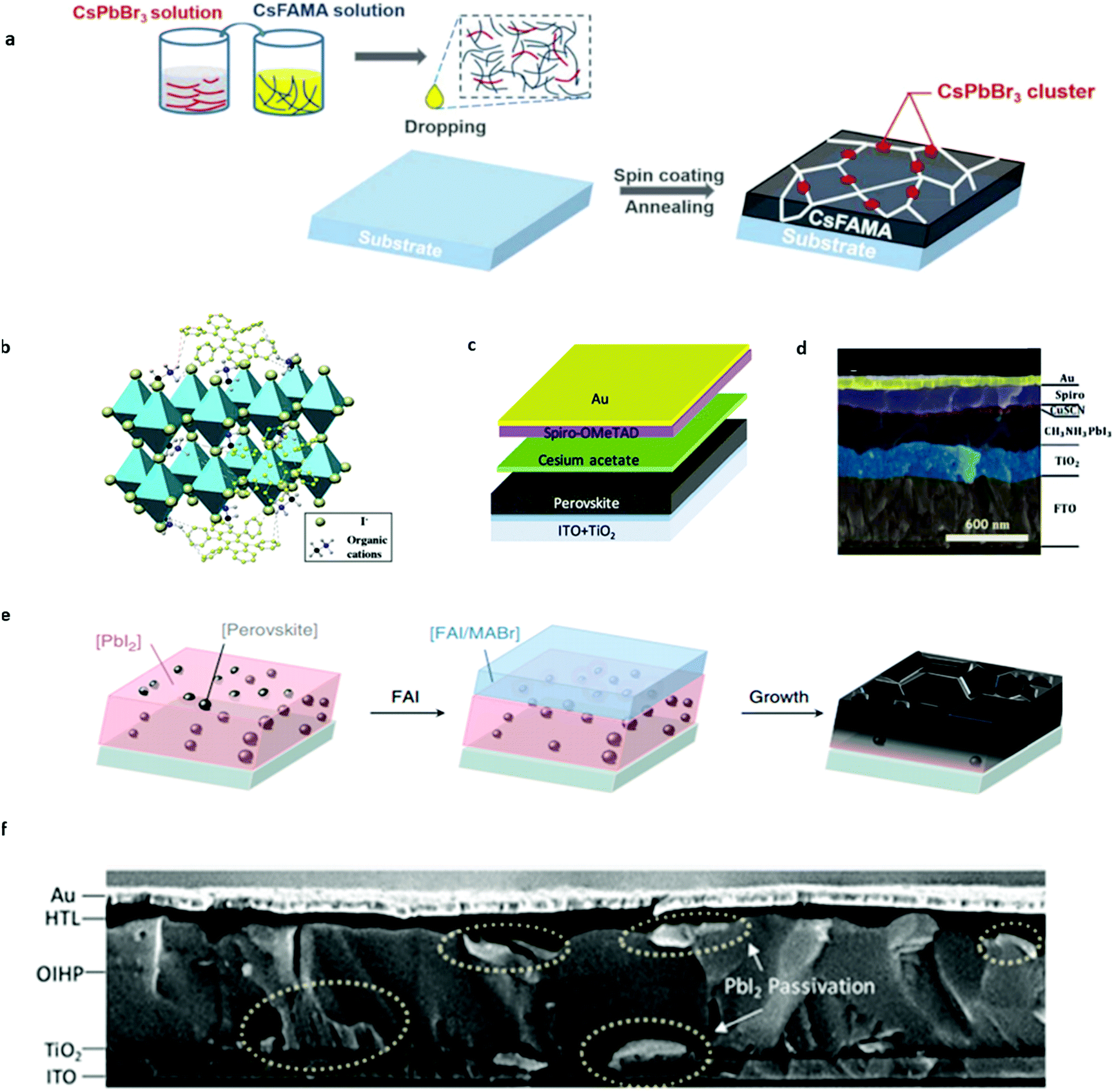 | ||
| Fig. 7 (a) Scheme of the device fabrication process of CsPbBr3 passivated perovskite films, which involves precursor solution preparation, subsequent spin coating and the final annealing step to form perovskite films. Adapted with permission from ref. 74. Copyright 2019 John Wiley & Sons, Inc. (b) Schematic illustration of the cation–π interaction between rubrene and organic cations of perovskites. Adapted with permission from ref. 77. Copyright 2018 John Wiley & Sons, Inc. (c) Schematic illustration of cesium acetate interfacial modification for PSCs. Adapted with permission from ref. 94. Copyright 2018 American Chemical Society. (d) Cross-sectional SEM image illustration of the CuSCN-Spiro-OMeTAD double layered hole transport layer for PSCs. Adapted with permission from ref. 95. Copyright 2017 Royal Society of Chemistry. (e) Schematic showing the perovskite seed-assisted growing method starting from perovskite seeds to perovskite crystallization. Adapted with permission from ref. 116. Copyright 2018 Springer Nature. (f) Cross-sectional SEM image of double-side passivated perovskite solar cells. Adapted with permission from ref. 117. Copyright 2019 John Wiley & Sons, Inc. | ||
Xu et al.76 suppressed ion migration via the Sb2S3 modification, which increased the energy barrier for migrating ions and improved the stability of PSCs. Cao et al.77 added extrinsic alkali cations (Rb+, K+, Na+, and Li+) into CsMAFA perovskite films, and found that the incorporation of proper size alkali cations can reduce the I–V hysteresis of PSCs, weaken the poling effects, and improve the photostability of wide-bandgap mixed-halide perovskites, all pointing to the suppressed ion migration.
4.2 Organic addition
Organic addition within the bulk perovskite material was found to have the ability of suppressing ion migration to increase the stability of PSCs. Wei et al.78 introduced the cation–π interaction rubrene compound into perovskite materials, which is effective for suppressing the migration of organic cations in PSCs (Fig. 7b). The binding energy of the cation–π interaction between the rubrene and perovskite is around 1.5 eV according to calculations, which is enough to immobilize the organic cations in PSCs. An obvious reduction of defects in perovskite films and the improvement of stability was observed in their PSCs. Time of flight secondary ion mass spectrometry (TOF-SIMS) results suggested that the CN− ion, which originates from the tracer compound, would penetrate into the PbI2 layer after operation for 72 h in pristine perovskite devices, while the modified devices shows no CN− signal, indicating the inhibited ion migration effect. The modified device can maintain its original PCE after 30 days of operation, much better than the control device. The much improved stability is attributed to the suppressed ion migration in perovskite films.4.3 Interface passivation
The migrated ions accumulating around interfaces (HTL/perovskite and ETL/perovskite) normally account for the fast degradation of PSCs. By interface passivation, the migrated ions would not likely to be trapped at the interface and thus the stability of PSCs can be improved. Tan et al.79 reported a contact-passivation strategy using a chlorine-capped TiO2 colloidal nanocrystal film that mitigates interfacial recombination and improves interface binding in low-temperature processed planar solar cells. The passivated solar cells have a certified PCE of 20.1% and retain 90% of their initial performance after 500 h of continuous room-temperature operation at MPP under 1 sun illumination.Based on Tan's job, Zhao et al.80 invented a water-based TiO2 nanoparticle dispersion method with chloroform ions attached on it by titanium tetrachloride hydrolysis. This method not only is environmentally friendly but also prevents the attachment of organic molecules onto TiO2 directly, which would induce more defects at the interface. The PSCs based on a water-based TiO2 ETL gained up to a 20.5% PCE with reduced hysteresis and maintained 80% of their initial performance after 500h of continuous operation under 1sun illumination at MPP. The much improved operational stability originates from the clean interface and Cl passivation, which would prevent I− ions from being trapped under operation and lead to the reduced defect concentration deduced from the impedance spectra. Moreover, π-conjugated Lewis based compound passivation, CuI passivation, PCBM passivation and Al2O3 passivation methods have also been proposed to reduce hysteresis and increase the stability of PSCs.81–84
In addition, suppression of ion penetration from the perovskite layer into the hole transport layer or substitution to the ion immune hole transport layer material is also very meaningful to the stability improvement of perovskite solar cells.78,85–94 Along this way of thinking, Zhao et al.95 incorporated cesium acetate as a buffer layer into PSCs to prevent organic ion migration into the Spiro-OMeTAD HTL of PSCs, in which cesium acetate is sandwiched between the perovskite and HTL (Fig. 7c). The mobile ions that migrate toward the organic transport layer (e.g., MA+) are gradually consumed by cesium acetate, resulting in cesium-rich perovskites at the interface. This in situ reaction and the subsequent Cs incorporation greatly enhance the operational stability of PSCs without efficiency loss. The optimized PSC presents a PCE of 20.9% with an open-circuit voltage of 1.18 V, maintaining ∼80% of its initial efficiency after 4500 min continuous operation at MPP.
Li et al.96 combined the CuSCN layer and Spiro-OMeTAD layer as the double hole transportation layer to prevent ion migration induced degradation in organic HTLs (Fig. 7d). The PSCs’ stability was significantly enhanced compared to those of single HTL layer devices. After applying double HTL configuration, PSC can maintain ∼90% of their initial PCE after 10 hours of continuous operation. They ascribed the long-term stability enhancement to the suppression of methylamine migration by the inorganic CuSCN layer. Guo et al.97 also developed a double HTL layer structure by combining conjugated polymer FBT-Th4 and copper oxide as the HTL and the resulting device gained a certified PCE of 22.1% with negligible hysteresis and improved stability for 500 h.
4.4 Grain boundary passivation or grain size management
Apart from the bulk perovskite materials, grain boundaries exhibit an even more intense ion migration effect.98–103 From the result of Yuan et al.,50 it can be deduced that the ion migration activation energy increases along with the increase in the crystal grain size, which suggested that disordered arrangement of molecules in grain boundaries would act as the starting point for ion migration. Shao et al.90 also found that ion migration in polycrystalline perovskites dominates through grain boundaries by atomic force microscopy measurements. They observed a stronger hysteresis both for photocurrent and dark current at the grain boundaries than in the crystalline grain, which is ascribed to faster ion migration at the grain boundaries due to the unordered arrangement of induced molecules thereby reducing ion activation energy. The enhanced ion migration results in the redistribution of ions along the grain boundaries after electric poling, in contrast to the intact grain area. The perovskite single-crystal devices without grain boundaries show negligible current hysteresis and no ion-migration signal. Therefore, increasing the grain size to decrease the grain boundary areas48,104–108 and direct passivation of the grain boundaries with passivation agents109–116 are both effective strategies toward better operational stability of PSCs.To increase the grain size, it is particularly important to control the nucleation density artificially. Most of the previous work focused on a random nucleation process, and few research studies have been carried out on the control of nucleation in the fabrication of perovskite films. Zhao et al.28 managed to obtain extremely large grain size perovskites and incorporate more Cs cations into perovskites simultaneously by a developed seed-assisted sequential deposition method. They introduced perovskite seeds into the PbI2 precursor solution in the first step and the perovskite seeds serve as cesium sources and act as nuclei to facilitate crystallization during the formation of perovskites (Fig. 7e). The seeding growth method does not need to overcome the nucleation free energy that needs to be overcome in random nucleation and could directly proceed to the seed crystal-induced growth in the second step. The grain size can be finely tuned by the added seed concentration into the PbI2 precursor solution. It is found that the seeding induced perovskite film has >3 μm grain size, much larger than the pristine perovskite films. Perovskite films with perovskite seeding growth exhibit a lowered trap density due to the decreased grain boundary. The resulting planar solar cells achieve a stabilized efficiency of 21.5% with a high open-circuit voltage of 1.13 V and a fill factor exceeding 80%. The seed induced perovskite devices also show significant improvement in operational stability by retaining 60% of their initial efficiency after 140 h of operation, much longer than other reported PSCs with the same composition. The significantly improved stability originates from Cs ion incorporation and much bigger grain size, thus further consolidating the positive effect of Cs addition and bigger grain size on the stability of PSCs.
As mentioned in previous discussion, direct passivation upon the grain boundary could also improve the performance of PSCs. The common method is to passivate the grain boundary at a single interface. Zhao et al.117 developed a simple way to passivate both sides of perovskite solar cells by PbI2. In the first step of the sequential deposition method, excessive lead iodide was added. After the perovskite film was prepared, IPA cleaning and long-term annealing were utilized to form PbI2 at the interface between the HTL/perovskite and ETL/perovskite interface (Fig. 7f). Moreover, PbI2 was also found to distribute at grain boundaries in perovskite films to act as effective passivators. The double-sided PbI2 passivated PSCs show negligible hysteresis, with a record potential loss of 0.38 V for 1.53 eV-bandgap perovskites due to the passivated grain boundaries.
5. Current challenges and outlook
In this review, we have overviewed the intrinsic factor that influences the operation stability of PSCs, ion migration. Ion migration originates from the loose perovskite structure and ionic conductor nature of the perovskite material, which will cause the hysteresis and degradation of PSCs. The characterization methods were summarized to identify ion migration in perovskite films, and then the influence of light on ion migration in perovskite materials was discussed. The effects of ion migration on the perovskite photovoltaic field were next summarized including hysteresis in I–V measurements, light-induced halide segregation and stability degradation of PSCs. Finally we have discussed several strategies to improve the operational stability of PSCs in terms of suppressing ion migration, such as the introduction of inorganic ions and organic molecules to immobilize ion migration, interface passivation, grain boundary passivation and grain size management.These solutions to PSCs’ stability are great, but are not enough for the commercialization of perovskite solar cells because of the strict stability requirement. It is necessary to inhibit the ion migration to ensure the stability of PSCs; however, some methods are too expensive or add highly toxic chemicals, which is actually not feasible for the mass production of PSCs. So, environmental protection and cost are very important factors that constrict the application of the modification method. Therefore, for example, when preparing the electron transport layer, it is better to choose nanoparticles that can be dispersed in water for spin coating. This is not only environmentally friendly, but also water can dissolve a variety of inorganic salts, which can be used as an additive for interface modification. Moreover, the use of spin coating methods to prepare the electron transport layer can reduce the traditional high temperature process, reduce the complexity of the fabrication and thus reduce the cost of production.
Moreover, thermal stability should be paid attention to, since the solar cell works under sunlight, where the temperature can reach 80 °C. Therefore, the thermal stability of the passivation additive should be considered. In addition, organic hole transport layer materials are easily damaged by MA ion migration, so a better method should not modify the interface between the hole transport layer and the perovskite light absorption layer, but should replace the organic hole transport layer with the inorganic one that is not sensitive to ion migration. Ligand engineering which has the ability to anchor ions or finding new effective passivators to make a more stable interface/grain boundary can be a very promising strategy to achieve the desired stability for PSCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program of China (No. 2019YFA0707003), the National Natural Science Foundation of China (NSFC 91733301 and 51872007) and Beijing Municipal Natural Science Foundation (No. 7202094).Notes and references
- J. R. Davis, A. Rohatgi, R. H. Hopkins, P. D. Blais, P. Rai-Choudhury, J. R. McCormick and H. C. Mollenkopf, IEEE Trans. Electron Devices, 1980, 27, 677–687 Search PubMed
.
- K. Masuko, M. Shigematsu, T. Hashiguchi, D. Fujishima, M. Kai, N. Yoshimura, T. Yamaguchi, Y. Ichihashi, T. Mishima, N. Matsubara, T. Yamanishi, T. Takahama, M. Taguchi, E. Maruyama and S. Okamoto, IEEE J. Photovolt., 2014, 4, 1433–1435 Search PubMed
- T. Dullweber and J. Schmidt, IEEE J. Photovolt., 2016, 6, 1366–1381 Search PubMed
- Z. Zhang, Y. Zeng, C.-S. Jiang, Y. Huang, M. Liao, H. Tong, M. Al-Jassim, P. Gao, C. Shou, X. Zhou, B. Yan and J. Ye, Sol. Energy Mater. Sol. Cells, 2018, 187, 113–122 CrossRef CAS
- E. M. Hutter, M. C. Gélvez-Rueda, A. Osherov, V. Bulović, F. C. Grozema, S. D. Stranks and T. J. Savenije, Nat. Mater., 2017, 16, 115–120 CrossRef CAS PubMed
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519 CrossRef CAS
- D. Meggiolaro, S. G. Motti, E. Mosconi, A. J. Barker, J. Ball, C. Andrea Riccardo Perini, F. Deschler, A. Petrozza and F. De Angelis, Energy Environ. Sci., 2018, 11, 702–713 RSC
- I. M. Reaney, E. L. Colla and N. Setter, Jpn. J. Appl. Phys., 1994, 33, 3984–3990 CrossRef CAS
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS PubMed
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed
- B. r.-c. e. c. w. n. g. p. a. p. b.-c.-e. p. National Renewable Energy Laboratory.
- J. S. Shaikh, N. S. Shaikh, S. S. Mali, J. V. Patil, K. K. Pawar, P. Kanjanaboos, C. K. Hong, J. H. Kim and P. S. Patil, Nanoscale, 2018, 10, 4987–5034 RSC
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS
- G. Divitini, S. Cacovich, F. Matteocci, L. Cinà, A. Di Carlo and C. Ducati, Nat. Energy, 2016, 1, 15012 CrossRef CAS
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu, P. M. Lahti, T. P. Russell, M. T. Tuominen and D. Venkataraman, J. Am. Chem. Soc., 2015, 137, 13130–13137 CrossRef CAS PubMed
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, J. Am. Chem. Soc., 2015, 137, 10048–10051 CrossRef CAS
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., 2014, 126, 3215–3221 CrossRef
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed
- Y. Zhao, W. Zhou, W. Ma, S. Meng, H. Li, J. Wei, R. Fu, K. Liu, D. Yu and Q. Zhao, ACS Energy Lett., 2016, 1, 266–272 CrossRef CAS
- Y.-C. Zhao, W.-K. Zhou, X. Zhou, K.-H. Liu, D.-P. Yu and Q. Zhao, Light: Sci. Appl., 2017, 6, e16243 CrossRef CAS PubMed
- A. Tumbul, F. Aslan, A. Göktaş and I. H. Mutlu, J. Alloys Compd., 2019, 781, 280–288 CrossRef CAS
- Y. Zhao, H. Tan, H. Yuan, Z. Yang, J. Z. Fan, J. Kim, O. Voznyy, X. Gong, L. N. Quan, C. S. Tan, J. Hofkens, D. Yu, Q. Zhao and E. H. Sargent, Nat. Commun., 2018, 9, 1607 CrossRef PubMed
- A. Göktaş, A. Tumbul and F. Aslan, J. Sol–Gel Sci. Technol., 2016, 78, 262–269 CrossRef
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. McDowell, P. Kanjanaboos, J.-P. Sun, X. Lan, L. N. Quan, D. H. Kim, I. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 7081 CrossRef CAS PubMed
- A. T. Barrows, A. J. Pearson, C. K. Kwak, A. D. F. Dunbar, A. R. Buckley and D. G. Lidzey, Energy Environ. Sci., 2014, 7, 2944–2950 RSC
- S. Das, B. Yang, G. Gu, P. C. Joshi, I. N. Ivanov, C. M. Rouleau, T. Aytug, D. B. Geohegan and K. Xiao, ACS Photonics, 2015, 2, 680–686 CrossRef CAS
- M. M. Tavakoli, L. Gu, Y. Gao, C. Reckmeier, J. He, A. L. Rogach, Y. Yao and Z. Fan, Sci. Rep., 2015, 5, 14083 CrossRef PubMed
- J. H. Kim, S. T. Williams, N. Cho, C.-C. Chueh and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1401229 CrossRef
- K. Cao, Z. Zuo, J. Cui, Y. Shen, T. Moehl, S. M. Zakeeruddin, M. Grätzel and M. Wang, Nano Energy, 2015, 17, 171–179 CrossRef CAS
- H.-S. Kim and N.-G. Park, J. Phys. Chem. Lett., 2014, 5, 2927–2934 CrossRef CAS PubMed
- C.-G. Wu, C.-H. Chiang, Z.-L. Tseng, M. K. Nazeeruddin, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2015, 8, 2725–2733 RSC
- H. Yoon, S. M. Kang, J.-K. Lee and M. Choi, Energy Environ. Sci., 2016, 9, 2262–2266 RSC
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed
- M. S. Islam, Solid State Ionics, 2002, 154–155, 75–85 CrossRef CAS
- J. Mizusaki, K. Arai and K. Fueki, Solid State Ionics, 1983, 11, 203–211 CrossRef CAS
- A. Walsh, C. R. A. Catlow, A. G. H. Smith, A. A. Sokol and S. M. Woodley, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 220301 CrossRef
-
D. Lan, Prog. Photovoltaics, 2019 Search PubMed
- J. Xing, Q. Wang, Q. Dong, Y. Yuan, Y. Fang and J. Huang, Phys. Chem. Chem. Phys., 2016, 18, 30484–30490 RSC
- H. Yuan, E. Debroye, K. Janssen, H. Naiki, C. Steuwe, G. Lu, M. Moris, E. Orgiu, H. Uji-i, F. De Schryver, P. Samorì, J. Hofkens and M. Roeffaers, J. Phys. Chem. Lett., 2016, 7, 561–566 CrossRef CAS PubMed
- R. S. Sanchez, V. Gonzalez-Pedro, J.-W. Lee, N.-G. Park, Y. S. Kang, I. Mora-Sero and J. Bisquert, J. Phys. Chem. Lett., 2014, 5, 2357–2363 CrossRef CAS PubMed
- A. Dualeh, T. Moehl, N. Tétreault, J. Teuscher, P. Gao, M. K. Nazeeruddin and M. Grätzel, ACS Nano, 2014, 8, 362–373 CrossRef CAS PubMed
- T.-Y. Yang, G. Gregori, N. Pellet, M. Grätzel and J. Maier, Angew. Chem., 2015, 127, 8016–8021 CrossRef
- Y. Yuan, J. Chae, Y. Shao, Q. Wang, Z. Xiao, A. Centrone and J. Huang, Adv. Energy Mater., 2015, 5, 1500615 CrossRef
- Y. Yuan, Q. Wang, Y. Shao, H. Lu, T. Li, A. Gruverman and J. Huang, Adv. Energy Mater., 2016, 6, 1501803 CrossRef
- C. Bischoff, K. Schuller, S. P. Beckman and S. W. Martin, Phys. Rev. Lett., 2012, 109, 075901 CrossRef CAS PubMed
- J. B. Goodenough, Rep. Prog. Phys., 2004, 67, 1915–1993 CrossRef CAS
- G. Y. Kim, A. Senocrate, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Nat. Mater., 2018, 17, 445–449 CrossRef CAS PubMed
- S. Meloni, G. Palermo, N. Ashari-Astani, M. Grätzel and U. Rothlisberger, J. Mater. Chem. A, 2016, 4, 15997–16002 RSC
- W.-J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef
- A. I. Popov, E. A. Kotomin and J. Maier, Solid State Ionics, 2017, 302, 3–6 CrossRef CAS
- S. Loftager, P. García-Fernández, J. A. Aramburu, M. Moreno and J. M. Garcia-Lastra, J. Phys. Chem. C, 2016, 120, 8509–8524 CrossRef CAS
- T. Brudevoll, E. A. Kotomin and N. E. Christensen, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 7731–7735 CrossRef CAS PubMed
- R. B. Murray and F. J. Keller, Phys. Rev., 1967, 153, 993–1000 CrossRef CAS
- D. J. Wilson, A. A. Sokol, S. A. French and C. R. A. Catlow, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 064115 CrossRef
- C. Li, S. Tscheuschner, F. Paulus, P. E. Hopkinson, J. Kießling, A. Köhler, Y. Vaynzof and S. Huettner, Adv. Mater., 2016, 28, 2446–2454 CrossRef CAS PubMed
- J. Wei, Y. Zhao, H. Li, G. Li, J. Pan, D. Xu, Q. Zhao and D. Yu, J. Phys. Chem. Lett., 2014, 5, 3937–3945 CrossRef CAS PubMed
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC
- W. Tress, N. Marinova, T. Moehl, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2015, 8, 995–1004 RSC
- B. C. O'Regan, P. R. Barnes, X. Li, C. Law, E. Palomares and J. M. Marin-Beloqui, J. Am. Chem. Soc., 2015, 137, 5087–5099 CrossRef
- D. Yang, R. Yang, K. Wang, C. Wu, X. Zhu, J. Feng, X. Ren, G. Fang, S. Priya and S. F. Liu, Nat. Commun., 2018, 9, 3239 CrossRef
- P. Calado, A. M. Telford, D. Bryant, X. Li, J. Nelson, B. C. O’Regan and P. R. F. Barnes, Nat. Commun., 2016, 7, 13831 CrossRef CAS PubMed
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC
- T. Leijtens, E. T. Hoke, G. Grancini, D. J. Slotcavage, G. E. Eperon, J. M. Ball, M. De Bastiani, A. R. Bowring, N. Martino, K. Wojciechowski, M. D. McGehee, H. J. Snaith and A. Petrozza, Adv. Energy Mater., 2015, 5, 1500962 CrossRef
- X. Xiao, J. Dai, Y. Fang, J. Zhao, X. Zheng, S. Tang, P. N. Rudd, X. C. Zeng and J. Huang, ACS Energy Lett., 2018, 3, 684–688 CrossRef CAS
- Y. Zhao, W. Zhou, H. Tan, R. Fu, Q. Li, F. Lin, D. Yu, G. Walters, E. H. Sargent and Q. Zhao, J. Phys. Chem. C, 2017, 121, 14517–14523 CrossRef CAS
- W. Zhou, Y. Zhao, X. Zhou, R. Fu, Q. Li, Y. Zhao, K. Liu, D. Yu and Q. Zhao, J. Phys. Chem. Lett., 2017, 8, 4122–4128 CrossRef CAS
- W. Zhou, S. Chen, Y. Zhao, Q. Li, Y. Zhao, R. Fu, D. Yu, P. Gao and Q. Zhao, Adv. Funct. Mater., 2019, 29, 1809180 CrossRef
- Z. Xu, L. Wang, Q. Han, Y. Kamata and T. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 12867–12873 CrossRef
- J. Cao, S. X. Tao, P. A. Bobbert, C.-P. Wong and N. Zhao, Adv. Mater., 2018, 30, 1707350 CrossRef PubMed
- D. Wei, F. Ma, R. Wang, S. Dou, P. Cui, H. Huang, J. Ji, E. Jia, X. Jia, S. Sajid, A. M. Elseman, L. Chu, Y. Li, B. Jiang, J. Qiao, Y. Yuan and M. Li, Adv. Mater., 2018, 30, 1707583 CrossRef PubMed
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722 CrossRef CAS PubMed
- Y. Zhao, Z. Han, W. Zhou, Q. Li, R. Fu, D. Yu and Q. Zhao, Sol. RRL, 2019, 3, 1900167 CrossRef
- Y. Lin, L. Shen, J. Dai, Y. Deng, Y. Wu, Y. Bai, X. Zheng, J. Wang, Y. Fang, H. Wei, W. Ma, X. C. Zeng, X. Zhan and J. Huang, Adv. Mater., 2017, 29, 1604545 CrossRef PubMed
- Y.-N. Zhang, B. Li, L.-Y. Zhang and L.-W. Yin, Sol. Energy Mater. Sol. Cells, 2017, 170, 187–196 CrossRef CAS
- J. Seo, S. Park, Y. Chan Kim, N. J. Jeon, J. H. Noh, S. C. Yoon and S. I. Seok, Energy Environ. Sci., 2014, 7, 2642–2646 RSC
- M. M. Byranvand, T. Kim, S. Song, G. Kang, S. U. Ryu and T. Park, Adv. Energy Mater., 2018, 8, 1702235 CrossRef
- P. Qin, S. Tanaka, S. Ito, N. Tetreault, K. Manabe, H. Nishino, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2014, 5, 3834 CrossRef CAS PubMed
- M. I. Hossain, F. H. Alharbi and N. Tabet, Sol. Energy, 2015, 120, 370–380 CrossRef CAS
- B. Abdollahi Nejand, V. Ahmadi and H. R. Shahverdi, ACS Appl. Mater. Interfaces, 2015, 7, 21807–21818 CrossRef CAS
- P.-K. Kung, M.-H. Li, P.-Y. Lin, Y.-H. Chiang, C.-R. Chan, T.-F. Guo and P. Chen, Adv. Mater. Interfaces, 2018, 5, 1800882 CrossRef
- J. A. Christians, R. C. M. Fung and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 758–764 CrossRef CAS
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC
- B. Abdollahi Nejand, P. Nazari, S. Gharibzadeh, V. Ahmadi and A. Moshaii, Chem. Commun., 2017, 53, 747–750 RSC
- Z. H. Bakr, Q. Wali, A. Fakharuddin, L. Schmidt-Mende, T. M. Brown and R. Jose, Nano Energy, 2017, 34, 271–305 CrossRef CAS
- S. Chatterjee and A. J. Pal, J. Phys. Chem. C, 2016, 120, 1428–1437 CrossRef CAS
- W. Sun, S. Ye, H. Rao, Y. Li, Z. Liu, L. Xiao, Z. Chen, Z. Bian and C. Huang, Nanoscale, 2016, 8, 15954–15960 RSC
- Y. Zhao, Y. Zhao, W. Zhou, Q. Li, R. Fu, D. Yu and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 33205–33213 CrossRef CAS PubMed
- Q. Li, Y. Zhao, R. Fu, W. Zhou, Y. Zhao, F. Lin, S. Liu, D. Yu and Q. Zhao, J. Mater. Chem. A, 2017, 5, 14881–14886 RSC
- Y. Guo, H. Lei, L. Xiong, B. Li and G. Fang, J. Mater. Chem. A, 2018, 6, 2157–2165 RSC
- T. Niu, J. Lu, R. Munir, J. Li, D. Barrit, X. Zhang, H. Hu, Z. Yang, A. Amassian, K. Zhao and S. Liu, Adv. Mater., 2018, 30, 1706576 CrossRef
- D.-Y. Son, J.-W. Lee, Y. J. Choi, I.-H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N.-G. Park, Nat. Energy, 2016, 1, 16081 CrossRef CAS
- T. Zhang, N. Guo, G. Li, X. Qian and Y. Zhao, Nano Energy, 2016, 26, 50–56 CrossRef CAS
- C. Bi, Q. Wang, Y. Shao, Y. Yuan, Z. Xiao and J. Huang, Nat. Commun., 2015, 6, 7747 CrossRef CAS PubMed
- W. Nie, H. Tsai, R. Asadpour, J.-C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H.-L. Wang and A. D. Mohite, Science, 2015, 347, 522 CrossRef CAS
- Y. Zong, Y. Zhou, Y. Zhang, Z. Li, L. Zhang, M.-G. Ju, M. Chen, S. Pang, X. C. Zeng and N. P. Padture, Chem, 2018, 4, 1404–1415 CAS
- H. Tsai, W. Nie, P. Cheruku, N. H. Mack, P. Xu, G. Gupta, A. D. Mohite and H.-L. Wang, Chem. Mater., 2015, 27, 5570–5576 CrossRef CAS
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58 CrossRef CAS PubMed
- N. D. Pham, V. T. Tiong, D. Yao, W. Martens, A. Guerrero, J. Bisquert and H. Wang, Nano Energy, 2017, 41, 476–487 CrossRef CAS
- M. Long, T. Zhang, W. Xu, X. Zeng, F. Xie, Q. Li, Z. Chen, F. Zhou, K. S. Wong, K. Yan and J. Xu, Adv. Energy Mater., 2017, 7, 1601882 CrossRef
- S. Li, P. Zhang, H. Chen, Y. Wang, D. Liu, J. Wu, H. Sarvari and Z. D. Chen, J. Power Sources, 2017, 342, 990–997 CrossRef CAS
- J. Xi, K. Xi, A. Sadhanala, K. H. L. Zhang, G. Li, H. Dong, T. Lei, F. Yuan, C. Ran, B. Jiao, P. R. Coxon, C. J. Harris, X. Hou, R. V. Kumar and Z. Wu, Nano Energy, 2019, 56, 741–750 CrossRef CAS
- C. Liu, Z. Huang, X. Hu, X. Meng, L. Huang, J. Xiong, L. Tan and Y. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 1909–1916 CrossRef CAS PubMed
- J. Cao, C. Li, X. Lv, X. Feng, R. Meng, Y. Wu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 11577–11580 CrossRef CAS PubMed
- A. A. Mamun, T. T. Ava, H. J. Jeong, M. S. Jeong and G. Namkoong, Phys. Chem. Chem. Phys., 2017, 19, 9143–9148 RSC
- P. Guo, Q. Ye, X. Yang, J. Zhang, F. Xu, D. Shchukin, B. Wei and H. Wang, J. Mater. Chem. A, 2019, 7, 2497–2506 RSC
- Y. Yan, W.-J. Yin, Y. Wu, T. Shi, N. R. Paudel, C. Li, J. Poplawsky, Z. Wang, J. Moseley, H. Guthrey, H. Moutinho, S. J. Pennycook and M. M. Al-Jassim, J. Appl. Phys., 2015, 117, 112807 CrossRef
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683 CrossRef CAS PubMed
- L. Liu, S. Huang, Y. Lu, P. Liu, Y. Zhao, C. Shi, S. Zhang, J. Wu, H. Zhong, M. Sui, H. Zhou, H. Jin, Y. Li and Q. Chen, Adv. Mater., 2018, 30, 1800544 CrossRef
- Y. Zhao, Q. Li, W. Zhou, Y. Hou, Y. Zhao, R. Fu, D. Yu, X. Liu and Q. Zhao, Sol. RRL, 2019, 3, 1800296 CrossRef
| This journal is © the Owner Societies 2021 |