
Chemical reactions on surfaces for applications in catalysis, gas sensing, adsorption-assisted desalination and Li-ion batteries: opportunities and challenges for surface science
Danil W.
Boukhvalov†
ab,
Valentina
Paolucci†
 c,
Gianluca
D’Olimpio
d,
Carlo
Cantalini
c and
Antonio
Politano
*de
c,
Gianluca
D’Olimpio
d,
Carlo
Cantalini
c and
Antonio
Politano
*de
aCollege of Science, Institute of Materials Physics and Chemistry, Nanjing Forestry University, Nanjing 210037, P. R. China
bTheoretical Physics and Applied Mathematics Department, Ural Federal University, Mira Street 19, 620002 Ekaterinburg, Russia
cDepartment of Industrial and Information Engineering and Economics, Via G. Gronchi 18, 7 University of L’Aquila, I-67100 L’Aquila, Italy
dDepartment of Physical and Chemical Sciences, University of L’Aquila, via Vetoio, 67100 L’Aquila, AQ, Italy. E-mail: antonio.politano@univaq.it
eCNR-IMM Istituto per la Microelettronica e Microsistemi, VIII Strada 5, I-95121 Catania, Italy
First published on 14th September 2020
Abstract
The study of chemical processes on solid surfaces is a powerful tool to discover novel physicochemical concepts with direct implications for processes based on chemical reactions at surfaces, largely exploited by industry. Recent upgrades of experimental tools and computational capabilities, as well as the advent of two-dimensional materials, have opened new opportunities and challenges for surface science. In this Perspective, we highlight recent advances in application fields strictly connected to novel concepts emerging in surface science. Specifically, we show for selected case-study examples that surface oxidation can be unexpectedly beneficial for improving the efficiency in electrocatalysis (the hydrogen evolution reaction and oxygen evolution reaction) and photocatalysis, as well as in gas sensing. Moreover, we discuss the adsorption-assisted mechanism in membrane distillation for seawater desalination, as well as the use of surface-science tools in the study of Li-ion batteries. In all these applications, surface-science methodologies (both experimental and theoretical) have unveiled new physicochemical processes, whose efficiency can be further tuned by controlling surface phenomena, thus paving the way for a new era for the investigation of surfaces and interfaces of nanomaterials. In addition, we discuss the role of surface scientists in contemporary condensed matter physics, taking as case-study examples specific controversial debates concerning unexpected phenomena emerging in nanosheets of layered materials, solved by adopting a surface-science approach.
1 Introduction
Chemical reactions at surfaces are the basis of heterogeneous catalysis,1,2 an industrial process exploited for the production of most chemicals.3–6 Definitely, the investigation of the adsorption of reactants on catalyst surfaces was the original goal of surface science, an interdisciplinary research field at the boundary between condensed matter physics, physical chemistry and materials science.7–14 In the last century, surface science has contributed to unraveling mechanisms ruling heterogeneous catalysis,15–18 starting with the pioneering work of Nobel Prize laureate Irving Langmuir on adsorption at surfaces19 up to the recent achievements of Nobel Prize laureate Gerhard Ertl, affording a detailed understanding of the surface chemistry of the Haber–Bosch process.20,21 The advent of surface science not only increased our fundamental understanding of the properties of condensed matter, but also significantly contributed to the description of complex technologically important phenomena, where the properties of surfaces play a major role. In particular, the investigation of interatomic interactions at surfaces by vibrational spectroscopies, such as high-resolution electron energy loss spectroscopy (HREELS)22–24 and inelastic helium atom scattering (HAS),25–37 was decisive in the identification of the physicochemical mechanisms governing chemical reactions at surfaces, i.e., adsorption, diffusion, combination of reactants, and desorption.38–40 Moreover, scanning tunneling microscopy (STM) enabled the manipulation of atoms and molecules on surfaces,41 with the fascinating prospect to trigger chemical reactions.42 Surface science has gradually transformed its original phenomenological characteristics into rational tailoring of materials and processes. Experimental methods have drastically improved in their resolution and capabilities, with the possibility to follow in real-time surface chemical reactions using both microscopic43,44 and spectroscopic45,46 tools. Similarly, the advent of operando techniques47–51 is abating the long-standing hurdle related to the “pressure gap”52 between in situ experiments in an ultrahigh vacuum and industrial catalytic processes.53 Correspondingly, theoretical models accomplished a huge leap with the unceasing upgrading of computational capabilities,54–58 including corrections for taking into account weak forces,59,60 now enabling the prediction of chemical properties of surfaces and interfaces.61–64The advent of two-dimensional (2D) materials had a dramatic impact on research of materials,65–71 so that most groups working on surface science moved their activities to match the current interests of the scientific community. Nevertheless, surface science should remain at the vanguard of both technological and scientific advances, also considering the higher relevance of surface phenomena in nanostructures.72 Though nanoscience inherently represents a giant opportunity and challenge for the surface-science community, actually the abundant amount of work produced so far on 2D materials often fails in reproducibility, owing to (i) the progressively reduced attention paid to experimental procedures to secure surface cleanliness (including environmental contamination73), (ii) the use of spectroscopic tools without surface sensitivity, and (iii) the predominance of poor-quality commercial samples.74 Therefore, the role of the surface-science community nowadays is particularly demanding. Explicitly, surface scientists should provide convincing and unambiguous proof of the necessity to adopt the rigid protocols of surface science to study surface-related processes on recently emerged nanomaterials, in order to afford their own contribution to avoid misleading interpretations.
Here, we provide a Perspective on recent developments of investigations of chemical processes on surfaces of novel 2D materials and their direct implications in application fields, such as catalysis, batteries, gas sensing and desalination. Especially, we highlight the possibilities arising from the formation of self-assembled heterostructures formed exploiting the natural interaction with air of surfaces of 2D materials or topological semimetals. Unexpectedly, surface oxidation is beneficial for both catalysis and gas sensing. Moreover, we define the role of surface science in application fields, such as distillation technologies for seawater desalination and Li-ion batteries.
2 The origin of the electrocatalytic performance in GaSe nanosheets: quantum size effects or surface oxidation?
The comprehension of electrocatalytic reactions had highly profited from the study of model systems with surface-science spectroscopies.75 Especially, the combination of in situ spectroscopic experiments with density functional theory calculations was decisive to establish (i) the mechanisms controlling the reaction paths and (ii) the influence of acidic and alkaline media.76Recently, exfoliation of GaSe in nanosheets has been shown to be beneficial for improving the performance of the hydrogen evolution reaction (HER),77 photoelectrochemical water splitting,78 and gas sensing.79 However, GaSe undergoes oxidation in ambient air with the formation of Ga2O3.80–88 Precisely, most researchers reported complete degradation of GaSe flakes with a thickness of 10–50 nm, although the time required for oxidation is quite controversial, ranging from 30 minutes82 up to some days (4 in ref. 89, 5 in ref. 90, 8 in ref. 91, and >14 in ref. 92). On the other hand, Rahaman et al.86 reported that the penetration of the oxidation has a self-limited depth of only three layers after five hours in air.
As evident from the analysis of X-ray photoelectron spectroscopy (XPS) data in Fig. 1b, GaSe is stable in a vacuum, while air exposure induces the emergence of spectral features related to the Ga+3 oxidation state in Ga2O3 for both the bulk87,93 and nanosheets.81,93 Definitely, oxidation proceeds with an intermediate Ga2Se3-like configuration, with the ultimate emergence of Ga2O3 for prolonged storage in air.93
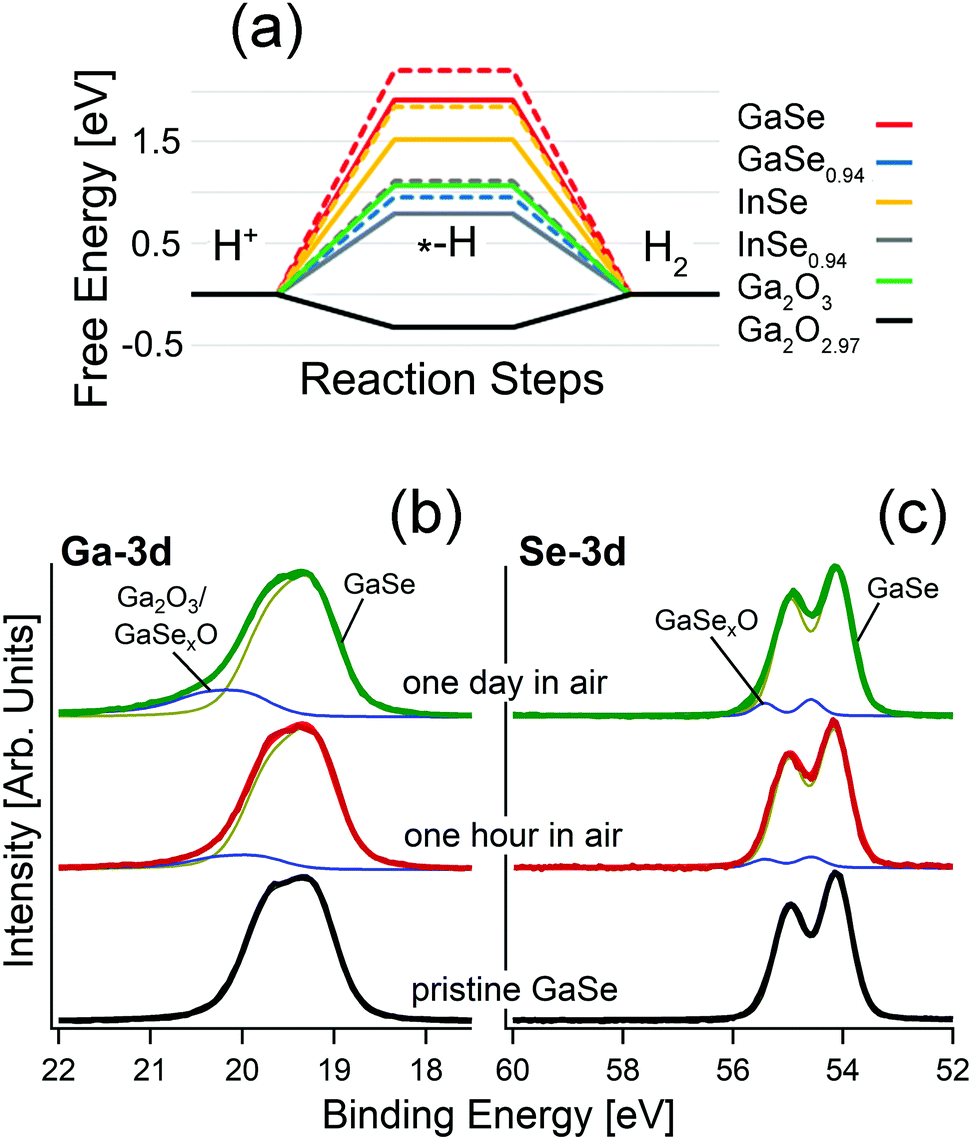 | ||
| Fig. 1 (a) Free energy diagram for the HER in acidic media over the surface of bulk and monolayer GaSe, GaSe0.94, InSe and InSe0.94 and for the HER over the surface of Ga2O3 and Ga2O2.97. Results for monolayers are depicted by dashed lines. Note that the line for the free energy for GaSe0.94 is overlapped with that associated with InSe0.94. Adapted with permission from data in ref. 93. Panels (b) and (c) report core-level spectra for (b) Ga-3d and (c) Se-3d for pristine GaSe and the same surface kept in air for one hour and one day, respectively. | ||
Actually, the incomplete picture of the physicochemical mechanisms ruling GaSe oxidation should be attributed to the insufficient surface sensitivity of the spectroscopic tools used so far. Indeed, the study of the oxidation state is usually performed using Raman spectroscopy,80,82 regardless of its low surface sensitivity, in consideration of its probing depth in the 300–650 nm range.94,95 Accordingly, the early steps of oxidation of GaSe-based systems, as well as their corresponding impact on surface chemical reactions in electrochemical, photocatalytic and gas-sensing processes, are generally undetected, owing to insufficient surface sensitivity.
Another important issue is related to the impact of the chemical reactivity of Se vacancies.96 Actually, most single crystals of GaSe are far from being stoichiometric. Typically, the [Ga]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Se] ratio ranges from 50.4:49.697 up to 59.1:40.978 even for nominal GaSe samples. Subsequently, the abundant amount of Se-vacancy sites could – at least in principle – play an important role in electrocatalytic and photocatalytic performances reported in ref. 78 and attributed to the exfoliation in nanosheets.
:[Se] ratio ranges from 50.4:49.697 up to 59.1:40.978 even for nominal GaSe samples. Subsequently, the abundant amount of Se-vacancy sites could – at least in principle – play an important role in electrocatalytic and photocatalytic performances reported in ref. 78 and attributed to the exfoliation in nanosheets.
Similar argumentation is also valid for the case of the parental compound InSe.93
Remarkably, calculated values of the free energies for the HER (Fig. 1a) evidence unsuitability of bulk GaSe and its parental compound InSe for this reaction, with energy barriers as high as 1.9 and 1.5 eV/H+, respectively. The presence of Se-vacancies in both GaSe and InSe significantly decreases the energy cost of the process (1.5 and 0.7 eV/H+, respectively), but the magnitude is still quite larger than for the Pt(111) surface (0.1 eV/H+), usually taken as a standard reference (Fig. 1a).98 In the case of free-standing monolayers, the values of the energy cost of the HER are even larger: 2.2 and 1.8 eV for GaSe and InSe, respectively. Therefore, the common picture that liquid-phase exfoliation of GaSe and InSe favors the improvement of the HER performance on the basis of the higher amounts of edges behaving as active sites, recently proposed in ref. 78 and 99, should be revised. On the contrary, the calculated values for Ga2O3 and the sub-stoichiometric oxide Ga2O2.97 are 1.1 and −0.3 eV/H+. Therefore, for Ga2O2.97 the Heyrovsky step (Hads + H+ + e− → H2) of the HER is exothermic.
In the case of the oxygen evolution reaction (OER), in both acidic and alkali media, the second step of the OER, which involves the formation of –O groups on the substrate, has highly negative values of free energy (−5.7 and −7.4 eV for GaSe and GaSe0.94), owing to the favorability of GaSe oxidation. Thus, the surface of bulk GaSe will be irreversibly oxidized during the OER in both acidic and alkali environments.93
Therefore, the literature data on electrocatalysis and photocatalysis based on GaSe should be re-interpreted. Especially, we note that, in the survey of the electrochemical properties of GaSe by Tan et al.,100 the authors assume from XPS analysis that the surface composition of GaSe is more complex with respect to the theoretical one, considering the non-stochiometric [Ga]:[Se] ratio and the presence of oxygen contaminants. The lowest Tafel slope (150 mV dec−1) in the HER was found in nominally reduced GaSe, for which the authors claimed the removal of the passivation layer of Ga2O3, supposed to poison active sites of the GaSe surface. However, surface treatments such as those in ref. 100 are expected to promote the formation of sub-stoichiometric Ga2O3−x phases, which represent a good platform for the HER.
Recently, liquid-phase exfoliated GaSe nanosheets have been used78 to produce photoelectrodes exhibiting catalytic activity toward water splitting reactions, i.e., the HER and OER. Based on the results in Fig. 1a, it is evident that the role of GaSe in photochemical water splitting is just to generate electron–hole pairs via light harvesting, while the Ga2O3 skin represents the only chemically active part of the photoelectrode. On the other hand, the presence of Ga2O3 in GaSe samples in ref. 78 can be clearly identified from the analysis of Raman, ultraviolet photoemission spectroscopy (UPS) and XPS experiments, with oxidation particularly favored by the largely non-stoichiometric [Ga]:[Se] ratio of 59.1:40.9.
3 The origin of the HER performance in PtSn4: topological states or surface oxidation?
The connection between topologically nontrivial electronic states and catalytic activity has been recently claimed by different groups.101–106 Among various topological materials, Pt-based topological systems are particularly relevant for catalytic applications, due to the wide use of Pt in several catalytic reactions, including the HER107 and OER.108 Recently, it has been reported that PtSn4 has outstanding performance in the HER,104 ascribed to (i) the existence of topological Dirac node arcs reported in ref. 109 and (ii) the supposed occurrence of a Pt skin, i.e. an atomic Pt layer as surface termination, which is just assumed by the authors to exist in analogy with other Pt-based alloys.110–112Considering the standard pathway of the HER, i.e. the (i) Volmer (H+ + e− → Hads), and (ii) Heyrovsky (Hads + H+ + e− → H2) steps, theoretical results (Fig. 2a) demonstrate that the energy cost of the Heyrovsky step is rather large for both Pt- and Sn-terminated PtSn4 surfaces (0.75 and 0.84 eV), in contrast with experimental results.104
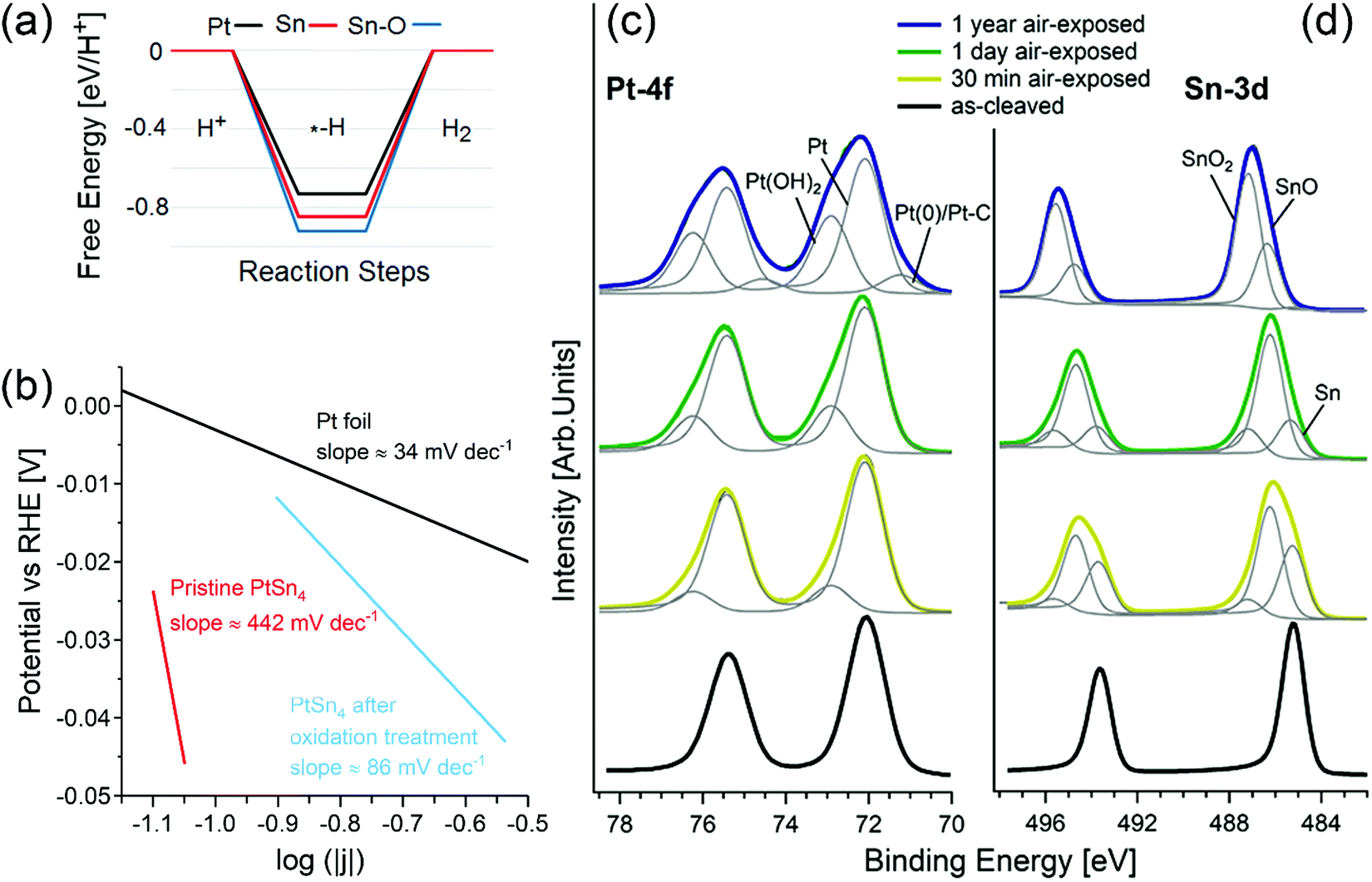 | ||
| Fig. 2 (a) Free energy diagrams for the HER on pristine, oxidized Pt-terminated and Sn-terminated surfaces of PtSn4. (b) Tafel plot of pristine PtSn4 and after the treatment at 1.035 V vs. RHE, registered at 1 mV s−1. The reference curve for Pt is also reported. Panels (c) and (d) show core-level spectra for (c) Pt-4f and (d) Sn-3d for pristine PtSn4 and the same surface kept in air for 30 min, 24 h, and one year. The photon energy is 800 eV and the spectra are normalized to the maximum. Adapted with permission from ref. 113. | ||
A completely different scenario is enabled by surface oxidation. Given the combination of the (i) Volmer and (ii) Tafel (2Hads → H2) steps, in the case of an oxidized Sn-terminated surface, hydrogen atoms bond to oxygen atoms with the formation of hydroxyl groups. In this process, the Tafel step does not represent the migration of the unbound adatom on the surface, but instead it is related to the migration of hydrogen from one oxygen atom to another. Considering (i) that the distance between oxygen atoms in the oxidized Sn-terminated surface is about 0.2 nm (less than in any other metal) and (ii) the existence of charge transfer from the Pt-sublayer, the energy cost of hydrogen migration results to be only 0.2 eV, i.e., comparable with the energy cost of the Heyrovsky reaction for Pt(111). Thus, one can conclude that the contribution from topological states in the electro-catalytic performance of PtSn4 is negligible,113,114 while surface oxidation plays an unexpectedly beneficial role for the HER.113,114
Correspondingly, the Tafel slope measured in electrocatalytic tests is reduced from ∼442 to ∼86 mV dec−1 upon oxidation treatment (Fig. 2b).
XPS experiments (Fig. 2c and d) confirm the transformation of the surface upon air exposure with two spectral components appearing in the Sn-3d core-level spectra (Fig. 2d), arising from SnO (binding energy, BE, of ∼486.2 eV) and SnO2 (BE of ∼487.2 eV) species.115 Especially, the SnO2 component (Fig. 2b) gradually increases from 5% (after 30 min in air) to 13% (after 24 h in air) and finally to 65% (after 1 year in air) with the corresponding disappearance of Sn(0) species. On the other hand, the spectrum of Pt-4f core levels (Fig. 2c) acquired for the PtSn4 surface kept 30 minutes in air shows a new component at BE 72.95 eV ascribable to Pt(OH)2116,117 or Pt–O units115 in an early stage oxidation of Pt atoms. Correspondingly, the thickness of the tin-oxide layer (SnO + SnO2) formed on the sample left in an ambient atmosphere increases from 9 ± 1 Å (30 min in air) up to 36 ± 5 Å (1 year in air).
4 Materials engineering and photocatalysis: the role of surface science
Surface science gave a huge contribution in the comprehension of bulk, surface and interfacial phenomena in photocatalysis.118,119 A typical application of photocatalysis is related to solar energy harnessing,120 which uses semiconductors for solar energy conversion into chemical energy,121 promoting redox reactions at over-surface catalytic sites by means of the separation of photogenerated electron–hole pairs.122 Although a large variety of photocatalysts were explored,123–125 some crucial drawbacks typically drop down their performances. For example, the light absorption provided by the most studied photocatalytic semiconductor, TiO2, is limited in the range of the ultraviolet spectrum, owing to its bandgap of 3.0–3.4 eV.126 Moreover, another typical problem arising from traditional photocatalysts is represented by the recombination of electron–hole pairs during charge transport.127 It can be related either to the low mobility of photoinduced carriers or to the charge carriers not being consumed by surface reactions, because of the low number of catalytic sites at the surface.128Especially, photocatalysis with 2D materials has attracted considerable interest for their peculiar physicochemical properties,129–131 considering the high surface-to-volume ratio of nanosheets, with intrinsically high amounts of active sites for catalytic reactions. Moreover, when the thickness is reduced to the nanoscale, the photo-induced charge carriers can move from the bulk to the surface in a short distance. This reduction in traveling length inhibits the electron–hole recombination during charge transport and allows more charge carriers to be preserved for surface reactions.
Nevertheless, the interpretation of the mechanisms governing the photocatalytic activities was traditionally limited to electrochemical properties and band-structure models.132 On the other hand, the use of surface-science techniques could provide useful insights for a detailed comprehension of the photocatalytic processes.133 Researchers focused their attention on the modification of 2D materials for improving the photocatalytic efficiency, including the design of the structure and morphology,134,135 doping and vacancy engineering,136 and integration with other semiconductors or metals.137,138 Surface science can play a pivotal role in terms of the analysis of photoactive sites and to unveil reaction mechanisms and intermediates, as well as the optimal operational conditions to get the final conversion.118
Recently, several studies have been published regarding the influence of the controlled oxidation of transition-metal dichalcogenides on photocatalysis, especially in the field of solar energy production.139–141 In particular, Patra et al.141 synthesized a MoS2/GO (with GO being graphene oxide) composite by a hydrothermal process and with subsequent MoS2 oxidation by calcination at different temperatures under an N2 atmosphere, which promotes the diffusion of atomic oxygen from the GO layer to the 2D MoS2 layer, resulting in a MoO3−x/MoS2 heterojunction, with the non-stoichiometric 2D MoO3−x exhibiting a surface plasmon resonance (SPR) with a frequency matching the solar spectrum.142 The observed enhanced photocatalytic activity is ascribed to the oxidation of Mo4+ cations of MoS2 to higher oxidation states (Mo5+ and Mo6+ in MoO3−x), which also modifies the valence-band (VB) electronic structure and the work function of the MoO3−x/MoS2/rGO composite (with rGO being reduced GO), as demonstrated by XPS and UPS experiments. The presence of 2D MoO3−x layers with a large amount of Mo5+ oxidation states allows easier carrier generation due to SPR generation, while MoS2 offers active sites for catalysis (Fig. 3). Solar H2 generation (SHG) was proved in the near-infrared and the visible region by combining plasmonic and catalytic effects in the composite.
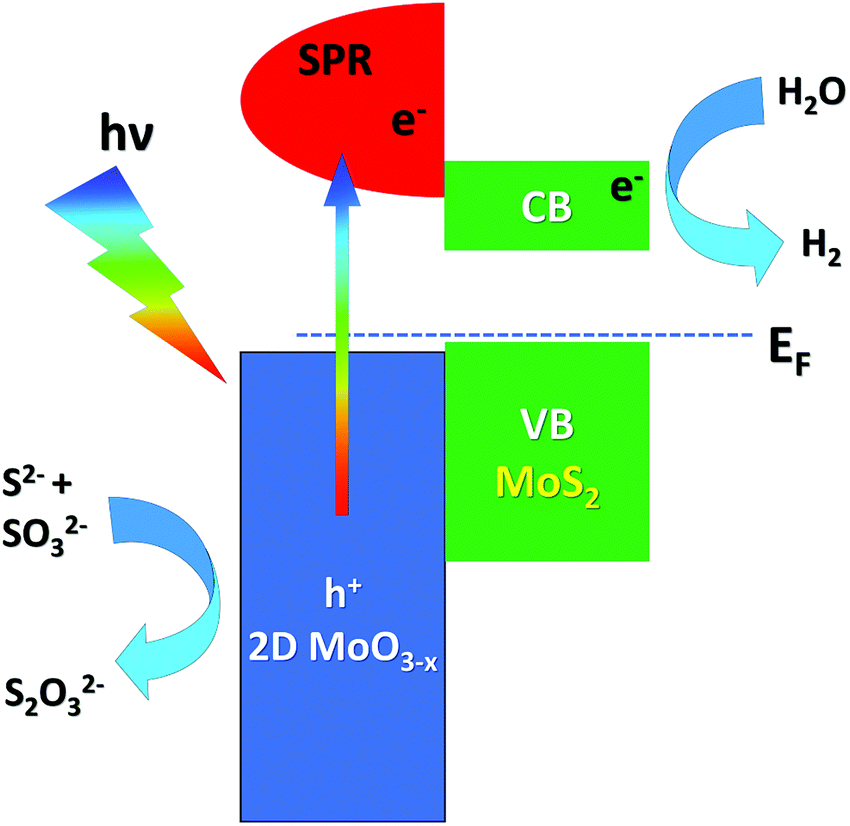 | ||
| Fig. 3 Energy-level alignment and the mechanism of electron transfer at the MoS2/MoO3 interface. Adapted with permission ref. 141. CB and VB stand for the conduction and valence band, respectively. | ||
5 Gas sensing in van der Waals semiconductors: the role of surface oxidation
Gas sensors for environmental detection of sub-ppb concentrations of noxious gas like CO, NO2, H2S, and H2 are based on catalytic materials, which modulate their electrical properties, due to complex redox reactions taking place between the target gas and the surface termination of the investigated sensing materials.143 Van der Waals semiconductors promise new perspectives for gas sensing applications beyond state-of-the-art porous metal–oxide sensors,144 due to their (i) high surface-to-volume ratio, (ii) intrinsically large amount of active sites for catalytic reactions, (iii) no residual porosity and (iv) reduced charge transfer travelling length, which enhance the amount of charge carriers for surface reactions.145 As for metal–oxide sensors,144 van der Waals semiconductor-based sensors are generally heated at mild operating temperatures (T < 150 °C) to avoid irreversible desorption of the target gas at low temperature, leading to surface oxidation in the case of metal chalcogenides, including transition-metal dichalcogenides.146 Remarkably, this issue has been regarded so far as a significant drawback, limiting the application of van der Waals semiconductor-based sensors. However, unexpectedly, the spontaneous oxidation of metal chalcogenides147 opens new outlooks for gas sensing applications. Here, we focus on the case-study example of tin diselenide (SnSe2), a van der Waals semiconductor with high electron mobility (462.6 cm2 V−1 s−1 at T = 300 K148), which has been recently reported to be a suitable material for the detection of NO2149,150 and NH3,151,152 in a nitrogen background carrier gas, as well as CH4153 and humidity in dry air.151,152 The presence of Se vacancies triggers oxidation of the surface in oxidative environments (including air), with the emergence of a SnO2 surface layer, with sub-nanometric thickness,154 whose presence is evident in HREELS analysis (Fig. 4). The formed SnO2 skin is amorphous, as indicated by the disappearance of diffraction peaks in the low-energy electron diffraction pattern.154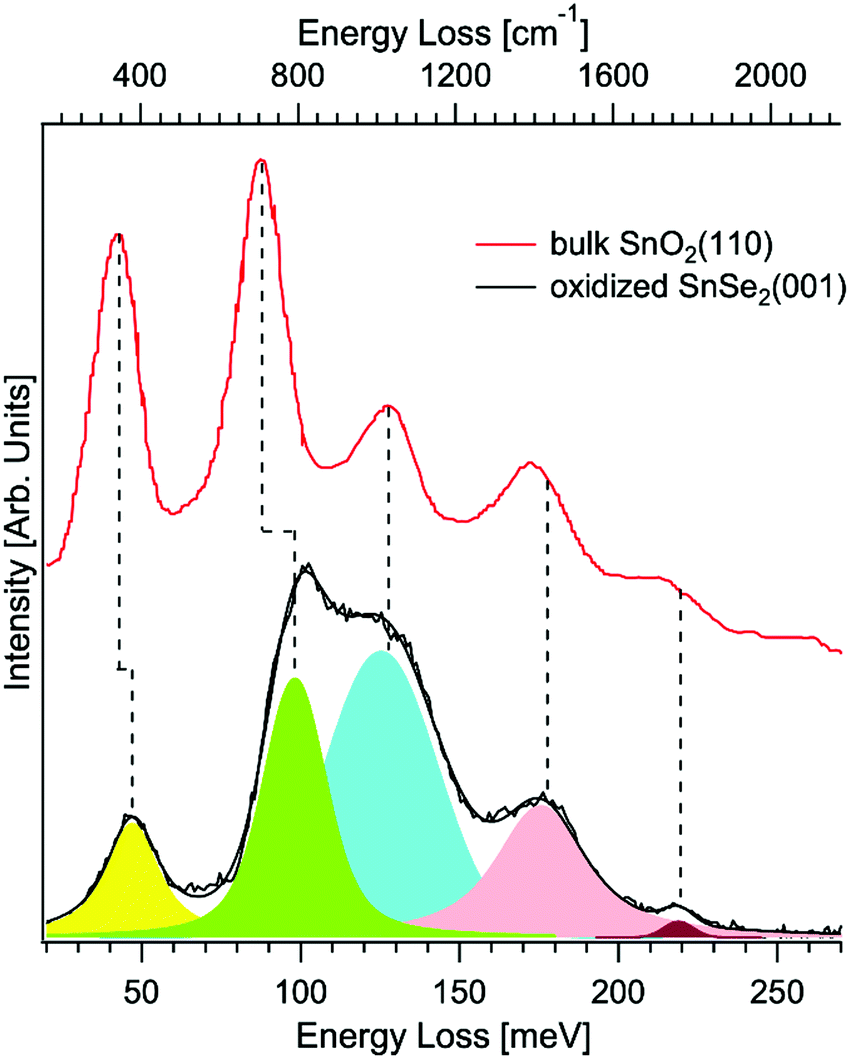 | ||
| Fig. 4 Vibrational spectra for the oxidized surface of SnSe2(001) taken by HREELS. The primary electron beam energy is 4 eV. Experiments were carried out at room temperature. For the sake of comparison, we report also data for bulk SnO2(110) (data taken from ref. 155). Reproduced with permission ref. 154. | ||
Density functional theory results indicate that SnO2/SnSe2 is more sensitive to chemisorbed species with respect to the pristine SnSe2.154 As a matter of fact, H2O, NO2 and H2 adsorption yield no substantial effect on both the electronic structure and charge densities (Fig. 5) of pristine SnSe2, which turns out to be unsuitable for gas sensing applications. Conversely, adsorption of H2O and NO2 molecules on the SnO2/SnSe2 heterostructure shows opposite effects on the electronic structure and charge densities, thus validating the SnO2/SnSe2 heterostructure for humidity and gas sensing applications.
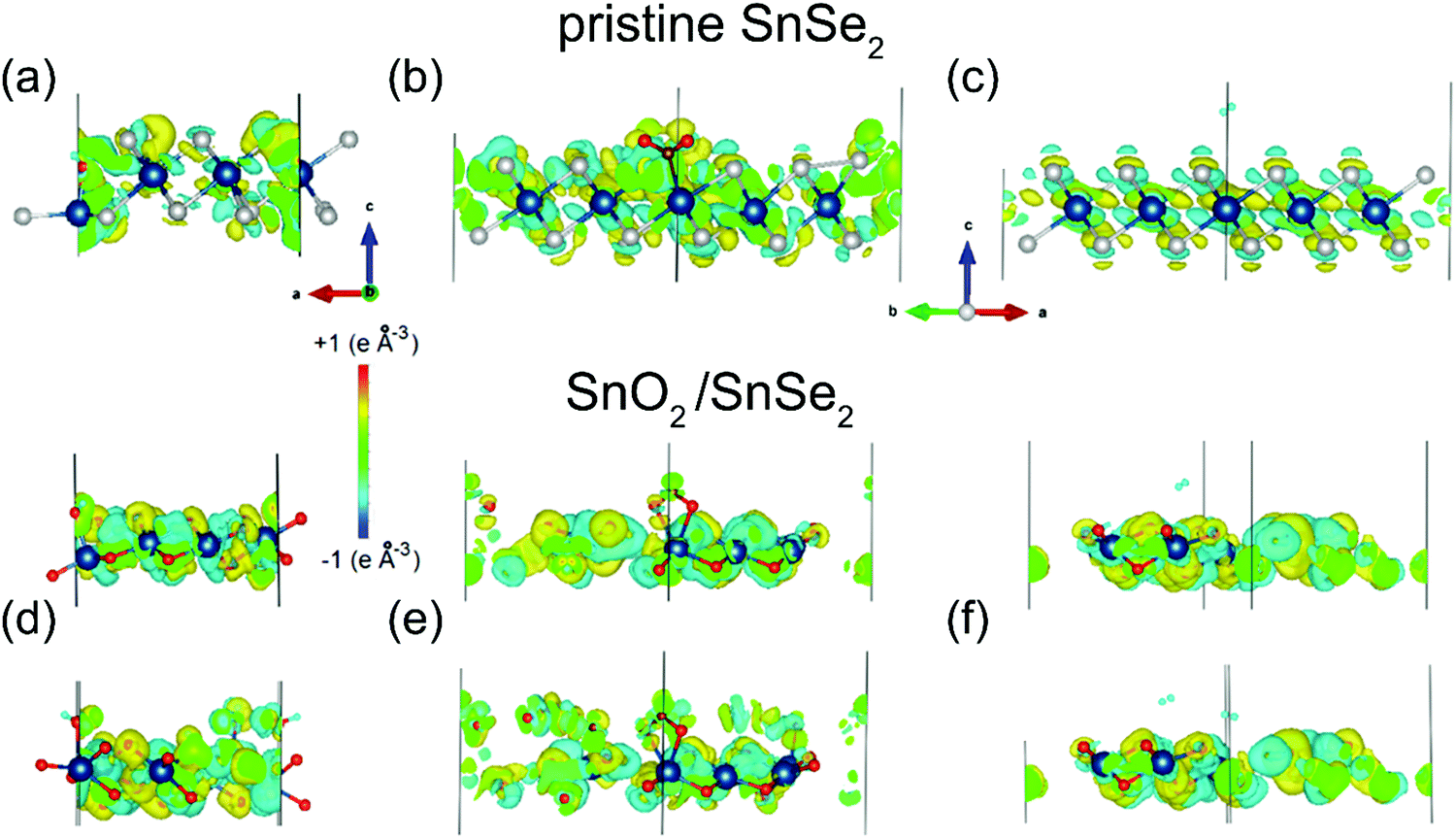 | ||
| Fig. 5 Changes in the charge densities in (a–c) SnSe2 or (d–f) SnO2 surfaces after adsorption of (a and d) water, (b and e) nitrogen dioxide and (c and f) hydrogen. For the SnO2 skin, results for adsorption of a single molecule are shown in the top layer and for the pair of molecules in the bottom layers of the panels. Blue, white and red balls denote Sn, Se, and O atoms, respectively. Reproduced with permission ref. 154. | ||
To validate the theoretical calculations, the SnO2/SnSe2 heterostructure was exposed to NO2 (400 ppb–1.5 ppm range) and H2 (5–100 ppm range) under a dry air carrier gas (to simulate conditions for environmental sensing) at an operational temperature of 150 °C (Fig. 6), which enables complete and fast baseline recovery after gas exposure/release cycles. Specifically, SnO2/SnSe2 shows detection limits of 400 ppb and 5 ppm with sensitivities of (1.06 ± 0.03) and (0.43 ± 0.02) [ppm]−1 for NO2 and H2, respectively.154
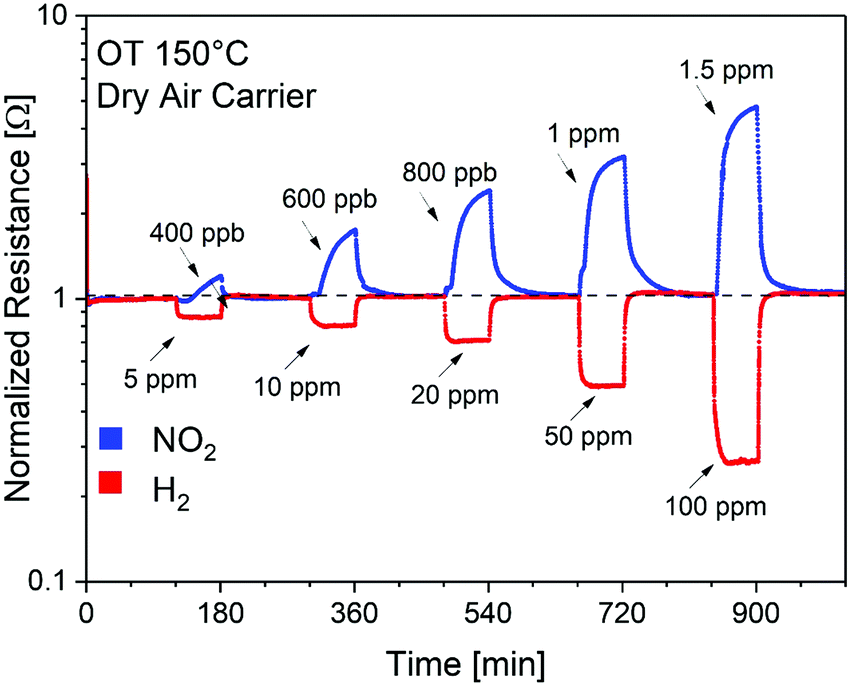 | ||
| Fig. 6 Dynamic gas responses of the SnO2/SnSe2 heterostructure for gas concentrations of NO2 (400 ppb–1.5 ppm) and H2 (5–100 ppm) in a dry air carrier gas at an operational temperature of 150 °C. OT stands for operational temperature. Reproduced with permission ref. 154. | ||
6 Adsorption-assisted desalination: membrane technology meets surface science through adsorption
Here, we will highlight the relevance of surface phenomena in emerging desalination techniques. Freshwater availability is one of the major challenges that our society is facing, due to the progressive demographic expansion, climate change and desertification.156 Correspondingly, the World Health Organization (WHO) evidenced an alarming scenario: ca. 50% of the world's population will be living in water-stressed regions by 2025.157 Evidently, seawater desalination represents the most reliable and economically sustainable way to produce drinking and reusable water, considering that seawater constitutes more than 97% (∼1.4 × 109 km3) of the total water resources.158–160 Among the different existing desalination technologies, it is possible to differentiate processes based on (i) evaporation (phase change) and (ii) membranes (non-phase change).161 However, the widespread applications of these desalination processes, such as multi-effect distillation (MED) and seawater reverse osmosis (SWRO), require significant energy power, thus limiting their viability in developed countries. In recent years, membrane distillation (MD) has emerged as a promising alternative or as a complementary unit to SWRO. MD is a thermally-driven membrane operation based on the use of microporous hydrophobic membranes.162 The hydro-repellent nature of the membrane prevents the permeation of liquids, while sustaining a vapour–liquid interface at the entrance of each pore, where water evaporation takes place. Under a temperature gradient, a net diffusive flux of vapour is observed from the hot feed (Fig. 7a). Here, we take graphene as a case-study example to highlight the role of surface science in the adsorption-assisted MD in polyvinylidene fluoride (PVDF) membranes with graphene fillers.163 Explicitly, penetration and diffusion of water molecules through free gaps between graphene platelets and the polymer take place via intermolecular interactions at defect sites (Fig. 7b and c). On the other hand, the local arrangement of platelets can affect nearby interactions, causing increased or reduced vapour diffusion. Even with a low amount of filler in the polymer matrix, adhesive interactions are activated at the water/graphene platelet interface, resulting in assisted vapour transport. A further increase in the concentration of the filler can cause massive water uptake over the space between the graphene platelets and the polymer network, thus resulting in potential competition between adhesive and cohesive forces, which causes the penetrant to extend its permanence inside of the membrane. This hypothesis is verified by the occurrence of a transient state in each experiment in ref. 163, carried out with membranes at different loading of graphene platelets. When the concentration of graphene platelets is significantly increased, a longer time is required for collecting water at the permeate side. Even four running hours are required in ref. 163 to observe the onset of permeation of water vapour through the membrane, thus suggesting a longer permanence of the penetrant inside the matrix. A reasonable explanation could arise from the occurrence of intermolecular interactions (Fig. 7c), assisting water to form a wetting layer covering the free gaps between the polymer and the graphene platelets. Consequently, one can conclude that intermolecular water–graphene interactions may assist or inhibit diffusion of water molecules through the membranes depending on the concentration of graphene platelets. Based on experimental findings,163 a graphene concentration of 0.5% is able to promote well-balanced kinetics of adsorption–desorption of the penetrant, leading to assisted transport. A massive concentration of graphene platelets yields a considerable amount of stagnant water, resulting in slowed down transport. As a result, graphene platelets could bring benefits to water vapour transport through membranes equipped in MD plants when their amount and rearrangement in the matrix are well equilibrated. | ||
| Fig. 7 (a) Schematic illustration of the membrane distillation process. Water vapour passes through the pores of a hydrophobic microporous membrane. (b) Representative SEM micrographs of the cross section of PVDF–graphene composite membranes. Reproduced with permission ref. 163. (c) Schematic representation of the working mechanisms of adsorption-assisted MD in PVDF–graphene nanocomposite membranes. Reproduced with permission ref. 164. | ||
7 A surface science approach for Li-ion batteries
Li-ion batteries generate and store electric power from the electrochemical redox reactions between the electrode materials. In Li-ion batteries, the interface between the active material constituting the electrode and the electrolyte is particularly relevant, due to the transfer of lithium ions causing the electrode redox reaction.165 Especially, the chemical reactions and charge transfer at cathode/electrolyte interfaces influence the performance and the stability of Li-ion cells. Definitely, the corrosion of the active electrode and the decomposition of the electrolyte are closely coupled to charge transfer reactions at the electrode/electrolyte interfaces, which are connected to the energy barriers for electrons and ions.The comprehension of the surface structure, the electronic structure, and chemical reactions at the electrode–electrolyte interface is crucial in order to improve the battery performance. However, the interface is located between the electrode and electrolyte materials, hindering the experimental analysis of the interface. Thus, the physicochemical properties and processes remain largely unclear.
In addition, the investigation of the structural properties of the surface of electrode materials is also particularly relevant. As a matter of fact, the break in crystalline periodicity at the interface strongly influences the interfacial physicochemical properties. The stability of the surface structure of the electrode material strongly depends on the plane orientation. Moreover, the electronic structure is significantly influenced by the termination structure. Experimentally, surface structures were studied only for LiCoO2(001),166 LiFePO4(010),167 Li4Ti5O12(111),168 LiTi2O4(111),169 and the basal plane of graphite. The investigation of electronic structures is just limited to the cases of LiCoO2(001),166 and LiTi2O4(111).169 The physicochemical properties of the surfaces of electrode materials with high activity for Li insertion and extraction, such as LiCoO2(104) and the edge plane of graphite, remain poorly understood. Recently, calculations demonstrated that the electronic structures of several LiCoO2 surface facets are significantly different from those of bulk LiCoO2, due to altered spin states of surface Co3+ atoms.170
Another issue related to Li-ion batteries that can be addressed only by means of surface-science experiments is related to the initial stages of the interactions between lithium and metal oxides. The study of the initial stages of adsorption of Li on the TiO2 surface by STM,171 metastable-induced electron spectroscopy (MIES) and UPS172 and the electron-stimulated desorption approach173 evidences the occurrence of metastable adsorption on the defect-free (110) surface and the importance of over-surface defect sites for further penetration of Li atoms in bulk titania.
Moreover, storage and operation of Li-ion battery electrodes result in electrode surface film formation (solid electrolyte interphase, SEI), which plays an important role in the cell properties and performance.174 Such surface films arise from the reactions at the electrode/electrolyte interface (side reactions) and significantly influence the lithium ion transfer. Therefore, surface-science techniques are particularly suitable for investigations of the SEI.
Finally, it is worth mentioning that photoemission spectroscopies can be employed to study174 the formation of contact potentials and the alignment of energy levels (for electrons and ions) at interfaces.
8 Conclusions and outlook
Here, we selected some case-study examples among 2D materials and novel topological phases of matter to discuss the crucial role of surface science in contemporary research on materials and their related applications. Specifically, we have demonstrated that the inevitable interaction with air should be taken into account when reporting unusual phenomena, erroneously ascribed to quantum size effects arising from exfoliation in nanosheets or to possible connections with the topological band structure. As a matter of fact, the catalytic properties of GaSe and PtSn4 are actually driven by the self-assembled heterostructure formed upon oxidation, while pristine surfaces of GaSe and PtSn4 are even unsuitable for catalytic applications. Definitely, the metal–oxide skin plays a pivotal role in all processes related to surface chemical reactions. In photocatalysis, the underlying bulk affords electron–hole pairs, while catalytic reactions are uniquely determined by the metal–oxide skin. Similarly, gas sensing in SnSe2-based systems is determined by the formation of an ultrathin SnO2 surface layer.Moreover, we have elucidated the role of surface-science tools in the analysis of Li-ion batteries and adsorption-mediated distillation techniques in membrane technology for seawater desalination, thus paving the way for a surface-science approach to application fields apparently far from traditional targets of this discipline.
Indeed, surface science is facing an important challenge related to its engagement in contemporary research on physical chemistry, condensed matter physics and materials science. Three obvious possibilities are available for bringing surface science at the vanguard of the scientific community: (i) to use surface systems to model physical concepts relevant for open issues in the literature, concerning both fundamental and applied research; (ii) to adopt experimental methodologies and techniques to avoid misleading interpretation of data in the literature related to poor control of surface phenomena; and (iii) to grow novel interfaces and heterostructures exhibiting exotic effects able to attract the interest of a wide community.
Conflicts of interest
There are no conflicts to declare.References
- H. J. Freund, N. Ernst, T. Risse, H. Hamann and G. Rupprechter, Phys. Status Solidi A, 2001, 187, 257–274 CrossRef CAS
.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS
- J. Niemantsverdriet, A. Engelen, A. de Jong, W. Wieldraaijer and G. Kramer, Appl. Surf. Sci., 1999, 144, 366–374 CrossRef
- J. Serafin, A. Liu and S. Seyedmonir, J. Mol. Catal. A: Chem., 1998, 131, 157–168 CrossRef CAS
- P. C. Thüne and J. H. Niemantsverdriet, Surf. Sci., 2009, 603, 1756–1762 CrossRef
- I. E. Wachs, Surf. Sci., 2003, 544, 1–4 CrossRef CAS
-
K. W. Kolasinski, Surface science: foundations of catalysis and nanoscience, John Wiley & Sons, 2012 Search PubMed
-
J. Hudson, Surface science: an introduction, Elsevier, 2013 Search PubMed
-
H. Lüth, Solid surfaces, interfaces and thin films, Springer, 2001 Search PubMed
-
G. Benedek and J. P. Toennies, Helium Atom Scattering Spectroscopy of Surface Phonons, Springer, Berlin, 2012 Search PubMed
- V. Aquilanti, D. Cappelletti, F. Pirani, L. Y. Rusin, M. B. Sevryuk and J. P. Toennies, J. Phys. Chem., 1991, 95, 8248–8255 CrossRef CAS
- J. P. Toennies, J. Phys.: Condens. Matter, 1993, 5, A25–A40 CrossRef CAS
- P. Nieto, E. Pijper, D. Barredo, G. Laurent, R. A. Olsen, E.-J. Baerends, G.-J. Kroes and D. Farías, Science, 2006, 312, 86–89 CrossRef CAS
- D. Maccariello, D. Campi, A. Al Taleb, G. Benedek, D. Farías, M. Bernasconi and R. Miranda, Carbon, 2015, 93, 1–10 CrossRef CAS
- G. A. Somorjai, J. Phys. Chem., 1990, 94, 1013–1023 CrossRef CAS
- H. Bonzel, Surf. Sci., 1977, 68, 236–258 CrossRef CAS
- G. A. Somorjai, Surf. Sci., 1994, 299, 849–866 CrossRef
- M. Sterrer and H.-J. Freund, Catal. Lett., 2013, 143, 375–385 CrossRef CAS
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS
- G. Ertl, Angew. Chem., Int. Ed. Engl., 1990, 29, 1219–1227 CrossRef
- A. Politano and G. Chiarello, J. Phys. Chem. C, 2011, 115, 13541–13553 CrossRef CAS
- H. Ibach, Surf. Sci., 1994, 299, 116–128 CrossRef
- H. Steininger, S. Lehwald and H. Ibach, Surf. Sci., 1982, 123, 1–17 CrossRef CAS
- M. F. Bertino, S. Miret-Artes and J. P. Toennies, Chem. Phys. Lett., 1998, 287, 663–670 CrossRef CAS
- P. Brault, H. Range, J. P. Toennies and C. Wöll, Z. Phys. Chem., 1997, 198, 1–17 CrossRef CAS
- J. Braun, J. P. Toennies and G. Witte, Surf. Sci., 1995, 340, 265–280 CrossRef CAS
- L. W. Bruch, A. Glebov, J. P. Toennies and H. Weiss, J. Chem. Phys., 1995, 103, 5109–5120 CrossRef CAS
- A. P. Graham, M. F. Bertino, F. Hofmann and J. P. Toennies, J. Chem. Soc., Faraday Trans., 1996, 92, 4749–4757 RSC
- A. P. Graham, A. Menzel and J. P. Toennies, J. Chem. Phys., 1999, 111, 1169–1174 CrossRef CAS
- A. P. Graham and J. P. Toennies, J. Chem. Phys., 2003, 118, 2879–2885 CrossRef CAS
- F. Hofmann, U. Svenson and J. P. Toennies, Surf. Sci., 1997, 371, 169–182 CrossRef CAS
- D. Farías and K. H. Rieder, Rep. Prog. Phys., 1998, 61, 1575–1664 CrossRef
- D. Farías, M. Patting and K. H. Rieder, J. Chem. Phys., 2002, 117, 1797–1803 CrossRef
- D. Farías, P. Schilbe, M. Patting and K. H. Rieder, J. Chem. Phys., 1999, 110, 559–569 CrossRef
- D. Farías, H. Troger, M. Patting and K. H. Rieder, Surf. Sci., 1996, 352, 155–160 CrossRef
- R. Apel, D. Farías, H. Troger and K. H. Rieder, Surf. Sci., 1995, 331, 57–61 CrossRef
- A. G. C. Ros, J.-S. McEwen and P. Gaspard, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 021604 CrossRef
- H. Arnolds, Prog. Surf. Sci., 2011, 86, 1–40 CrossRef CAS
- Y. Morikawa, J. J. Mortensen, B. Hammer and J. K. Nørskov, Surf. Sci., 1997, 386, 67–72 CrossRef CAS
- L. Grill, J. Phys.: Condens. Matter, 2010, 22, 084023 CrossRef
- N. Pavliček and L. Gross, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef
- J. Wintterlin, J. Trost, S. Renisch, R. Schuster, T. Zambelli and G. Ertl, Surf. Sci., 1997, 394, 159–169 CrossRef CAS
- T. Duchoň, J. Hackl, J. Höcker, K. Veltruská, V. Matolín, J. Falta, S. Cramm, S. Nemšák, C. M. Schneider and J. I. Flege, Ultramicroscopy, 2017, 183, 84–88 CrossRef
- M. Bowker, R. A. Bennett and I. Z. Jones, Top. Catal., 2004, 28, 25–30 CrossRef CAS
- A. F. Lee, D. E. Gawthrope, N. J. Hart and K. Wilson, Surf. Sci., 2004, 548, 200–208 CrossRef CAS
- J. Kim, W. H. Doh, H. Kondoh, K. Mase, J.-J. Gallet, F. Bournel, B. S. Mun and J. Y. Park, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146857 CrossRef CAS
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, J. Am. Chem. Soc., 2017, 139, 8960–8970 CrossRef CAS
- S. Nappini, A. Matruglio, D. Naumenko, S. Dal Zilio, F. Bondino, M. Lazzarino and E. Magnano, Nanoscale, 2017, 9, 4456–4466 RSC
- M. Amati, V. Bonanni, L. Braglia, F. Genuzio, L. Gregoratti, M. Kiskinova, A. Kolmakov, A. Locatelli, E. Magnano, A. A. Matruglio, T. O. Menteş, S. Nappini, P. Torelli and P. Zeller, J. Electron Spectrosc. Relat. Phenom., 2019, 146902, DOI:10.1016/j.elspec.2019.146902
- M. Fracchia, P. Ghigna, T. Pozzi, U. Anselmi Tamburini, V. Colombo, L. Braglia and P. Torelli, J. Phys. Chem. Lett., 2020, 11, 3589–3593 CrossRef CAS
- P. Stoltze and J. Nørskov, Phys. Rev. Lett., 1985, 55, 2502 CrossRef CAS
- H. Oosterbeek, Phys. Chem. Chem. Phys., 2007, 9, 3570–3576 RSC
-
K. Reuter and H. Metiu, Handbook of Materials Modeling: Applications: Current and Emerging Materials, 2020, pp. 1309–1319 Search PubMed
- O. Mamun, K. T. Winther, J. R. Boes and T. Bligaard, Sci. Data, 2019, 6, 1–9 CAS
- J. M. Kweun, C. Li, Y. Zheng, M. Cho, Y. Y. Kim and K. Cho, Appl. Surf. Sci., 2016, 370, 279–290 CrossRef CAS
- T. Hu, M. Hu, B. Gao, W. Li and X. Wang, J. Phys. Chem. C, 2018, 122, 18501–18509 CrossRef CAS
- M. G. Vergniory, L. Elcoro, C. Felser, N. Regnault, B. A. Bernevig and Z. Wang, Nature, 2019, 566, 480–485 CrossRef CAS
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS
- D. W. Boukhvalov, Y. W. Son and R. S. Ruoff, ACS Catal., 2014, 4, 2016–2021 CrossRef CAS
- C. Stampfl, M. V. Ganduglia-Pirovano, K. Reuter and M. Scheffler, Surf. Sci., 2002, 500, 368–394 CrossRef CAS
- S. Heiden, D. Usvyat and P. Saalfrank, J. Phys. Chem. C, 2019, 123, 6675–6684 CrossRef CAS
- T. Hirai, M. Okoshi, A. Ishikawa and H. Nakai, Surf. Sci., 2019, 686, 58–62 CrossRef CAS
- P. Miro, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537–6554 RSC
- A. K. Geim, Rev. Mod. Phys., 2011, 83, 851–862 CrossRef CAS
- K. S. Novoselov, Rev. Mod. Phys., 2011, 83, 837–849 CrossRef CAS
- M. S. Vitiello, JPhys Mater., 2019, 3, 014008 CrossRef
- L. Viti, D. G. Purdie, A. Lombardo, A. C. Ferrari and M. S. Vitiello, Nano Lett., 2020, 20, 3169–3177 CrossRef CAS
- M. Vanni, M. Serrano-Ruiz, F. Telesio, S. Heun, M. Banchelli, P. Matteini, A. M. Mio, G. Nicotra, C. Spinella, S. Caporali, A. Giaccherini, F. D’Acapito, M. Caporali and M. Peruzzini, Chem. Mater., 2019, 31, 5075–5080 CrossRef CAS
- A. Al Taleb, G. Anemone, R. Miranda and D. Farías, 2D Mater., 2018, 5, 045002 CrossRef CAS
- E. Plummer, R. Matzdorf, A. Melechko, J. Pierce and J. Zhang, Surf. Sci., 2002, 500, 1–27 CrossRef CAS
- H. K. Jeong, H. J. Noh, J. Y. Kim, L. Colakerol, P. A. Glans, M. H. Jin, K. E. Smith and Y. H. Lee, Phys. Rev. Lett., 2009, 102, 099701 CrossRef
- A. P. Kauling, A. T. Seefeldt, D. P. Pisoni, R. C. Pradeep, R. Bentini, R. V. Oliveira, K. S. Novoselov and A. H. Castro Neto, Adv. Mater., 2018, 30, 1803784 CrossRef
- N. M. Marković and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef
- B. Braunchweig, D. Hibbitts, M. Neurock and A. Wieckowski, Catal. Today, 2013, 202, 197–209 CrossRef CAS
- S. M. Tan, C. K. Chua, D. Sedmidubský, Z. B. Sofer and M. Pumera, Phys. Chem. Chem. Phys., 2016, 18, 1699–1711 RSC
- M. I. Zappia, G. Bianca, S. Bellani, M. Serri, L. Najafi, R. Oropesa-Nuñez, B. Martín-García, D. Bouša, D. Sedmidubský, V. Pellegrini, Z. Sofer, A. Cupolillo and F. Bonaccorso, Adv. Funct. Mater., 2020, 30, 1909572 CrossRef CAS
- Y. Wu, D. Zhang, K. Lee, G. S. Duesberg, A. Syrlybekov, X. Liu, M. Abid, M. Abid, Y. Liu, L. Zhang, C. Ó. Coileáin, H. Xu, J. Cho, M. Choi, B. S. Chun, H. Wang, H. Liu and H. C. Wu, Adv. Mater. Technol., 2017, 2, 1600197 CrossRef
- O. A. Balitskii, V. P. Savchyn and V. O. Yukhymchuk, Semicond. Sci. Technol., 2002, 17, L1–L4 CrossRef CAS
- T. E. Beechem, B. M. Kowalski, M. T. Brumbach, A. E. McDonald, C. D. Spataru, S. W. Howell, T. Ohta, J. A. Pask and N. G. Kalugin, Appl. Phys. Lett., 2015, 107, 173103 CrossRef
- A. Bergeron, J. Ibrahim, R. Leonelli and S. Francoeur, Appl. Phys. Lett., 2017, 110, 241901 CrossRef
- E. Filippo, M. Siciliano, A. Genga, G. Micocci, A. Tepore and T. Siciliano, Mater. Res. Bull., 2013, 48, 1741–1744 CrossRef CAS
- H. Iwakuro, C. Tatsuyama and S. Ichimura, Jpn. J. Appl. Phys., 1982, 21, 94 CrossRef CAS
- B. M. Kowalski, N. Manz, D. Bethke, E. A. Shaner, A. Serov and N. G. Kalugin, Mater. Res. Express, 2019, 6, 085907 CrossRef CAS
- M. Rahaman, R. D. Rodriguez, M. Monecke, S. A. Lopez-Rivera and D. R. T. Zahn, Semicond. Sci. Technol., 2017, 32, 105004 CrossRef
- T. Siciliano, M. Tepore, A. Genga, G. Micocci, M. Siciliano and A. Tepore, Vacuum, 2013, 92, 65–69 CrossRef CAS
- N. K. Tovstyuk, J. Phys. Stud., 2004, 8, 47–54 CrossRef CAS
- D. Pozo-Zamudio, S. Schwarz, J. Klein, R. Schofield, E. Chekhovich, O. Ceylan, E. Margapoti, A. Dmitriev, G. Lashkarev and D. Borisenko, 2015, arXiv:1506.05619.
- D. Andres-Penares, A. Cros, J. P. Martínez-Pastor and J. F. Sánchez-Royo, Nanotechnology, 2017, 28, 175701 CrossRef
- Q. Zhao, R. Frisenda, P. Gant, D. Perez de Lara, C. Munuera, M. Garcia-Hernandez, Y. Niu, T. Wang, W. Jie and A. Castellanos-Gomez, Adv. Funct. Mater., 2018, 28, 1805304 CrossRef
- J. Susoma, J. Lahtinen, M. Kim, J. Riikonen and H. Lipsanen, AIP Adv., 2017, 7, 015014 CrossRef
- G. D'Olimpio, S. Nappini, M. Vorokhta, L. Lozzi, F. Genuzio, T. O. Menteş, V. Paolucci, B. Gürbulak, S. Duman, L. Ottaviano, A. Locatelli, F. Bondino, D. W. Boukhvalov and A. Politano, Adv. Funct. Mater., 2020 DOI:10.1002/adfm.202005466
- J. Kim, S. M. Han and W. S. Yoo, J. Micro/Nanolithogr., MEMS, MOEMS, 2014, 13, 011205 CrossRef
- P. Borowicz, J. Spectrosc., 2016, 2016, 1617063 Search PubMed
- L. Shi, Q. Zhou, Y. Zhao, Y. Ouyang, C. Ling, Q. Li and J. Wang, J. Phys. Chem. Lett., 2017, 8, 4368–4373 CrossRef CAS
- Y. Ni, H. Wu, C. Huang, M. Mao, Z. Wang and X. Cheng, J. Cryst. Growth, 2013, 381, 10–14 CrossRef CAS
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS
- E. Petroni, E. Lago, S. Bellani, D. W. Boukhvalov, A. Politano, B. Gürbulak, S. Duman, M. Prato, S. Gentiluomo, R. Oropesa-Nuñez, J. K. Panda, P. S. Toth, A. E. Del Rio Castillo, V. Pellegrini and F. Bonaccorso, Small, 2018, 14, 1800749 CrossRef
- S. M. Tan, C. K. Chua, D. Sedmidubský, Z. Sofer and M. Pumera, Phys. Chem. Chem. Phys., 2016, 18, 1699–1711 RSC
- Q. Yang, G. Li, K. Manna, F. Fan, C. Felser and Y. Sun, Adv. Mater., 2020, 32, 1908518 CrossRef CAS
- A. Politano, G. Chiarello, Z. Li, V. Fabio, L. Wang, L. Guo, X. Chen and D. W. Boukhvalov, Adv. Funct. Mater., 2018, 28, 1800511 CrossRef
- C. R. Rajamathi, U. Gupta, N. Kumar, H. Yang, Y. Sun, V. Suss, C. Shekhar, M. Schmidt, H. Blumtritt, P. Werner, B. Yan, S. Parkin, C. Felser and C. N. R. Rao, Adv. Mater., 2017, 29, 1606202 CrossRef
- G. Li, C. Fu, W. Shi, L. Jiao, J. Wu, Q. Yang, R. Saha, M. E. Kamminga, A. K. Srivastava, E. Liu, A. N. Yazdani, N. Kumar, J. Zhang, G. R. Blake, X. Liu, M. Fahlman, S. Wirth, G. Auffermann, J. Gooth, S. Parkin, V. Madhavan, X. Feng, Y. Sun and C. Felser, Angew. Chem., Int. Ed., 2019, 58, 13107–13112 CrossRef CAS
- G. Li, Q. Xu, W. Shi, C. Fu, L. Jiao, M. E. Kamminga, M. Yu, H. Tüysüz, N. Kumar and V. Süβ, Sci. Adv., 2019, 5, eaaw9867 CrossRef CAS
- G. Li and C. Felser, Appl. Phys. Lett., 2020, 116, 070501 CrossRef CAS
- Z. P. Xiang, H. Q. Deng, P. Peljo, Z. Y. Fu, S. L. Wang, D. Mandler, G. Q. Sun and Z. X. Liang, Angew. Chem., Int. Ed., 2018, 130, 3522–3526 CrossRef
- J. Greeley, I. Stephens, A. Bondarenko, T. P. Johansson, H. A. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS
- Y. Wu, L.-L. Wang, E. Mun, D. D. Johnson, D. Mou, L. Huang, Y. Lee, S. L. Bud’ko, P. C. Canfield and A. Kaminski, Nat. Phys., 2016, 12, 667–671 Search PubMed
- V. R. Stamenković, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Marković, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Marković and V. R. Stamenković, Angew. Chem., Int. Ed., 2012, 124, 3193–3196 CrossRef
- Z. Yang, J. Wang and X. Yu, Chem. Phys. Lett., 2010, 499, 83–88 CrossRef CAS
- D. W. Boukhvalov, A. Marchionni, J. Filippi, R. Edla, S. Nappini, J. Fuji, C. N. Kuo, G. D’Olimpio, L. Ottaviano, C. S. Lue, P. Torelli, F. Vizza and A. Politano, J. Mater. Chem. A, 2020, 8, 2349–2355 RSC
- G. D'Olimpio, D. W. Boukhvalov, J. Fujii, P. Torelli, A. Marchionni, J. Filippi, C.-N. Kuo, R. Edla, L. Ottaviano and C. S. Lue, Appl. Surf. Sci., 2020, 145925 CrossRef
- Y. Jugnet, D. Loffreda, C. L. Dupont, F. O. Delbecq, E. Ehret, F. J. Cadete Santos Aires, B. S. Mun, F. Aksoy Akgul and Z. Liu, J. Phys. Chem. Lett., 2012, 3, 3707–3714 CrossRef CAS
- I. G. Casella and E. Desimoni, Electroanalysis, 1996, 8, 447–453 CrossRef CAS
- K. I. B. Eguiluz, G. R. P. Malpass, M. M. S. Pupo, G. R. Salazar-Banda and L. A. Avaca, Energy Fuels, 2010, 24, 4012–4024 CrossRef CAS
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS
- L. Mino, A. Zecchina, G. Martra, A. M. Rossi and G. Spoto, Appl. Catal., B, 2016, 196, 135–141 CrossRef CAS
- H. Cheng, J. Hou, O. Takeda, X.-M. Guo and H. Zhu, J. Mater. Chem. A, 2015, 3, 11006–11013 RSC
- N. Khalid, Z. Israr, M. Tahir and T. Iqbal, Int. J. Hydrogen Energy, 2020, 45, 8479–8489 CrossRef CAS
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS
- W. Wang, G. Li, D. Xia, T. An, H. Zhao and P. K. Wong, Environ. Sci.: Nano, 2017, 4, 782–799 RSC
- P. Prasannalakshmi and N. Shanmugam, Mater. Sci. Semicond. Process., 2017, 61, 114–124 CrossRef CAS
- D. Bahnemann, Sol. Energy, 2004, 77, 445–459 CrossRef CAS
- M. Landmann, E. Rauls and W. G. Schmidt, J. Phys.: Condens. Matter, 2012, 24, 6 CrossRef
- B. Ohtani, Catalysts, 2013, 3, 942–953 CrossRef CAS
- Y. Li, C. Gao, R. Long and Y. Xiong, Mater. Today Chem., 2019, 11, 197–216 CrossRef CAS
- Z. Xie, Y.-P. Peng, L. Yu, C. Xing, M. Qiu, J. Hu and H. Zhang, Sol. RRL, 2020, 4, 1900400 CrossRef
- W. Peng, Y. Li, F. Zhang, G. Zhang and X. Fan, Ind. Eng. Chem. Res., 2017, 56, 4611–4626 CrossRef CAS
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC
- C. A. Walenta, M. Tschurl and U. Heiz, J. Phys.: Condens. Matter, 2019, 31, ab351a CrossRef
- C. A. Walenta, C. Courtois, S. L. Kollmannsberger, M. Eder, M. Tschurl and U. Heiz, ACS Catal., 2020, 10, 4080–4091 CrossRef CAS
- X. Yang, Z. Chen, J. Xu, H. Tang, K. Chen and Y. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 15285–15293 CrossRef CAS
- A. Alagarasi, P. U. Rajalakshmi, K. Shanthi and P. Selvam, Catal. Today, 2018, 309, 202–211 CrossRef CAS
- J. Xiong, J. Di, J. Xia, W. Zhu and H. Li, Adv. Funct. Mater., 2018, 28, 1801983 CrossRef
- M. Wang, L. Cai, Y. Wang, F. Zhou, K. Xu, X. Tao and Y. Chai, J. Am. Chem. Soc., 2017, 139, 4144–4151 CrossRef CAS
- L. Jing, Y. Xu, Z. Chen, M. He, M. Xie, J. Liu, H. Xu, S. Huang and H. Li, ACS Sustainable Chem. Eng., 2018, 6, 5132–5141 CrossRef CAS
- F. J. Zhang, X. Li, X. Y. Sun, C. Kong, W. J. Xie, Z. Li and J. Liu, Appl. Surf. Sci., 2019, 487, 734–742 CrossRef CAS
- D. Yim, F. Raza, J. H. Park, J. H. Lee, H. I. Kim, J. K. Yang, I. J. Hwang and J. H. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 36960–36969 CrossRef CAS
- K. K. Patra, M. K. Ghosalya, H. Bajpai, S. Raj and C. S. Gopinath, J. Phys. Chem. C, 2019, 123, 21685–21693 CrossRef CAS
- H. Cheng, X. Qian, Y. Kuwahara, K. Mori and H. Yamashita, Adv. Mater., 2015, 27, 4616–4621 CrossRef CAS
- L. E. Ocola, Y. Wang, R. Divan and J. Chen, Sensors, 2019, 19, 2061 CrossRef CAS
- N. Barsan and U. Weimar, J. Electroceram., 2001, 7, 143–167 CrossRef CAS
- R. Kumar, N. Goel, M. Hojamberdiev and M. Kumar, Sens. Actuators, A, 2020, 303, 111875 CrossRef CAS
- Q. Li, Q. Zhou, L. Shi, Q. Chen and J. Wang, J. Mater. Chem. A, 2019, 7, 4291–4312 RSC
- Y. Guo, S. Zhou and J. Zhao, ChemNanoMat, 2020, 6, 838–849 CrossRef CAS
- A. Shafique, A. Samad and Y.-H. Shin, Phys. Chem. Chem. Phys., 2017, 19, 20677–20683 RSC
- O. S. L. Camargo Moreira, W.-Y. Cheng, H.-R. Fuh, W.-C. Chien, W. Yan, H. Fei, H. Xu, D. Zhang, Y. Chen and Y. Zhao, ACS Sens., 2019, 4, 2546–2552 CrossRef CAS
- X. Wang, Y. Liu, J. Dai, Q. Chen, X. Huang and W. Huang, Chem. – Eur. J., 2020, 26, 3870–3876 CrossRef CAS
- A. S. Pawbake, A. Date, S. R. Jadkar and D. J. Late, ChemistrySelect, 2016, 1, 5380–5387 CrossRef CAS
- M. Pawar, S. Kadam and D. J. Late, ChemistrySelect, 2017, 2, 4068–4075 CrossRef CAS
- M. Chen, Z. Li, W. Li, C. Shan, W. Li, K. Li, G. Gu, Y. Feng, G. Zhong and L. Wei, Nanotechnology, 2018, 29, 455501 CrossRef
- V. Paolucci, G. D’Olimpio, C.-N. Kuo, C. S. Lue, D. W. Boukhvalov, C. Cantalini and A. Politano, ACS Appl. Mater. Interfaces, 2020, 12, 34362–34369 CrossRef CAS
- P. Cox, R. Egdell, W. Flavell and R. Helbig, Vacuum, 1983, 33, 835–838 CrossRef CAS
- C. Tortajada and A. K. Biswas, Curr. Opin. Environ. Sustainability, 2018, 34, 21–25 CrossRef
- J. Willet, K. Wetser, J. Vreeburg and H. H. Rijnaarts, Water Resources and Industry, 2019, 21, 100110 CrossRef
- A. Politano, G. Di Profio, E. Fontananova, V. Sanna, A. Cupolillo and E. Curcio, Desalination, 2019, 451, 192–199 CrossRef CAS
- A. Politano, P. Argurio, G. Di Profio, V. Sanna, A. Cupolillo, S. Chakraborty, H. A. Arafat and E. Curcio, Adv. Mater., 2017, 29, 1603504 CrossRef
- L. Eykens, K. De Sitter, C. Dotremont, L. Pinoy and B. Van der Bruggen, Ind. Eng. Chem. Res., 2016, 55, 9333–9343 CrossRef CAS
- F. Calise, F. L. Cappiello, R. Vanoli and M. Vicidomini, Appl. Energy, 2019, 253, 113575 CrossRef
- E. Drioli, A. Ali and F. Macedonio, Desalination, 2015, 356, 56–84 CrossRef CAS
- E. Gontarek, F. Macedonio, F. Militano, L. Giorno, M. Lieder, A. Politano, E. Drioli and A. Gugliuzza, Nanoscale, 2019, 11, 11521–11529 RSC
- F. Perreault, A. Fonseca de
Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC
- T. Minato and T. Abe, Prog. Surf. Sci., 2017, 92, 240–280 CrossRef CAS
- K. Iwaya, T. Ogawa, T. Minato, K. Miyoshi, J. Takeuchi, A. Kuwabara, H. Moriwake, Y. Kim and T. Hitosugi, Phys. Rev. Lett., 2013, 111, 126104 CrossRef CAS
- S. Kobayashi, C. A. J. Fisher, T. Kato, Y. Ukyo, T. Hirayama and Y. Ikuhara, Nano Lett., 2016, 16, 5409–5414 CrossRef CAS
- M. Kitta, M. Kohyama and H. Onishi, Appl. Phys. Lett., 2014, 105, 111606 CrossRef
- Y. Okada, Y. Ando, R. Shimizu, E. Minamitani, S. Shiraki, S. Watanabe and T. Hitosugi, Nat. Commun., 2017, 8, 15975 CrossRef CAS
- L. Hong, L. Hu, J. W. Freeland, J. Cabana, S. Öğüt and R. F. Klie, J. Phys. Chem. C, 2019, 123, 8851–8858 CrossRef CAS
- H. Tatsumi, A. Sasahara and M. Tomitori, J. Phys. Chem. C, 2012, 116, 13688–13692 CrossRef CAS
- S. Krischok, J. Schaefer, O. Höfft and V. Kempter, Surf. Interface Anal., 2005, 37, 83–89 CAS
- V. Ageev and S. Solov’ev, Phys. Solid State, 2000, 42, 2159–2163 CrossRef CAS
- R. Hausbrand, D. Becker and W. Jaegermann, Prog. Solid State Chem., 2014, 42, 175–183 CrossRef CAS
Footnote |
| † These authors contributed equally. |
| This journal is © the Owner Societies 2021 |