
Ethanol–ethylene conversion mechanism on hydrogen boride sheets probed by in situ infrared absorption spectroscopy†
Asahi
Fujino
a,
Shin-ichi
Ito
 bc,
Taiga
Goto
a,
Ryota
Ishibiki
a,
Ryota
Osuga
d,
Junko N.
Kondo
d,
Tadahiro
Fujitani
ce,
Junji
Nakamura
c,
Hideo
Hosono
b and
Takahiro
Kondo
*bc
bc,
Taiga
Goto
a,
Ryota
Ishibiki
a,
Ryota
Osuga
d,
Junko N.
Kondo
d,
Tadahiro
Fujitani
ce,
Junji
Nakamura
c,
Hideo
Hosono
b and
Takahiro
Kondo
*bc
aGraduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1, Tennodai, Tsukuba, Ibaraki 305-8573, Japan
bMaterials Research Center for Element Strategy, Tokyo Institute of Technology, Yokohama 226-8503, Japan. E-mail: takahiro@ims.tsukuba.ac.jp
cDepartment of Materials Science and Tsukuba Research Center for Energy Materials Science, Faculty of Pure and Applied Sciences, University of Tsukuba, Tsukuba 305-8573, Japan
dInstitute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Yokohama, Kanagawa 226-8503, Japan
eInterdisciplinary Research Center, National Institute of Advanced Industrial Science and Technology, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
First published on 12th August 2020
Abstract
Two-dimensional hydrogen boride (HB) sheets were recently demonstrated to act as a solid acid catalyst in their hydrogen-deficient state. However, both the active sites and the mechanism of the catalytic process require further elucidation. In this study, we analyzed the conversion of ethanol adsorbed on HB sheets under vacuum during heating using in situ Fourier transform infrared (FT-IR) absorption spectroscopy with isotope labelling. Up to 450 K, the FT-IR peak associated with the OH group of the adsorbed ethanol molecule disappeared from the spectrum, which was attributed to a dehydration reaction with a hydrogen atom from the HB sheet, resulting in the formation of an ethyl species. At temperatures above 440 K, the number of BD bonds markedly increased in CD3CH2OH, compared to CH3CD2OH; the temperature dependence of the formation rate of BD bonds was similar to that of the dehydration reaction rate of ethanol on HB sheets under steady-state conditions. The rate-determining step of the dehydration of ethanol on HB was thus ascribed to the dehydrogenation of the methyl group of the ethyl species on the HB sheets, followed by the immediate desorption of ethylene. These results show that the catalytic ethanol dehydration process on HB involves the hydrogen atoms of the HB sheets. The obtained mechanistic insights are expected to promote the practical application of HB sheets as catalysts.
1. Introduction
Two-dimensional (2D) materials offer considerable potential for application as catalysts or catalyst supports because of their unique properties such as large surface areas and novel electronic states.1,2 For example, graphene has shown superior properties as a support,3,4 while nitrogen-doped graphene exhibits excellent electrocatalytic properties in various reactions.5,6 2D materials comprising boron are considered to possess unique properties owing to the large number of theoretically predicted polymorphs.7,8 Various 2D boron-containing compounds, including boron chalcogenides,9 boron oxides,10,11 boron hydrides,12,13 and boron phosphides,14 have also been theoretically predicted to have a large number of polymorphs. Depending on the structure, the bonding strengths of the boron materials with gas molecules are expected to be different, thereby leading to a wide variety of catalytic properties for 2D boron-related materials. However, the experimental synthesis of 2D boron-related materials has rarely been reported.15,16Hydrogen boride (HB) or hydrogenated borophene sheets are novel 2D boron materials that have been reported recently and are composed of two light elements, namely boron and hydrogen, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio.17 HB sheets are terminated by protons on the negatively charged hexagonal boron sheets and they are known to exhibit versatile properties, such as a large H2 content (8.5 wt%), semimetal electronic properties,18,19 metal ion reducibility,20 and a light-responsive hydrogen release function.21 Our recent study revealed that HB sheets in their hydrogen-deficient state (H:B ratio of approximately 1:2.5 ± 0.5) catalyze the conversion of ethanol to ethylene and water above 493 K with high selectivity independent of the contact time, and an apparent activation energy of 102.8 ± 5.5 kJ mol−1.22 The activation energy of this process is comparable to those reported for the catalytic dehydration of ethanol over Al2O3 (53–155 kJ mol−1),23,24 the Lewis acidic catalyst Zr-KIT-6 (79 kJ mol−1),25 silica–alumina (125.5 kJ mol−1),26 and the microporous Fe-ZSM-5 (137.7–271.1 kJ mol−1).27 The formation rate of ethylene on the HB catalyst is lower than that offered by state-of-the-art catalysts, but is of the same order as that on the commercial SynDol (Al2O3–MgO/SiO2) catalyst.22 Therefore, HB sheets can be regarded as a solid-acid catalyst. However, the mechanism of HB catalysis has not yet been elucidated, and the active sites have not been identified. In this study, to gain insights into the HB catalysis of the ethanol conversion process, we conducted in situ Fourier transform infrared (FT-IR) absorption spectroscopy with isotope labelling, since the observation and analysis of the vibrational frequency shifts upon isotope labelling of the intermediate species by in situ FT-IR could lead to determination of the conversion mechanism of the molecules on solid surfaces.28 Overall, we investigate the interactions between the adsorbed ethanol and the HB sheets during heating under vacuum through in situ FT-IR spectroscopy and isotope labelling.
:1 stoichiometric ratio.17 HB sheets are terminated by protons on the negatively charged hexagonal boron sheets and they are known to exhibit versatile properties, such as a large H2 content (8.5 wt%), semimetal electronic properties,18,19 metal ion reducibility,20 and a light-responsive hydrogen release function.21 Our recent study revealed that HB sheets in their hydrogen-deficient state (H:B ratio of approximately 1:2.5 ± 0.5) catalyze the conversion of ethanol to ethylene and water above 493 K with high selectivity independent of the contact time, and an apparent activation energy of 102.8 ± 5.5 kJ mol−1.22 The activation energy of this process is comparable to those reported for the catalytic dehydration of ethanol over Al2O3 (53–155 kJ mol−1),23,24 the Lewis acidic catalyst Zr-KIT-6 (79 kJ mol−1),25 silica–alumina (125.5 kJ mol−1),26 and the microporous Fe-ZSM-5 (137.7–271.1 kJ mol−1).27 The formation rate of ethylene on the HB catalyst is lower than that offered by state-of-the-art catalysts, but is of the same order as that on the commercial SynDol (Al2O3–MgO/SiO2) catalyst.22 Therefore, HB sheets can be regarded as a solid-acid catalyst. However, the mechanism of HB catalysis has not yet been elucidated, and the active sites have not been identified. In this study, to gain insights into the HB catalysis of the ethanol conversion process, we conducted in situ Fourier transform infrared (FT-IR) absorption spectroscopy with isotope labelling, since the observation and analysis of the vibrational frequency shifts upon isotope labelling of the intermediate species by in situ FT-IR could lead to determination of the conversion mechanism of the molecules on solid surfaces.28 Overall, we investigate the interactions between the adsorbed ethanol and the HB sheets during heating under vacuum through in situ FT-IR spectroscopy and isotope labelling.
2. Methods
HB sheets were prepared using a previously reported ion-exchange method.17–22 More specifically, MgB2 powder (1.0 g, 99%, Rare Metallic Co., Ltd, Tokyo, Japan) in acetonitrile (300 mL, 99.5%, Wako Pure Chemical Industries Ltd, Osaka, Japan) was added to a mixture of an ion-exchange resin (60 mL, Amberlite IR120B hydrogen, Organo Corp., Tokyo, Japan) and acetonitrile (200 mL) in a Schlenk flask under a nitrogen atmosphere. Water was carefully removed due to the facile hydrolysis of MgB2.29 The resulting mixture was then stirred with a magnetic stirrer at 400 rpm for 2–3 d at room temperature (∼300 K). After this time, the reaction mixture was allowed to settle for a sufficient time and the supernatant was collected and kept for 1 d at 255 K to physically separate the B(OH)3 byproduct. In samples containing unreacted materials, such as the oxides present in the starting materials, the reaction mixture was filtered through a 1.0 or 0.2 μm pore filter (Omnipore Membrane Filters, Merck Millipore, Billerica, MA, USA) and the filtrate was kept for 1 d at 255 K to physically separate the B(OH)3 byproduct. Dried HB sheets were prepared by heating the resulting liquid at 343 K, where the gas that evaporated during heating was pumped through a liquid-nitrogen trap. We rigorously characterized the product using X-ray photoelectron spectroscopy (XPS, JPS 9010 TR, JEOL, Ltd, Tokyo, Japan) to confirm the absence of Mg, the presence of negatively charged B, and the absence of oxidized B (a survey X-ray photoelectron spectrum of the HB sheets is shown in Fig. S1, ESI†).17–22For in situ FT-IR spectroscopy analysis, we used a homemade vacuum system (Fig. S2, ESI†) coupled with an FT-IR spectrometer (FT/IR-610, Jasco, Ltd, Tokyo, Japan) to observe the structural changes in the HB sheets during heating under vacuum (1.0 × 10−6 Torr) after exposure to ethanol vapor (∼44 Torr in 123 cc) for 1 h at room temperature (∼300 K). The FT-IR measurements were conducted in transmission mode, with the sample mounted on a CaF2 disk. Each spectrum was obtained by averaging 20 scans with a resolution of 4 cm−1. The IR absorbance A is defined as A = −ln(T/T0), where T and T0 are the transmitted intensities with and without the sample on the CaF2 disk, respectively. The heating rate was set as 7.2 K min−1; thus, the sample temperature increased by 2.4 K during the measurement time of 20 s for one spectrum (20 scans). The sample temperature was monitored using a K-type thermocouple close to the sample holder. To obtain details regarding the structural changes taking place in ethanol, the isotopes CD3CH2OH (ethanol-2,2,2-d3, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and CH3CD2OH (ethanol-1,1-d2, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) were used in addition to C2H5OH (99.5%, Wako Pure Chemical Industries Ltd, Osaka, Japan). To assign the FT-IR peaks, the vibrational modes of an isolated ethanol molecule were determined using density functional theory (DFT) calculations performed using Gaussian 16.30 The functional and basis set used for the calculations were B3LYP and 6-31++g(d,p), respectively.
To determine the catalytic activity, gaseous ethanol was introduced onto the HB sheets using argon as a carrier gas under atmospheric pressure in a homemade fixed-bed flow reactor, as reported previously.22 The product gas was then analyzed downstream using a gas chromatograph (GC-8A, Shimadzu, Kyoto, Japan) with a thermal conductivity detector equipped with 5A molecular sieves and Porapak Q columns. The catalytic conversion was estimated from the total amount of hydrocarbon produced as follows: ethanol conversion (%) = [(number of carbon atoms in detected hydrocarbon molecules) (mol min−1)/(number of introduced ethanol molecules × 2) (mol min−1)] × 100. The conversion estimated using this method has been confirmed to be similar to that estimated on the basis of the consumption of ethanol under our experimental conditions;22 therefore, the carbon accumulation on HB can be neglected. The W/F conditions (g min mmol−1), where W is the weight of the catalyst (g) and F is the flow rate of C2H5OH (mmol min−1), were controlled by adjusting the C2H5OH flow rate and the sample weight. Under our experimental conditions (W/F = 4–30), the product exhibited no apparent W/F dependence.22
3. Results and discussion
Fig. 1 shows the FT-IR spectra of the HB sheets measured under vacuum before and after exposure to ethanol vapor or isotope-labelled ethanol vapor for 1 h at room temperature (∼300 K). In each case, new absorbance peaks originating from ethanol were observed after exposure to ethanol vapor. To assign the absorbance peaks, we compared the peak positions in the experimentally obtained spectra with those from the DFT calculations for isolated ethanol and the reported values for ethanol under various conditions31–40 as shown in Table 1. In the case of C2H5OH (Fig. 1a), absorbance peaks corresponding to the OH group (νOH), CH3 group (νCH3), and CH2 group (νCH2) were observed at 3492, 2980, and 2937 cm−1, respectively. An absorbance peak at 2230 cm−1 attributable to the CD3 group (νCD3) was only observed in the case of CD3CH2OH (Fig. 1b), and an absorbance peak at 2120 cm−1 attributable to the CD2 group (νCD2) was only observed in the case of CH3CD2OH (Fig. 1c). These results suggest that ethanol is adsorbed on the HB sheet at room temperature (∼300 K) under vacuum.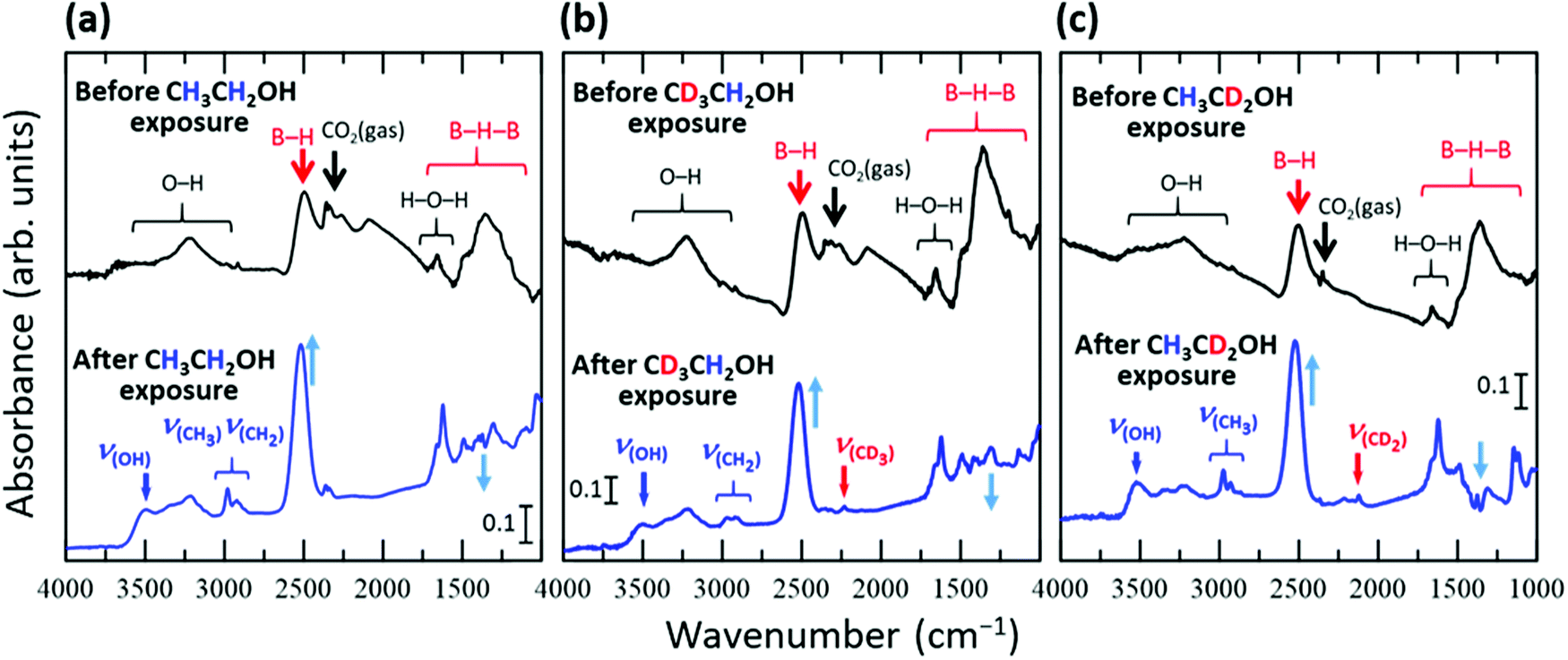 | ||
| Fig. 1 FT-IR spectra of the HB sheets obtained under vacuum before and after exposure to the vapour of ethanol or isotope-labelled ethanol for 1 h at room temperature (∼300 K); (a) CH3CH2OH, (b) CD3CH2OH, and (c) CH3CD2OH. | ||
| CH3CH2OH | CD3CH2OH | CH3CD2OH | Ref. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ν (OH) | ν as(CH3) | ν s(CH3) | ν s(CH2) | ν (OH) | ν as(CD3) | ν as(CH2) | ν s(CH2) | ν s(CD3) | ν (OH) | ν as(CD2) | ν as(CH3) | ν s(CD2) | ν s(CH3) | ||
| On HB | 3492 | 2980 | (2937) | (2937) | 3499 | (2232) | (2964) | (2964) | (2232) | 3525 | 2219 | (2975) | 2123 | (2975) | This work |
| Gas (Fig. S3, ESI) | 3676 | 2989 | (2900) | (2900) | 3677 | 2235 | 2948 | 2903 | 2096 | 3676 | 2195 | 2988 | 2112 | 2889 | This work |
| Free molecule B3-LYP trans-geometry | 3834 | 3117 | 3045 | 2989 | 3834 | 2309 | 3021 | 2989 | 2188 | 3834 | 2245 | 3116 | 2172 | 3045 | This work |
| Glass (80 K) | 3279 | 2987 | 2927 | 2872 | — | 2231 | 2925 | 2288 | 2124 | — | — | — | — | — | 31–33 |
| Multilayer on Si(100) (135 K) | 3267 | 2983 | 2928 | 2881 | 3286 | 2233 | 2889 | — | 2102 | — | — | — | — | — | 33 |
| Multilayer on HOPG (98 K) | 3280 | 2972 | 2927 | 2875, 2896 | — | — | — | — | — | — | — | — | — | — | 34 |
| Multilayer on CsI (10 K) | 3291 | 2971 | 2930 | 2884 | — | — | — | — | — | — | — | — | — | — | 35 |
| Matrix with H2O on CsI (10 K) CH3CH2OH/H2O = 1/20 | — | 2978 | 2904 | 2871 | — | — | — | — | — | — | — | — | — | — | 35 |
| Matrix with CO on CsI (10 K) CH3CH2OH/CO = 1/20 | 3283–3448 | 2982 | 2937 | 2886 | — | — | — | — | — | — | — | — | — | — | 35 |
| Multilayer on Cu(111) (100 K) | ∼3280 | 2975 | 2933 | 2895 | — | — | — | — | — | — | — | — | — | — | 36 |
| Matrix with Ar (20 K) | 3658 | 2996 | 2940 | 2901, 2954 | — | — | — | — | — | — | — | — | — | — | 37 |
| Free molecule B3-LYP | 3808 | 3106 | 3035 | 2987 | 3808 | 2297 | 3088 | 2987 | 2182 | — | — | — | — | — | 33 |
| 0.1 M in CCl4 | — | — | — | — | — | 2229 | 2928 | 2884 | 2123 | — | 2184–2196 | 2973 | 2095–2120 | 2932 | 38 |
| Raman | ν FR(CH2) + νas(CH3) | ν FR(CH3) + νas(CH2) | ν FR(CH3) | ν s(CH3) + νs(CH2) | ν FR(CH2) | ν as(CH2) | ν s(CH2) | ν FR(CH3) | ν as(CH3) | ν FR(CH3) | ν s(CH3) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas (experiment) | 2983 | 2938 | 2917 | 2882 | 2981 | 2945 | 2888 | 2938 | 2962–2988 | 2913 | 2878 | 39 |
| Calculation (× 0.973) | 2969 | 2924 | 2919 | 2884 | 2963 | 2926 | 2899 | 2951 | 3015 | 2922 | 2881 | |
| Intensity | s | vs | w | vs | s | vw | vs | vs | vw | w | s |
| IR and Raman band | ν as(CH3) + νips(CH2)-g | ν s(CH3) + νas(CH2)-t + νFR(CH2) | ν FR(CH3) + νs(CH2)-t + νops(CH2)-g | ν ips(CH2)-g | ν as(CH2)-t + νFR(CH2) | ν s(CH2)-t + νops(CH2)-g | ν as(CH3) | ν FR(CH3) | ν s(CH3) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Gas (theoretical) | 2973 | 2930 | 2876 | 2960 | 2900–2930 | 2876 | 2973 | 2850–2900 | 2933 | 40 |
With the appearance of the ethanol-related absorbance peaks, the intensity of the broad absorbance peak at ∼1300 cm−1 decreased, while those of the peaks at 1650 and 2500 cm−1 increased. Furthermore, following exposure of the HB sheets to ethanol vapor, the peak at 2500 cm−1 shifted to 2520 cm−1 in each case (Fig. 1). These results indicate that the structure of the HB sheets changes upon the adsorption of ethanol. The absorbance peak at 1300 cm−1 can be assigned to the B–H–B vibrational mode,19 whereas the peak at 1650 cm−1 can be assigned to the bending vibration of H–O–H and/or the vibration of B–H–B bonds at sites with a large strain in the HB sheet, or at the edge of the HB sheet.17,19 The absorbance peak at 2500 cm−1 can be attributed to the B–H stretching vibration of isolated BH bonds in the HB sheet.17 These results suggest that the HB sheets may be distorted by the adsorption of ethanol, and some of the BH bonds in the BHB bonds may dissociate to form isolated BH bonds, as indicated by the increase in intensity and shift of the peak corresponding to isolated BH bonds following ethanol adsorption. The complicated profile in the 1000–1500 cm−1 region is caused by not only this change in the BHB bonds of the HB sheets, but also the appearance of absorbance peaks corresponding to the bending, rocking, twisting, scissoring, and wagging modes of the (normal and/or deuterated) methyl and methylene groups of the adsorbed ethanol and/or the stretching mode of CO in the adsorbed ethanol.28,31–40 Therefore, we mainly focused on the absorbance peaks outside this overlapped region, although some of the peaks were assigned, as shown in Fig. S4–S6 (ESI†).
Fig. 2a shows the FT-IR spectra of the HB sheets with adsorbed C2H5OH (referred to as C2H5OH/HB) at 300 and 623 K under vacuum. As shown, the νOH, νCH3, and νCH2 peaks almost disappeared at 623 K, while a decrease in peak intensity was observed for the peak of the B–H stretching vibrational mode at 2520 cm−1. In contrast, the absorbance at 1000–1500 cm−1 (the region including the B–H–B vibrational mode) increased with heating. This tendency, in which the peak intensity of B–H decreases while that of B–H–B increases (Fig. 2a), is opposite to that observed during the ethanol adsorption process (Fig. 1). These results indicate that when heated to 623 K, ethanol is no longer adsorbed on the HB sheets and the original state of the HB sheets may somehow be recovered in terms of the B–H–B bonding configuration. Here, we note that the number of hydrogen atoms within the HB sheets should be lower at 623 K than at 300 K because hydrogen molecules are released during the decomposition of the HB sheets over the wide temperature range of 423–1473 K.17,19,22 Thus, the decrease in peak intensity at 2520 cm−1 may also be caused by the partial decomposition of the HB sheets.
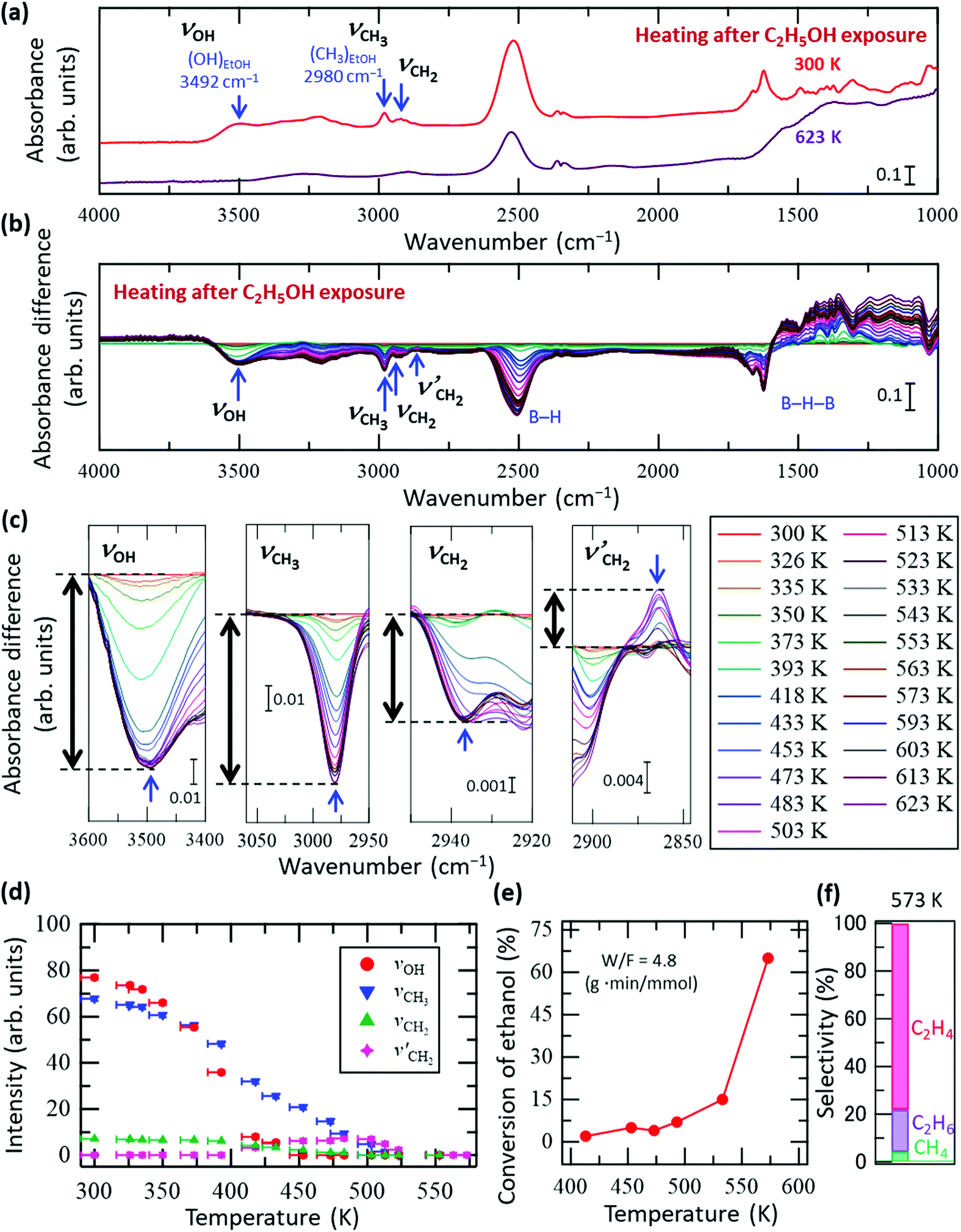 | ||
| Fig. 2 In situ FT-IR analysis results of the HB sheets with adsorbed C2H5OH (C2H5OH/HB) during heating under vacuum. (a) FT-IR spectra of C2H5OH/HB at 300 and 623 K under vacuum. (b) Absorbance difference spectra during the heating of C2H5OH/HB; the FT-IR spectrum at 300 K was subtracted from the spectrum at each temperature (all spectra before subtraction are shown in Fig. S4a, ESI†). (c) Magnified spectra in the OH group (νOH), CH3 group (νCH3), and CH2 group (νCH2 and νCH2′) regions (note that νCH2′ corresponds to a new peak that appeared during heating). (d) Intensity change of each peak in (c) as a function of temperature; the intensity of the final state is set as zero for each case. (e) Conversion of the ethanol reforming reaction investigated using a fixed-bed flow reactor at W/F = 4.8 g min mmol−1 in the presence of HB sheets without heating pre-treatment. The measurements were conducted sequentially while increasing the temperature from 413 to 575 K. (f) Product selectivity at 573 K for the results in (e). | ||
There are two possible reasons for the disappearance of ethanol from the HB sheets during heating, namely desorption and decomposition followed by desorption. To clarify the details of the process on C2H5OH/HB during heating, in situ FT-IR spectra of C2H5OH/HB were collected during heating under vacuum. Fig. 2b shows the absorbance difference spectra during heating of the sample, in which the FT-IR spectrum at 300 K was subtracted from the spectrum at each temperature; a negative peak indicates a decrease in absorbance with heating whereas a positive peak indicates an increase (all spectra before subtraction are shown in Fig. S4a, ESI†). Fig. 2c shows the magnified spectra in the νOH, νCH3, νCH2, and νCH2′ regions (note that νCH2′ is a new peak that appeared during heating). As shown by the intensity profile of each peak as a function of temperature in Fig. 2d, each peak clearly shows a different temperature dependence. More specifically, the νOH peak intensity decreases with heating and disappears at 453 K, whereas the νCH3 peak intensity decreases slowly up to 523 K. Meanwhile, the νCH2′ peak intensity increases initially from 400 to 453 K at the expense of the νCH2 peak intensity and then decreases from 503 K to 553 K. These results indicate that the disappearance of ethanol is not due to a simple desorption process but instead due to the conversion of ethanol to other species. Importantly, the results presented in Fig. 2a–d clearly show that the OH group of ethanol disappears first. This observation is consistent with previously reported results for the catalytic process under steady-state conditions, where the HB sheets in the hydrogen-deficient state catalyze the conversion of ethanol to ethylene and water through a dehydration reaction (C2H5OH → C2H4 + H2O) at temperatures above 493 K.22Fig. 2e shows the conversion of the ethanol reforming reaction in the presence of HB sheets without heating pre-treatment, as measured using a fixed-bed flow reactor at W/F = 4.8 g min mmol−1. The measurements were conducted sequentially while increasing the temperature from 413 to 575 K. The product selectivity at 573 K is shown in Fig. 2f. Although these measurement conditions differ from those used for the FT-IR measurements of C2H5OH/HB under vacuum, the results are consistent in terms of the disappearance of the OH group of ethanol on the HB sheets. Due to the fact that the νCH3 and νCH2′ peaks are still observed in the FT-IR spectra after the disappearance of the νOH peak (Fig. 2b–d), the intermediate decomposition product of ethanol likely remains on the HB sheets at 453 K.
There are two possible intermediate decomposition products of C2H5OH that may generate the νCH3 and νCH2′ peaks even after the disappearance of νOH: (i) ethyl species (CH3CH2–) and (ii) ethoxy species (CH3CH2O–). Herein, it can be interpreted that the intermediate decomposition product is an ethyl species rather than an ethoxy species, because the νOH peak did not reappear upon heating after its complete disappearance at 453 K (Fig. 2). Importantly, for the dehydration reaction (C2H5OH → C2H4 + H2O) to occur continuously on the HB sheets as a catalytic conversion, as shown in Fig. 2e and as previously reported,22 the OH group must be formed from the ethoxy species or from the decomposed product of the ethoxy species as an intermediate to form H2O on HB sheets. Furthermore, a continuous decrease in the peak intensities at 1070–1150 cm−1 was observed up to 453 K, ultimately leading to the saturation of the peak change (Fig. S4b, ESI†). This peak can be assigned to the CO stretching vibrational mode of ethanol (νCO).31–37 If the ethoxy species is formed, the νCO peak should be observed at a similar position even at temperatures above 453 K, and its intensity should decrease with temperature. Although νCO can be interpreted to be present even above 453 K, the intensity decrease is saturated between 453 and 623 K, as can be inferred from Fig. S4b (ESI†). Therefore, the ethoxy species may not be involved in the reaction, despite possibly being formed on the surface. Upon analysis, the ethanol isotope (CD3CH2OH and CH3CD2OH) results fail to explain this phenomenon (Fig. S5 and S6, ESI†). To sufficiently elucidate the possibility of ethoxy formation, experiments using isotope-labelled ethanol of C2H5OD are required, which will be carried out in our future work. However, the absence of the νOH peak at temperatures exceeding 453 K suggests that the intermediate decomposition product of ethanol present at temperatures above 453 K is not the ethoxy species but the ethyl species.
To clarify the conversion process of ethanol adsorbed on the HB sheets more in detail, we conducted the same FT-IR experiments with isotope-labelled ethanol (CD3CH2OH and CH3CD2OH), as shown in Fig. 3 and 4, respectively (all the original spectra before subtraction are shown in Fig. S5 and S6, ESI†). For both isotopes, the observed trends were the same as those for C2H5OH (Fig. 2), i.e., the νOH peak intensity decreased with heating and disappeared at ∼440 K, whereas the νCD3, νCH2, νCD2, and νCH3 peak intensities decreased slowly up to ∼550 K. Interestingly, in the case of CD3CH2OH, an absorbance peak attributable to the B–D stretching vibration (νBD) appeared at 1895 cm−1 at ∼440 K (together with the disappearance of νOH), and the absorbance intensity increased with temperature (Fig. 3c and e). This observation indicates that the CD3 group of CD3CH2OH is decomposed on HB to form a BD bond after the disappearance of the OH group. In other words, the hydrogen deficient HB sheets may undergo a dehydrogenation reaction with the deuterium atom from the CD3 group of the CD3CH2 species on HB. The product of this reaction, CD2CH2, might subsequently be desorbed into the gas phase because both νCH2 and νCD3 intensities decrease continuously upon heating to 550 K (Fig. 3a–d). Similarly, in the case of CH3CD2OH, an absorbance peak appeared at 1895 cm−1; however, the peak intensity was considerably lower than that in the case of CD3CH2OH (Fig. 3e and 4e). More specifically, the maximum intensity ratios for νOH(MAX)/νBD(MAX) were 14.0 for CD3CH2OH and 3.4 for CH3CD2OH. These results indicate that hydrogen abstraction during the dehydrogenation reaction occurs preferentially from the methyl group rather than the methylene group of the decomposed ethanol.
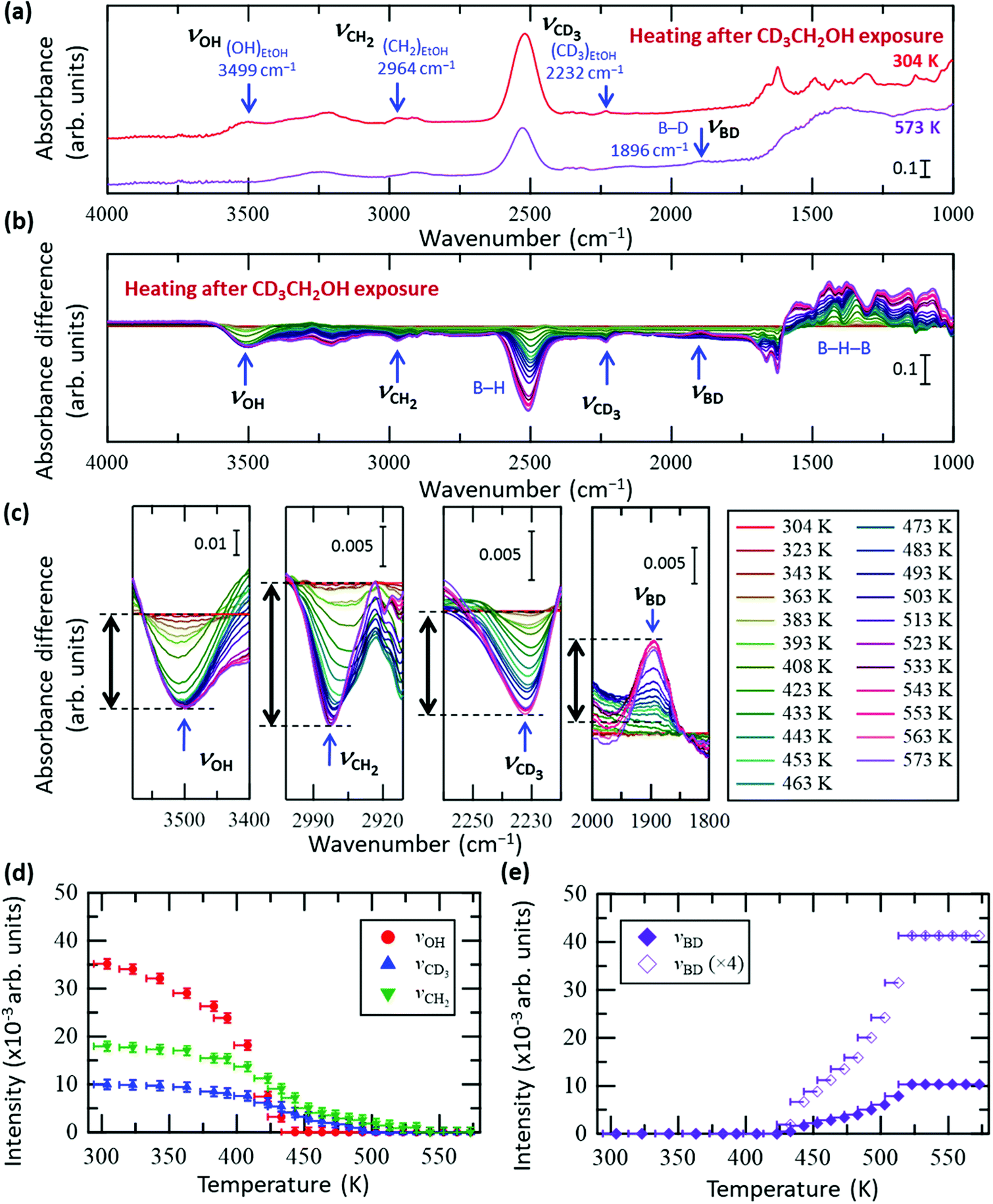 | ||
| Fig. 3 In situ FT-IR analysis results of the HB sheets with adsorbed CD3CH2OH (CD3CH2OH/HB) during heating under vacuum. (a) FT-IR spectra of CD3CH2OH/HB at 304 and 573 K under vacuum. (b) Absorbance difference spectra during heating of CD3CH2OH/HB; the FT-IR spectrum at 304 K was subtracted from the spectrum at each temperature (all spectra before subtraction are shown in Fig. S5a, ESI†). (c) Magnified spectra in the OH group (νOH), CH2 group (νCH2), CD3 group (νCD3), and BD group (νBD) regions. (d and e) Intensity change of each peak in (c) as a function of temperature. The intensity of the final state is set to zero in (d), whereas the intensity of the initial state is set to zero in (e). | ||
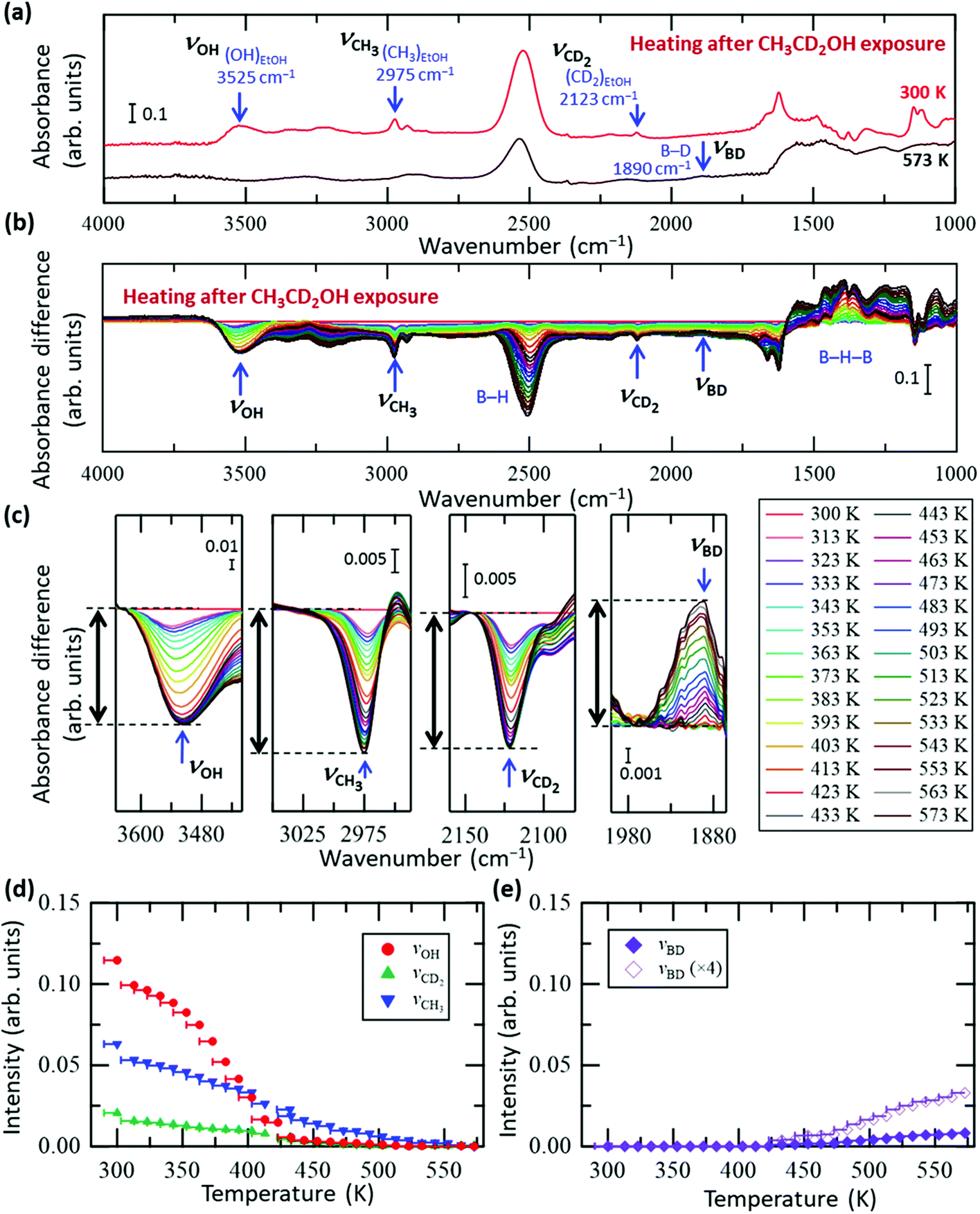 | ||
| Fig. 4 In situ FT-IR analysis results of the HB sheets with adsorbed CH3CD2OH (CH3CD2OH/HB) during heating under vacuum. (a) FT-IR spectra of CH3CD2OH/HB at 300 and 573 K under vacuum. (b) Absorbance difference spectra during the heating of CH3CD2OH/HB; the FT-IR spectrum at 300 K was subtracted from the spectrum at each temperature (all spectra before subtraction are shown in Fig. S6a, ESI†). (c) Magnified spectra in the OH group (νOH), CH3 group (νCH3), CD2 group (νCD2), and BD group (νBD) regions. (d and e) Intensity change of each peak in (c) as a function of temperature. The intensity of the final state is set to zero in (d), whereas the intensity of the initial state is set to zero in (e). | ||
Of the several possible dehydration mechanisms available to ethanol on the solid surface,28,41–46 two involve dehydrogenation of the intermediate species by the atoms of the solid surface. More specifically, in the case of CH3CD2OH, dehydrogenation may occur after the hydration reaction via the formation of an ethyl species (CH3CD2) bonded to boron as an intermediate (CH3CD2–B(inHB)), followed by dehydrogenation at the CH3 group with the concerted elimination of C–B and C–H bonds; in this case, the BH bond is expected to form without BD bond formation on the HB sheets (thus BD/BH = 0). Alternatively, it may occur via the formation of a free carbenium ion (carbocation) as a transition state, with or without an intermediate state, CH3CD2–B(inHB), followed by dehydrogenation; in this case, the carbocation formed is converted to a more stable symmetric species, in which the two C atoms are equivalent.28 Thus, all H or D atoms can be considered to have the same probability of remaining in the HB sheets when evolving into ethylene. The expected BD/BH ratio in the product sites for this mechanism is therefore 0.67. Using the experimentally obtained maximum intensity ratios for νOH(MAX)/νBD(MAX) (14.0 for CD3CH2OH and 3.4 for CH3CD2OH), the corresponding BD/BH ratio can be evaluated as 3.4/14 = 0.24, which is slightly larger than 0 but smaller than 0.67. If we consider the possible isotope exchange reaction for CH3CD2OH (for example, the isotope exchange reaction at the transition state from ethyl species to ethylene), the finite value of the ratio is reasonable; thus, dehydrogenation is suggested to occur at the methyl group rather than the methylene group of the ethyl species, without forming a carbocation. This is consistent with the fact that the positions of the νCH3 or νCD3 peaks do not shift (Fig. 2c, 3c and 4c), indicating that the bonding configuration of the methyl group is maintained as a part of the ethyl species until the methyl group is cleaved by the dehydrogenation reaction (concerted elimination), rather than the formation of another intermediate by sequential complete bond cleavages such as C–B bond cleavage followed by C–H bond cleavage. Concomitantly, the absorbance peak at 2864 cm−1 (νCH2′) observed in Fig. 2c can be assigned to the CH2 group in the ethyl species (CH3CH2–B(inHB)) generated by the decomposition of CH3CH2OH via dehydration on the HB sheets. As such, the νCH2′ peak intensity initially increases as the νOH intensity decreases, and subsequently, the νCH2′ intensity begins to decrease together with the νCH3 intensity (Fig. 2d) due to the dehydrogenation of the ethyl species.
Fig. 5 shows the Arrhenius plot of the reaction rate k (left axis), where k = (C2H5OH conversion [%])/100 × (C2H5OH flux [mol s−1])/(HB amount [mol]) was estimated from the results shown in Fig. 2e. The results for ln(ΔABD/Δt) are presented on the right axis to evaluate the formation rate of the BD bond, where ΔABD/Δt is the time derivative of the νBD intensity (ABD) for the data shown in Fig. 3e. It should be noted that while the values on the vertical axes are different, the width is the same to facilitate comparison of the slopes. Above 480 K, the slopes are similar between ln k and ln(ΔABD/Δt), indicating that the BD bond formation rate determines the reaction rate of the ethanol–ethylene conversion on the HB sheets under our experimental conditions, i.e., the rate-determining step is the dehydrogenation of the intermediate ethyl species (CH3CH2–B(inHB)). It is known that the activation energy for the bond cleavage of CD is higher than that for CH due to the zero-point energy difference; the zero-point energy of CH is approximately 1.36 times higher than that of CD, based on the vibrational energy of a simple classical harmonic oscillator with the same spring constant. The similar slopes of ln k (estimated from CH3CH2OH) and ln(ΔABD/Δt) (estimated for CD3CH2OH) in Fig. 5 indicate similar activation energies, which is consistent with the concerted mechanism (discussed in the previous paragraph), where the cleavage of both the C–B and C–H or C–D bonds proceeds simultaneously. Thus, the contribution of the C–H or C–D bond in the apparent activation energy is less for the concerted mechanism compared to that for the complete cleavage mechanism. As shown in Fig. 5, the slope of ln k is slightly less steep than those reported for the HB samples with heating pre-treatment at 573 K under a flow of Ar for 1 h (left axis)22 and a clear flat region can be seen below 480 K. These results indicate that the catalytic activity changes with heating due to the inevitable hydrogen release from HB sheets in the case of the sample without heating pre-treatment.22 However the differences in slopes are small at higher temperatures (above 480 K), suggesting that the rate-determining step of the ethanol–ethylene conversion is also the dehydrogenation of the intermediate ethyl group (CH3CH2–B(inHB)) for the HB sheets with heating pre-treatment. Since the νBD intensity is constant above 523 K (Fig. 3e), it is assumed that the formed BD species is not decomposed and released as HD or D2 but stays on the HB sheets at least at 523 K under our experimental conditions, i.e., the observed slope for ln(ΔABD/Δt) may not include the contribution of their decomposition.
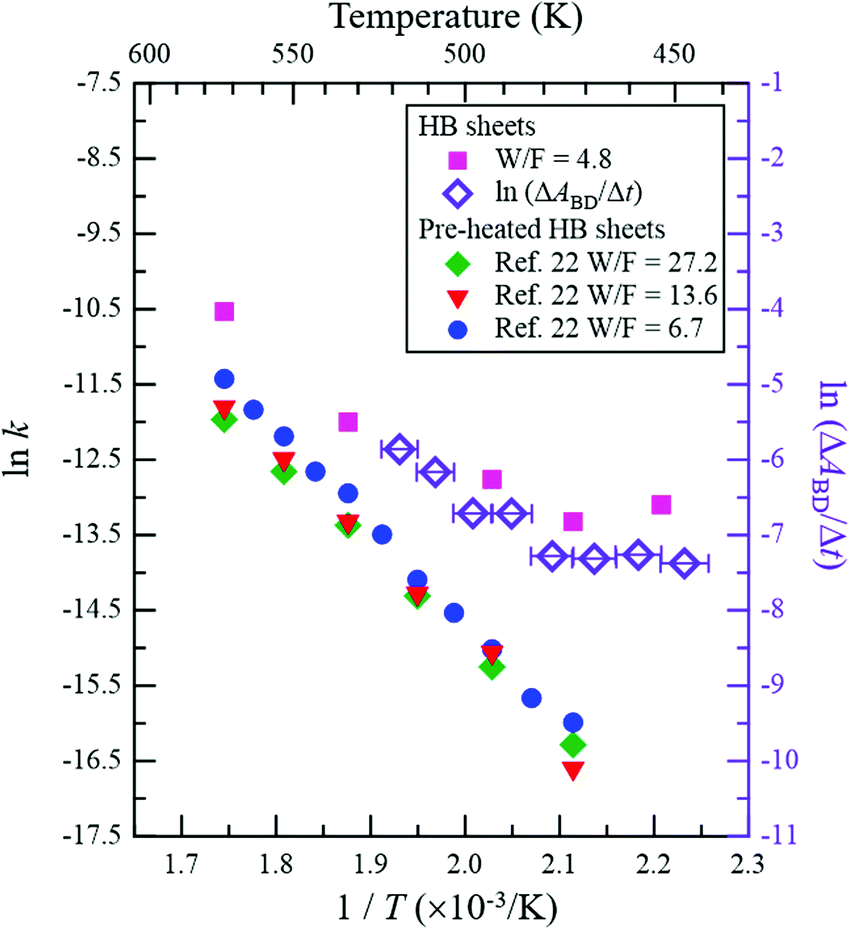 | ||
| Fig. 5 Arrhenius plot of reaction rate k (left axis). k = (C2H5OH conversion [%])/100 × (C2H5OH flux [mol s−1])/(HB amount [mol]) was estimated from the results shown in Fig. 2e (W/F = 4.8 g min mmol−1). ln(ΔABD/Δt) is plotted against 1/T (T is the temperature) on the right axis to evaluate the formation rate of the BD bond, where ΔABD/Δt is the time derivative of νBD intensity (ABD) for the data presented in Fig. 3e. The Arrhenius plots for the HB sheets with heating pre-treatment (at 573 K under Ar flow for 1 h) of the sample (W/F = 6.7, 13.6 and 27.2 g min mmol−1)22 are also shown for comparison (left axis). | ||
Finally, we proposed a mechanism for the ethanol dehydration reaction on HB sheets based on the in situ FT-IR analysis, as shown in Fig. 6 and the following chemical equations, using isotope-labelled CD3CH2OH:
| CD3CH2OH(g) + BHB(inHB) → CD3CH2OH(a)⋯B(inHB) + HB(inHB) | (1) |
| CD3CH2OH(a)⋯B(inHB) + H(inHB) → CD3CH2–B(inHB)′ + H2O(g) | (2) |
| CD3CH2–B(inHB)′ + B(inHB)′′ → (RDS)CD2CH2(g) + D–B(inHB)′′ | (3) |
 | ||
| Fig. 6 Schematic reaction mechanism for ethanol dehydration on the HB sheets. (a) HB model structure (note that the model structure shown here is the originally proposed local structure,17 whereas the real sample structure is not crystalline but amorphous and consists of a distorted hexagonal boron network structure with BH and BHB bonds19). (b) Schematic reaction mechanism for isotopically labelled ethanol (CD3CH2OH) dehydration on the HB sheets derived from the in situ FT-IR results. The adsorption structure of ethanol on the HB sheets is not clear, except for the local conversion of the structure from BHB to BH upon adsorption. Therefore, only one possible adsorption structure is shown in I. | ||
More specifically, at room temperature (∼300 K), ethanol vapor is adsorbed on the HB sheets under vacuum, which breaks some of the BHB bonds to form BH bonds as indicated by Fig. 1 (eqn (1) and Fig. 6bI). The adsorption structure of ethanol on the HB sheets is not clear, except for the local conversion of the structure from BHB to BH upon adsorption. Therefore, only one possible adsorption structure is shown in Fig. 6bI, which is assumed to be similar to the proposed adsorption structure of ethanol on H-ZSM5, in terms of the H–O–H configuration.47 Next, upon increasing the temperature, the OH group of ethanol may react with a hydrogen atom of the HB sheet, which results in the dehydration of ethanol and the formation of an ethyl group (CD3CH2–B(inHB)) as indicated in eqn (2) and Fig. 6bII, where C–O bond cleavage and C–B bond formation may occur as a concerted reaction. The reaction may be facilitated by tilting the molecular structure to promote contact between C and B (labeled 3 in Fig. 6a and bII). Meanwhile the bonds between the O and H atoms near the B atoms (labeled 1 and 2 in Fig. 6a and bI) are retained, along with a distortion in the hexagonal boron network. This is merely a hypothesis regarding the possible structures, and further examination is required to ascertain the structural change. Meanwhile, hydrogen molecules are released by the partial decomposition of the HB sheets. Finally, as the rate-determining step, the hydrogen-deficient sites of HB (i.e., boron) abstract deuterium from the methyl group of CD3CH2–B(inHB) in a dehydrogenation reaction, where the concerted elimination of C–B and C–D bonds occurs, and the desorption of CD2CH2 follows, as indicated by Fig. 2–5 (eqn (3) and Fig. 6bIII). The observed ethanol–ethylene conversion mechanism on the HB sheets is thus different from the so-called intramolecular dehydration reaction of ethanol.41 However, the mechanism may be similar to that in the case of the H-form zeolite28,42 in terms of the role of the Brønsted acidic sites and the mechanism of ethanol dehydration; the lattice oxygen and intermediate C2H5–O(lattice) species in the case of the H-form zeolite28,42 correspond to the lattice boron and C2H5–B(inHB) species, respectively, in the case of the HB sheets.
4. Conclusions
We studied the conversion process of ethanol adsorbed on HB sheets during heating under vacuum through in situ FT-IR absorption spectroscopy with isotope labelling. Up to a temperature of 450 K, the absorption band of OH in the ethanol adsorbed on HB disappeared, suggesting that the dehydration reaction may occur between the OH group of the adsorbed ethanol molecule and the hydrogen atom from the HB sheet. The decomposed ethanol then forms ethyl species on the HB sheets (CH3CH2–B(inHB)). Subsequently, in the rate-determining step, the dehydrogenation reaction forms a bond between the boron atom and a hydrogen atom from the methyl group of CH3CH2–B(inHB), rather than that of the methylene group, by a concerted elimination of the C–B and C–H bonds in CH3CH2–B(inHB) above 440 K. This is followed by the immediate desorption of the ethylene product. These results indicate that the ethanol dehydration process on HB involves the hydrogen atoms of the HB sheets. The obtained mechanistic insights are expected to aid the design of two-dimensional boron-containing materials as effective catalysts for various reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS Kakenhi (Grant No. 18K18989, 18H03874, 19H02551, and 19H05046:A01), the MEXT Element Strategy Initiative to Form Core Research Centre (JPMXP0112101001), the Ogasawara Foundation for the Promotion of Science & Engineering, and MHI Innovation Accelerator LLC. We thank Prof. Takaki Kanbara for useful discussion.References
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218 CrossRef CAS PubMed.
- E. Antolini, Appl. Catal., B, 2012, 123–124, 52 CrossRef CAS.
- N. M. Julkapli and S. Bagheri, Int. J. Hydrogen Energy, 2015, 40, 948 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790 CrossRef CAS PubMed.
- I. Boustani, Surf. Sci., 1997, 370, 355 CrossRef CAS.
- E. S. Penev, S. Bhowmick, A. Sadrzadeh and B. I. Yakobson, Nano Lett., 2012, 12, 2441 CrossRef CAS PubMed.
- B. Mortazavi and T. Rabczuk, Energies, 2018, 11, 1573 CrossRef.
- R. Zhang, Z. Li and J. Yang, J. Phys. Chem. Lett., 2017, 8, 4347 CrossRef CAS PubMed.
- F. M. Arnold, G. Seifert and J. Kunstmann, J. Phys. Commun., 2020, 4, 031001 CrossRef.
- Y. Jiao, F. Ma, J. Bell, A. Bilic and A. Du, Angew. Chem., Int. Ed., 2016, 55, 10292 CrossRef CAS PubMed.
- T. A. Abtew, B. Shih, P. Dev, V. H. Crespi and P. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094108 CrossRef.
- Z. Zhu, X. Cai, C. Niu, C. Wang and Y. Jia, Appl. Phys. Lett., 2016, 109, 153107 CrossRef.
- A. J. Mannix, B. Kiraly, M. C. Hersam and N. P. Guisinger, Nat. Rev. Chem., 2017, 1, 0014 CrossRef CAS.
- T. Kondo, Sci. Technol. Adv. Mater., 2017, 18, 780 CrossRef PubMed.
- H. Nishino, T. Fujita, N. T. Cuong, S. Tominaka, M. Miyauchi, S. Iimura, A. Hirata, N. Umezawa, S. Okada, E. Nishibori, A. Fujino, T. Fujimori, S. Ito, J. Nakamura, H. Hosono and T. Kondo, J. Am. Chem. Soc., 2017, 139, 13761 CrossRef CAS PubMed.
- I. Tateishi, N. T. Cuong, C. A. S. Moura, M. Cameau, R. Ishibiki, A. Fujino, S. Okada, A. Yamamoto, M. Araki, S. Ito, S. Yamamoto, M. Niibe, T. Tokushima, D. E. Weibel, T. Kondo, M. Ogata and I. Matsuda, Phys. Rev. Mater., 2019, 3, 024004 CrossRef CAS.
- S. Tominaka, R. Ishibiki, A. Fujino, K. Kawakami, K. Ohara, T. Masuda, I. Matsuda, H. Hosono and T. Kondo, Chem, 2020, 6, 406 CAS.
- S. Ito, T. Hirabayashi, R. Ishibiki, R. Kawamura, T. Goto, T. Fujita, A. Yamaguchi, H. Hosono, M. Miyauchi and T. Kondo, Chem. Lett., 2020, 49, 789 CrossRef CAS.
- R. Kawamura, N. T. Cuong, T. Fujita, R. Ishibiki, T. Hirabayashi, A. Yamaguchi, I. Matsuda, S. Okada, T. Kondo and M. Miyauchi, Nat. Commun., 2019, 10, 4880 CrossRef PubMed.
- A. Fujino, S. Ito, T. Goto, R. Ishibiki, J. N. Kondo, T. Fujitani, J. Nakamura, H. Hosono and T. Kondo, ACS Omega, 2019, 4, 14100 CrossRef CAS PubMed.
- D. N. Bakoyannakis, D. Zamboulis, G. A. Stalidis and E. A. Deliyanni, J. Chem. Technol. Biotechnol., 2001, 76, 1159 CrossRef CAS.
- M. A. Christiansen, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2013, 3, 1965 CrossRef CAS.
- Q. Pan, A. Ramanathan, W. Kirk Snavely, R. V. Chaudhari and B. Subramaniam, Top. Catal., 2014, 57, 1407 CrossRef CAS.
- F. Roca, L. De Mourgues and Y. Trambouze, J. Catal., 1969, 14, 107 CrossRef.
- T. Maihom, P. Khongpracha, J. Sirijaraensre and J. Limtrakul, ChemPhysChem, 2013, 14, 101 CrossRef CAS PubMed.
- J. N. Kondo, H. Yamazaki, R. Osuga, T. Yokoi and T. Tatsumi, J. Phys. Chem. Lett., 2015, 6, 2243 CrossRef CAS PubMed.
- H. Nishino, T. Fujita, A. Yamamoto, T. Fujimori, A. Fujino, S. Ito, J. Nakamura, H. Hosono and T. Kondo, J. Phys. Chem. C, 2017, 121, 10587 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-P. Perchard and M.-L. Josien, J. Chim. Phys. Phys.-Chim. Biol., 1968, 65, 1834 CrossRef CAS.
- J.-P. Perchard and M.-L. Josien, J. Chim. Phys. Phys.-Chim. Biol., 1968, 65, 1856 CrossRef CAS.
- J. Eng, Jr., K. Raghavachari, L. M. Struck, Y. J. Chabal, B. E. Bent, G. W. Flynn, S. B. Christman, E. E. Chaban, G. P. Williams, K. Radermacher and S. Mantl, J. Chem. Phys., 1997, 106, 9889 CrossRef.
- D. J. Burke, A. J. Wolff, J. L. Edridge and W. A. Brown, J. Chem. Phys., 2008, 128, 104702 CrossRef CAS PubMed.
- N. Boudin, W. A. Schutte and J. M. Greenberg, Astron. Astrophys., 1998, 331, 749 CAS.
- S. C. Street and A. J. Gellman, Colloids Surf., 1995, 105, 27 CrossRef CAS.
- A. J. Barnes and H. E. Hallam, Trans. Faraday Soc., 1970, 66, 1932 RSC.
- J. Xu, X. Zhang, R. Zenobi, J. Yoshinobu, Z. Xu and J. T. Yates, Jr., Surf. Sci., 1991, 256, 288 CrossRef CAS.
- Y. Yu, K. Lin, X. Zhou, H. Wang, S. Liu and X. Ma, J. Phys. Chem. C, 2007, 111, 8971 CrossRef CAS.
- L. Wang, T. Ishiyama and A. Morita, J. Phys. Chem. A, 2017, 121, 6701 CrossRef CAS PubMed.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505 CrossRef CAS.
- J. N. Kondo, K. Ito, E. Yoda, F. Wakabayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 10969 CrossRef CAS PubMed.
- S. Roy, G. Mpourmpakis, D. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846 CrossRef CAS.
- J. F. DeWilde, H. Chiang, D. A. Hickman, C. R. Ho and A. Bhan, ACS Catal., 2013, 3, 798 CrossRef CAS.
- J. F. DeWilde, C. J. Czopinski and A. Bhan, ACS Catal., 2014, 4, 4425 CrossRef CAS.
- K. Alexopoulos, M. John, K. Van der Borght, V. Galvita, M. Reyniers and G. B. Marin, J. Catal., 2016, 339, 173 CrossRef CAS.
- F. Haase and J. Sauer, J. Am. Chem. Soc., 1995, 117, 3780 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp03079a |
| This journal is © the Owner Societies 2021 |