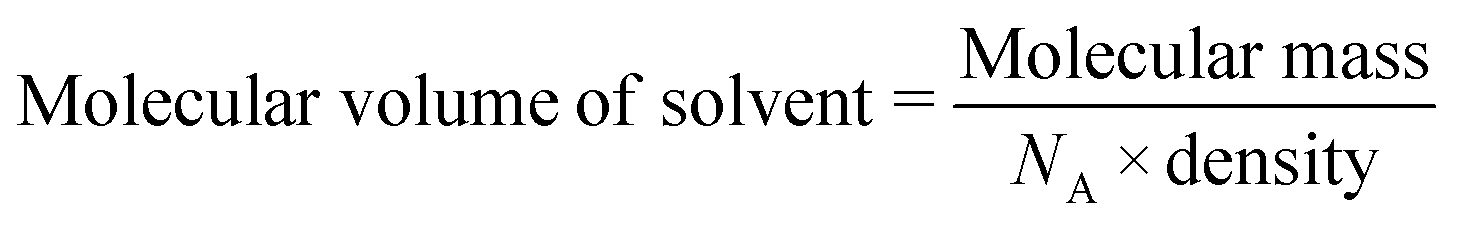DOI:
10.1039/D1CE01052B
(Paper)
CrystEngComm, 2021,
23, 7162-7170
Robust pyridylbenzoate metal–organic frameworks as sorbents for volatile solvents and gases†
Received
9th August 2021
, Accepted 20th September 2021
First published on 21st September 2021
Abstract
Two activated isostructural porous compounds, [Co(34pba)(44pba)]n (1d) and [Zn(34pba)(44pba)]n (2d) (where 34pba is 3-(pyridin-4-yl)benzoate, 44pba is 4-(pyridin-4-yl)benzoate, and d indicates the activated phase) were used for the adsorption of halogenated volatile organic compounds (VOCs), iodine, carbon dioxide (CO2), and hydrogen. Elucidation of single-crystal structures found that the desolvated phases 1d and 2d show rotational disorder of one ring on each linker. The crystal structure of {[Co(34pba)(44pba)]·1.6 I2}n (1dI2) was obtained by vapour sorption of iodine into 1d, and this could be desolvated to a phase isostructural with 1d. Thus the MOF is robust to cycling through sorption/desorption processes with an accompanying order/disorder phase transition. Activation energies for the desorption of dichloromethane, dibromomethane, and iodine were measured as 70, 60, and 77 kJ mol−1 respectively. Both activated compounds were tested as gas sorbents, with 1d showing higher adsorption capacity than 2d for both carbon dioxide and hydrogen.
1. Introduction
Porous metal–organic frameworks (MOFs) have been widely studied in applications such as gas sorption and storage, catalysis, luminescence, magnetism, and sensing.1–8 Features such as surface area, solvent-accessible void volume, or pore size play an important role in the applications of a given MOF. Sorption properties are governed by supramolecular interactions between the framework (host) and the adsorbed species (guest).9,10 The presence of a specific substituent in a guest molecule allows a given set of specific host–guest supramolecular interactions which may include hydrogen or halogen bonding.11,12 These interactions often involve structural changes which can reversibly induce pore opening and closing.13–15 Such reversibility in sorption/desorption in response to chemical and physical stimuli are important in the recovery of adsorbents. Thus, host–guest chemistry can be used to design adsorbents for specific purposes. One such application is the removal of harmful volatile organic compounds (VOCs) in the environment.16,17
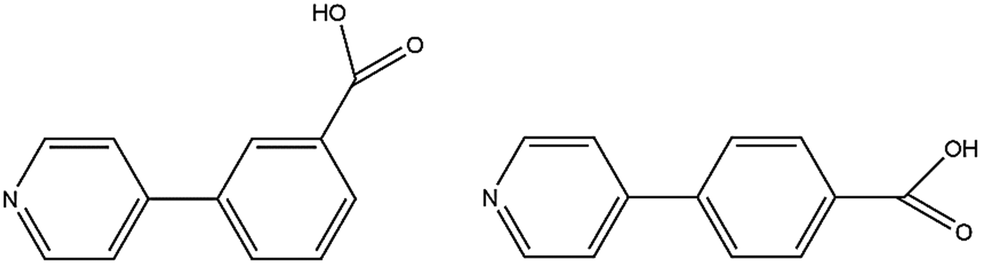
We previously reported isomorphous MOFs in which the channel double-walls are formed by the related linkers 3-(pyridin-4-yl)benzoic acid (H34pba) and 4-(pyridin-4-yl)benzoic acid (H44pba), Scheme 1. These MOFs showed selective adsorption for the chlorinated VOCs dichloromethane, chloroform, and chlorobenzene.18 Guest molecules could be removed with the retention of the framework. To further understand the robustness of this framework through the sorption of additional halogenated compounds, we now consider the use of [Co(34pba)(44pba)]n (1d) and [Zn(34pba)(44pba)]n (2d) (where 34pba is 3-(pyridin-4-yl)benzoate, 44pba is 4-(pyridin-4-yl)benzoate, and d indicates the activated phase) as adsorbents for the bromo- and iodo-derivatives of the chloro-compounds studied previously. Furthermore, the sorption of iodine vapour, and of carbon dioxide and hydrogen gases by the same adsorbents is also presented here.
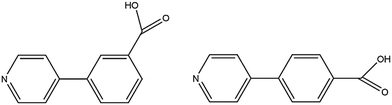 |
| | Scheme 1 3-(Pyridin-4-yl)benzoic acid (H34pba) and 4-(pyridin-4-yl)benzoic acid (H44pba). | |
2. Materials and methods
2.1 Preparation and activation of MOFs and sorption experiments
The preparation of the precursors of the activated adsorbents, {[Co(34pba)(44pba)]n·DMF} (1) and {[Zn(34pba)(44pba)]n·DMF} (2) were reported previously.18 To obtain the activated phases 1d and 2d, they were heated at 210 °C under vacuum for six hours. This protocol was selected to ensure complete desolvation but not decomposition of the MOFs. The corresponding crystallographic data of 1d and 2d are presented here.
Powdered activated samples of mass 5–7 mg were placed in narrow glass vials. The latter were placed into larger vials containing the relevant VOC and then sealed to allow vapour sorption at room temperature (Fig. S1†). The VOCs selected for the study were dibromomethane (CH2Br2), bromoform (CHBr3), bromobenzene (BrBenz), diiodomethane (CH2I2), iodoform (CHI3), iodobenzene (IBenz). Code names for the corresponding phases from 1d and 2d are provided in Table 1. Samples were analysed after exposure for between one and fourteen days depending on the vapour pressure of the solvent.
Table 1 Code names of the phases obtained from sorption experiments
| Halogenated compound |
Sorbent 1d |
Sorbent 2d |
| Dibromomethane, CH2Br2 |
1dCH2Br2 |
2dCH2Br2 |
| Bromoform, CHBr3 |
1dCHBr3 |
2dCHBr3 |
| Bromobenzene, BrBenz |
1dBrBenz |
2dBrBenz |
| Diiodomethane, CH2I2 |
1dCH2I2 |
2dCH2I2 |
| Iodoform, CHI3 |
1dCHI3 |
2dCHI3 |
| Iodobenzene, IBenz |
1dIBenz |
2dIBenz |
| Iodine, I2 |
1dI2 |
— |
The sorption for iodine in 1d was carried out in a similar way, using the sublimation of solid iodine to expose single crystals to iodine vapour, with samples taken after two hours, two days, eight days, and finally two weeks. Each sample was analyzed by thermogravimetry (TGA) to determine the mass loss owing to desorption of guests and by powder X-ray diffraction (PXRD) to identify changes in phase. A single crystal was selected for analysis and the structure reported as 1dI2. The desorption of iodine from 1dI2 was performed by soaking crystals of 1dI2 in methanol for three days, to recover the activated form (1dI2d). As this retained its crystallinity, the single crystal structure of 1dI2d is also reported here.
Gas sorption capacity of the 1d and 2d adsorbents was investigated for carbon dioxide (CO2) and hydrogen (H2) gases using a Micromeritics 3Flex Surface Area Analyzer. After grinding the sample, masses between 130–140 mg were prepared using a Micromeritics Flowprep using a flow of nitrogen over the samples for 2 h with continuous heating at 60 °C. Thereafter, samples were heated at 150 °C under vacuum for 2 h prior to the sorption analysis. The sorption for CO2 was carried out at various temperatures in order to determine the heat of adsorption (Qst), while the sorption capacity for H2 was only carried out at 77 K. Loading of gas into the samples was characterized by a pressure change from 0 mmHg to the maximum pressure equilibrium (between 600–1000 mmHg). The complete sorption corresponded to the equilibrium pressure which was followed by the desorption process.
2.2 Thermogravimetric analysis (TGA)
Mass losses attributable to desolvation of guest molecules were determined by thermogravimetric analysis (TGA) using a TA instrument TA-Q500 on 1–2 mg samples in open platinum pans under nitrogen gas flow (50 mL min−1) at a heating rate of 10 °C min−1.
2.3 Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) patterns were measured on a Bruker D8 Advance X-ray diffractometer operating in a DaVinci geometry equipped with a Lynxeye detector using Cu-Kα-radiation (λ = 1.5406 Å). X-rays were generated at 30 kV and 40 mA. Samples were placed on a zero-background sample holder and scanned over a range of 4–40° in 2θ with 0.016° step size per second.
2.4 Crystal structure determination
Single crystals were selected using optical microscopy under plane-polarized light. Intensity data were recorded based on a Bruker KAPPA APEX II DUO diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Data were corrected for Lorentz-polarization effects and for absorption (SADABS19). The structures were solved by direct methods in SHELXS and refined by full-matrix least-squares on F2 using SHELXL20 within the XSEED interface.21 Non-hydrogen atoms were located in difference electron density maps and were refined anisotropically while hydrogen atoms were placed in calculated positions and refined isotropically. Details of the crystal structures are given in Tables 2 and S1.†
Table 2 Crystallographic data
| Compound |
1
d
|
2
d
|
1
dI
2
|
1
dI
2
d
|
|
2
d was found to contain ca. 0.25 water molecules per Zn, which are disordered over five sites.
|
| Formula |
C24H16N2O4Co |
C24H16N2O4Zn·0.25H2O |
C24H16N2O4Co·1.6I2 |
C24H16N2O4Co |
| Formula mass (g mol−1) |
455.34 |
466.26 |
658.36 |
455.34 |
| Crystal size (mm3) |
0.080 × 0.090 × 0.14 |
0.030 × 0.060 × 0.090 |
0.080 × 0.12 × 0.18 |
0.080 × 0.12 × 0.18 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
P21/c |
|
a (Å) |
10.3931(14) |
10.2197(5) |
10.114(3) |
10.4014(11) |
|
b (Å) |
16.027(2) |
16.1263(8) |
16.501(4) |
16.1330(17) |
|
c (Å) |
14.996(2) |
14.8826(7) |
14.700(4) |
14.6878(15) |
|
β (°) |
98.243(2) |
98.059(2) |
97.159(4) |
98.482(2) |
|
V (Å3) |
2472.2(6) |
2428.5(2) |
2434.3(11) |
2437.7(4) |
|
T (K) |
100(2) |
100(2) |
293(2) |
293(2) |
|
Z
|
4 |
4 |
4 |
4 |
|
D
c (g cm−3) |
1.223 |
1.275 |
1.796 |
1.241 |
|
μ (Mo-Kα) (mm−1) |
0.722 |
1.042 |
2.766 |
0.733 |
|
F(000) |
932 |
954 |
1271 |
932 |
| Range scanned, θ (°) |
1.870–27.963 |
2.526–28.318 |
2.029–26.425 |
1.886–25.043 |
| No. reflections collected |
22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 903 903 |
139665 |
18540 |
13599 |
| No. unique reflections |
5889 |
6052 |
4970 |
4301 |
| No. reflections with I ≥ 2σ(I) |
3893 |
4793 |
3691 |
3067 |
| Parameters/restraints |
335/0 |
354/0 |
334/3 |
358/0 |
| Goodness of fit, S |
1.042 |
1.031 |
1.110 |
1.031 |
| Final R indices (I ≥ 2σ(I)) |
R
1 = 0.0655 |
R
1 = 0.0518 |
R
1 = 0.0687 |
R
1 = 0.0430 |
| wR2 = 0.1677 |
wR2 = 0.1329 |
wR2 = 0.1838 |
wR2 = 0.0959 |
| Final R indices (all data) |
R
1 = 0.1085 |
R
1 = 0.0715 |
R
1 = 0.0934 |
R
1 = 0.0749 |
| wR2 = 0.1878 |
wR2 = 0.1459 |
wR2 = 0.1956 |
wR2 = 0.1071 |
| Min, max e− density/(e Å−3) |
0.697, −0.585 |
0.608, −0.546 |
2.179, −0.643 |
0.488, −0.361 |
3. Results and discussion
3.1 Crystal structures of 1d and 2d
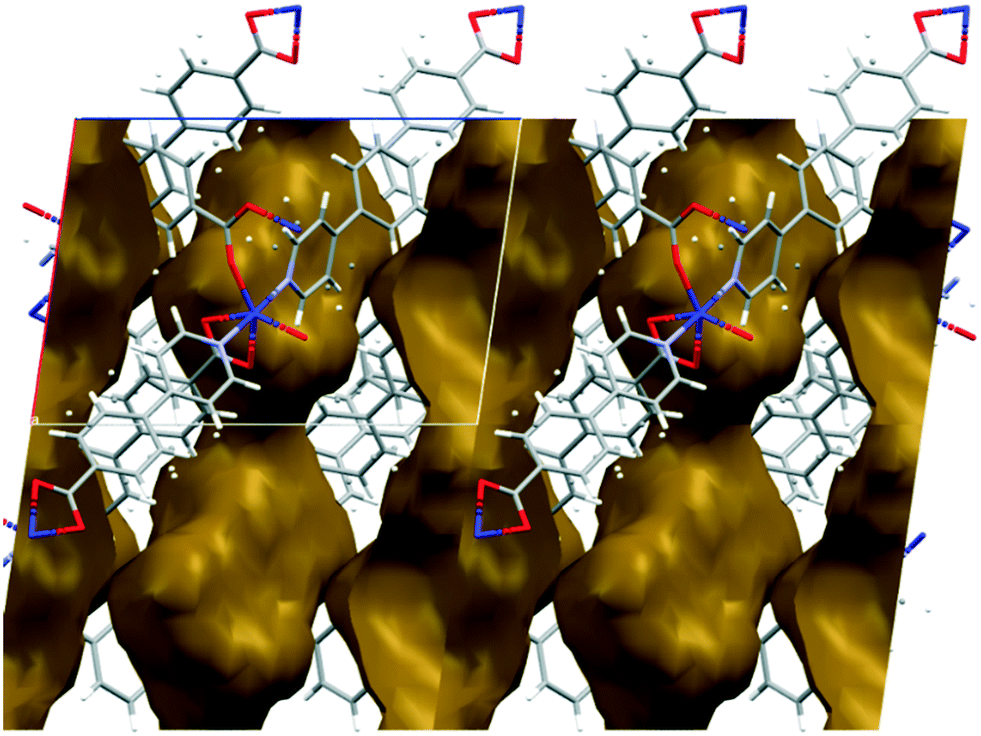
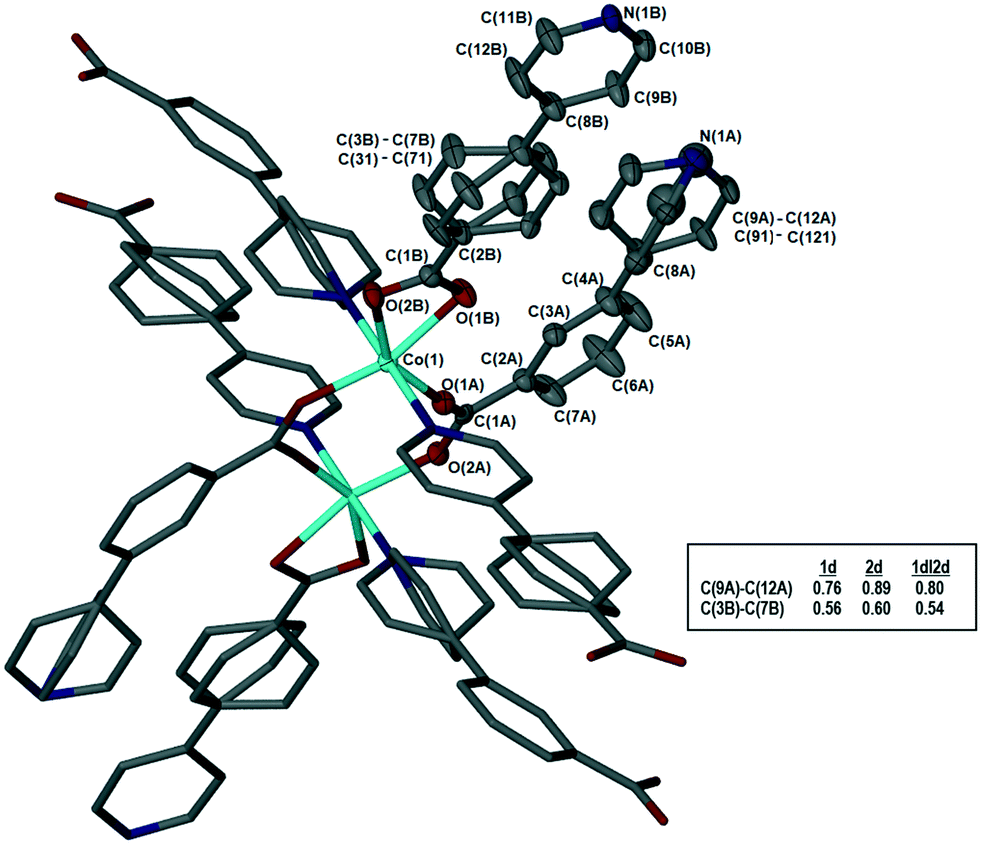
The metal–organic frameworks {[Co(34pba)(44pba)]n·DMF} (1) and {[Zn(34pba)(44pba)]n·DMF} (2) were reported previously.18 These form frameworks where each side is comprised of a 34pba and a 44pba linker, giving a double-walled MOF of bcu topology. Hour-glass channels (ca. 8 Å at the widest point and ca. 3.5 Å at the narrowest) run parallel to [100] (Fig. 1). At that time, we were unable to obtain a single crystal of these MOFs after desorption of the guest species, although PXRD showed minimal changes in phase on desorption. Now, after optimizing the desorption process, we are able to report the single crystal structures of both Co(II) (1d) and Zn(II) (2d) MOFs. As anticipated the frameworks do not change significantly, except that in both 1d and 2d, one ring on each of the 34pba and 44pba linkers shows rotational disorder. This is more pronounced for the 44pba moiety, in which the benzyl ring is disordered over two positions with almost equal site occupancies, while in the 34pba moiety the pyridyl ring is disordered over two positions with the major position having approximately 80% occupancy. This disorder is illustrated in Fig. 2.
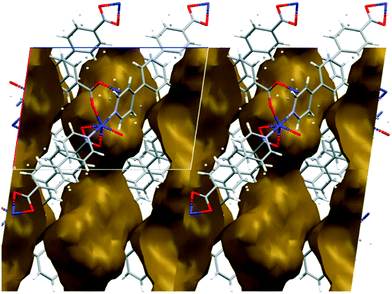 |
| | Fig. 1 Channels in the structure of 1d run parallel to [100] and have an hour-glass shape (widest point ca. 8 Å, narrowest point ca. 3.5 Å). | |
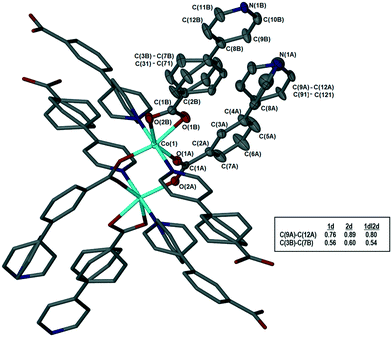 |
| | Fig. 2 Coordination geometry and atom labelling, illustrated for 1d. The asymmetric unit is shown with thermal ellipsoids at 50% probability; hydrogen atoms omitted for clarity. Inset table shows site occupancies of the major components for the disordered rings in 1d, 2d, and 1dI2d. | |
3.2 Sorption of halogenated VOCs by 1d and 2d
In this study, we have extended the range of halogenated volatile organic compounds which are included by 1d and 2d. In doing so, we expected to see differences in loading capacity of the frameworks based on differences in halogen substituent, owing to their differences in polarity, vapour pressure and ability to participate in non-covalent interactions such as halogen bonding.
The volumes of the solvent-accessible voids in 1d and 2d were estimated using Mercury22 with a probe radius of 1.2 Å and a grid spacing of 0.2 Å. These were estimated as 481 Å3 (20%) per unit cell for 1d and 571 Å3 (24%) per unit cell for 2d. Results of the sorption experiments are presented in Table 3. The loading capacity values (Lc) were calculated from TGA analysis (Fig. S2†) using the crystallographically derived void volume and the liquid density of the corresponding solvent. We approximated the maximum loading capacity (MLc) for the empty frameworks using eqn (1):
| | | MLc = (solvent accessible void volume)/(Z × molecular volume) | (1) |
The molecular volumes of the VOCs were calculated using their liquid density according to
eqn (2):
| | 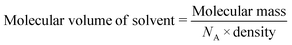 | (2) |
The loading capacity (
Lc) in the proposed formula {[M(34pba)(44pba)]·
x solvent}
n for both systems is lower than the maximum loading capacity. The dihalomethanes have lower molecular volumes and higher vapour pressures than their corresponding haloforms, which may account for their higher loading capacities. The aromatic halogenated VOCs have lower vapour pressures, but their loading capacity was less affected. In part, this is probably due to the additional interactions possible for these compounds (see next paragraph). In general, higher sorption (% loading capacity) was observed in
1d (over
2d) for each individual VOC.
Table 3 Uptake of selected solvents by the activated phases 1d and 2d
| VOC |
Experimental mass loss, TGA (%) |
Temperature range of mass loss (°C) |
Loading capacity, Lc (x in formula): {[M(34pba)(44pba)]·x solvent}n |
MLc |
% loading capacity |
|
Reported in ref. 18.
|
|
1
d
|
| CH2Cl2a |
14.0 |
60–154 |
0.9 |
1.3 |
69 |
| CH2Br2 |
23.1 |
70–200 |
0.8 |
1.2 |
67 |
| CH2I2 |
33.3 |
70–260 |
0.8 |
1.0 |
80 |
| CHCl3a |
17.1 |
118–285 |
0.8 |
1.0 |
80 |
| CHBr3 |
19.2 |
91–236 |
0.4 |
1.0 |
40 |
| CHI3 |
26.0 |
125–278 |
0.4 |
0.8 |
50 |
| ClBenza |
13.0 |
87–264 |
0.6 |
0.8 |
75 |
| BrBenz |
19.2 |
38–235 |
0.7 |
0.8 |
88 |
| IBenz |
19.1 |
40–277 |
0.5 |
0.7 |
71 |
|
2
d
|
| CH2Cl2a |
11.0 |
88–220 |
0.7 |
1.4 |
50 |
| CH2Br2 |
23.0 |
78–266 |
0.8 |
1.2 |
67 |
| CH2I2 |
10.0 |
87–290 |
0.2 |
1.1 |
18 |
| CHCl3a |
13.3 |
110–232 |
0.6 |
1.1 |
55 |
| CHBr3 |
12.7 |
109–233 |
0.3 |
1.0 |
30 |
| CHI3 |
10.0 |
105–248 |
0.1 |
0.9 |
11 |
| ClBenza |
11.0 |
61–252 |
0.5 |
0.9 |
55 |
| BrBenz |
14.0 |
58–278 |
0.5 |
0.8 |
63 |
| IBenz |
10.6 |
58–270 |
0.3 |
0.8 |
38 |
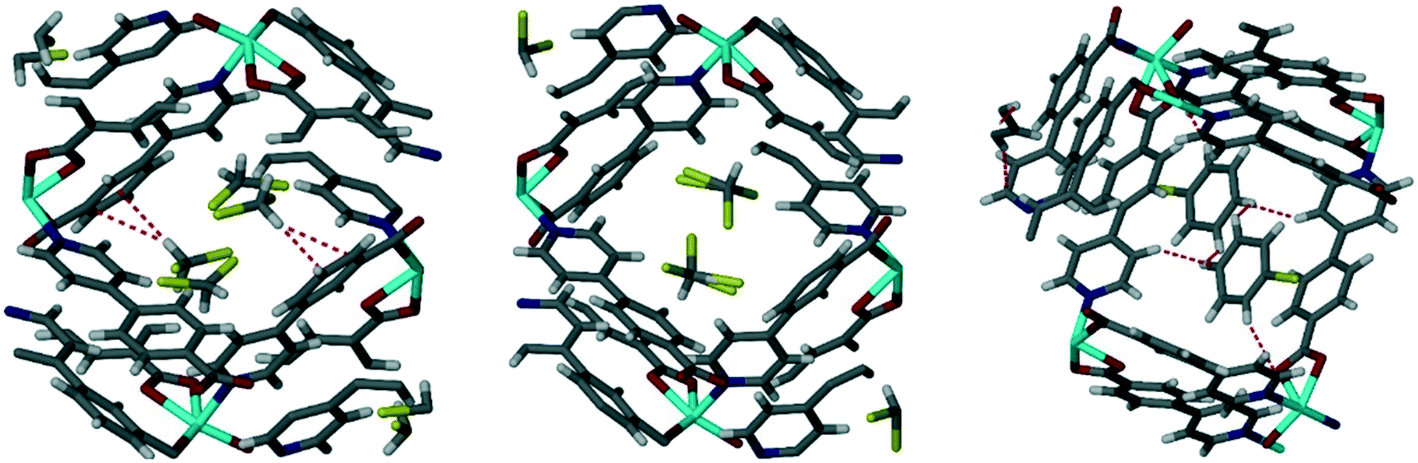
Single crystal data was obtainable for the inclusion of the chlorinated VOCs in Table 3 in the framework of 1d. These structures were previously reported18 and showed that the guest VOCs are included in the MOF channels and held in place by Cl⋯π and C–H⋯π interactions and by π⋯π interactions between chlorobenzene and the MOF walls (Fig. 3). The crystallographic information for these structures is reproduced in the ESI,† Table S1.
 |
| | Fig. 3 Single crystal structures of chlorinated VOCs in 1d, from left to right 1dCH2Cl2, 1dCHCl3, 1dClBenz. Non-covalent interactions are shown with dotted lines.18 | |
Unfortunately, single crystals could not be obtained for the remaining sorption products, but PXRD analysis confirms that only subtle changes in the frameworks are evident in all cases (Fig. S3†).
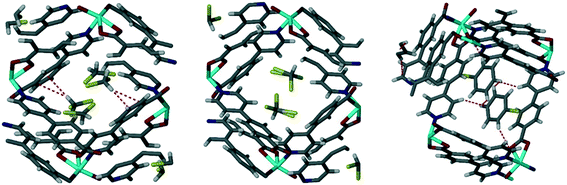
3.3 Sorption of iodine by 1d: structure and solvatochromism
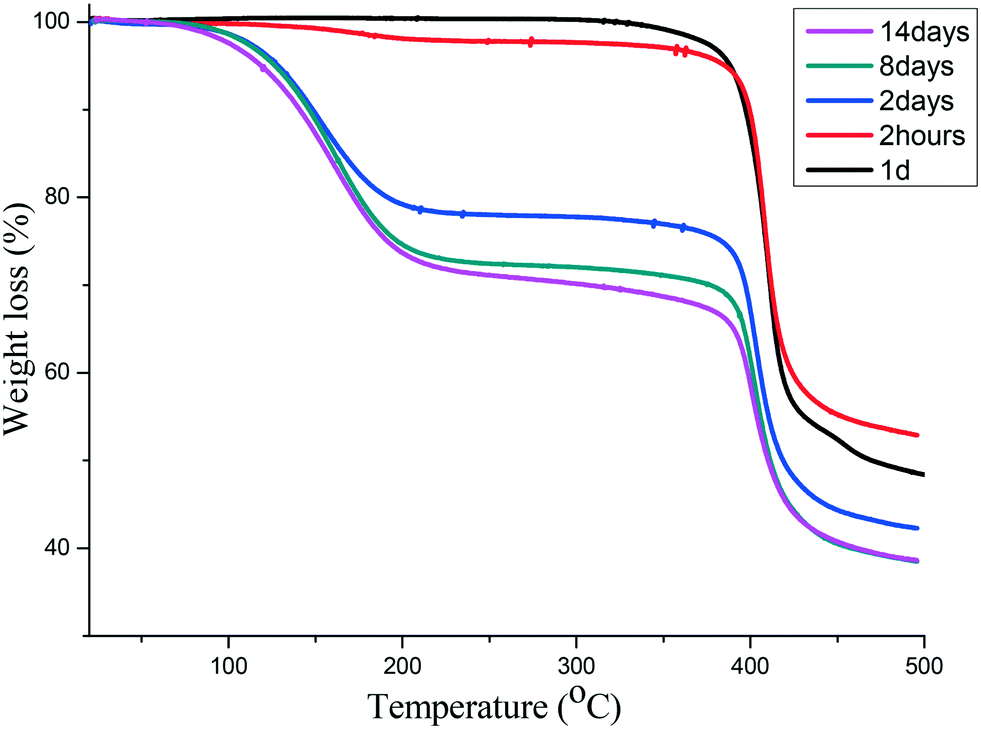
Using a similar process to that described above, we followed the sorption of iodine into 1d over a period of two weeks using TGA and found that the maximum mass loss of 31% was reached after 8 days (Fig. 4). This corresponds to 1.6 iodine molecules per [Co(34pba)(44pba)] unit.
 |
| | Fig. 4 TGA of iodine sorption by 1d at different time intervals. The maximum is reached after 8 days. | |
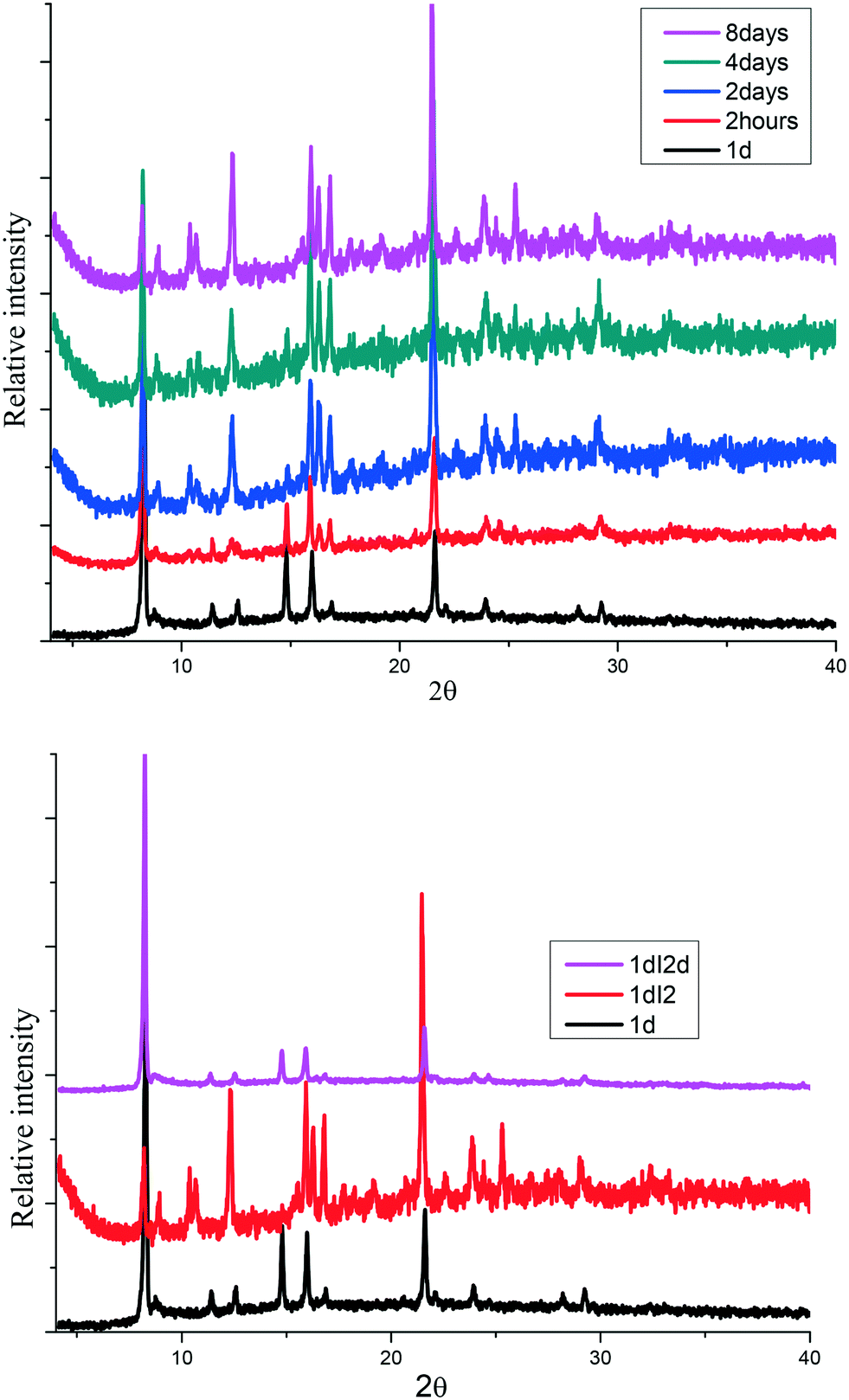
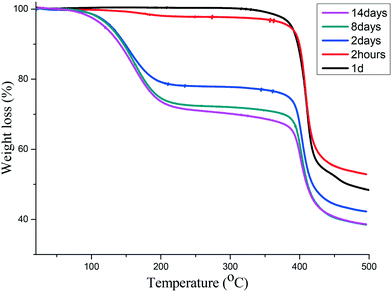
The sorption process was characterized using PXRD where gradual changes of patterns up to eight days were observed, Fig. 5(top). Most notable is a gradual decrease in peak intensities at 8° and 15° while there was a gradual increase in the peak intensities at 9 and 22.5°. New peaks appear at 10.5 and 25°. These changes can be attributed to the interaction of iodine molecule and the channels of the 1d framework (see crystal structure of 1dI2 which is described below). The uptake of iodine could be followed visually (Fig. S4†).
 |
| | Fig. 5 (top) Gradual phase changes related to the sorption of iodine in 1d to form 1dI2, (bottom) PXRD for iodine desorption from 1dI2 into methanol to form 1dI2d. | |
The desorption of iodine from 1dI2 into methanol could be monitored by following the colour changes that occur at a constant temperature of 22 °C (Fig. 6). The PXRD (Fig. 5 bottom) indicates the robustness of the phase changes during the recovery of iodine into methanol.
 |
| | Fig. 6 Photographs showing colour change on the release of iodine from 1dI2 into methanol. | |
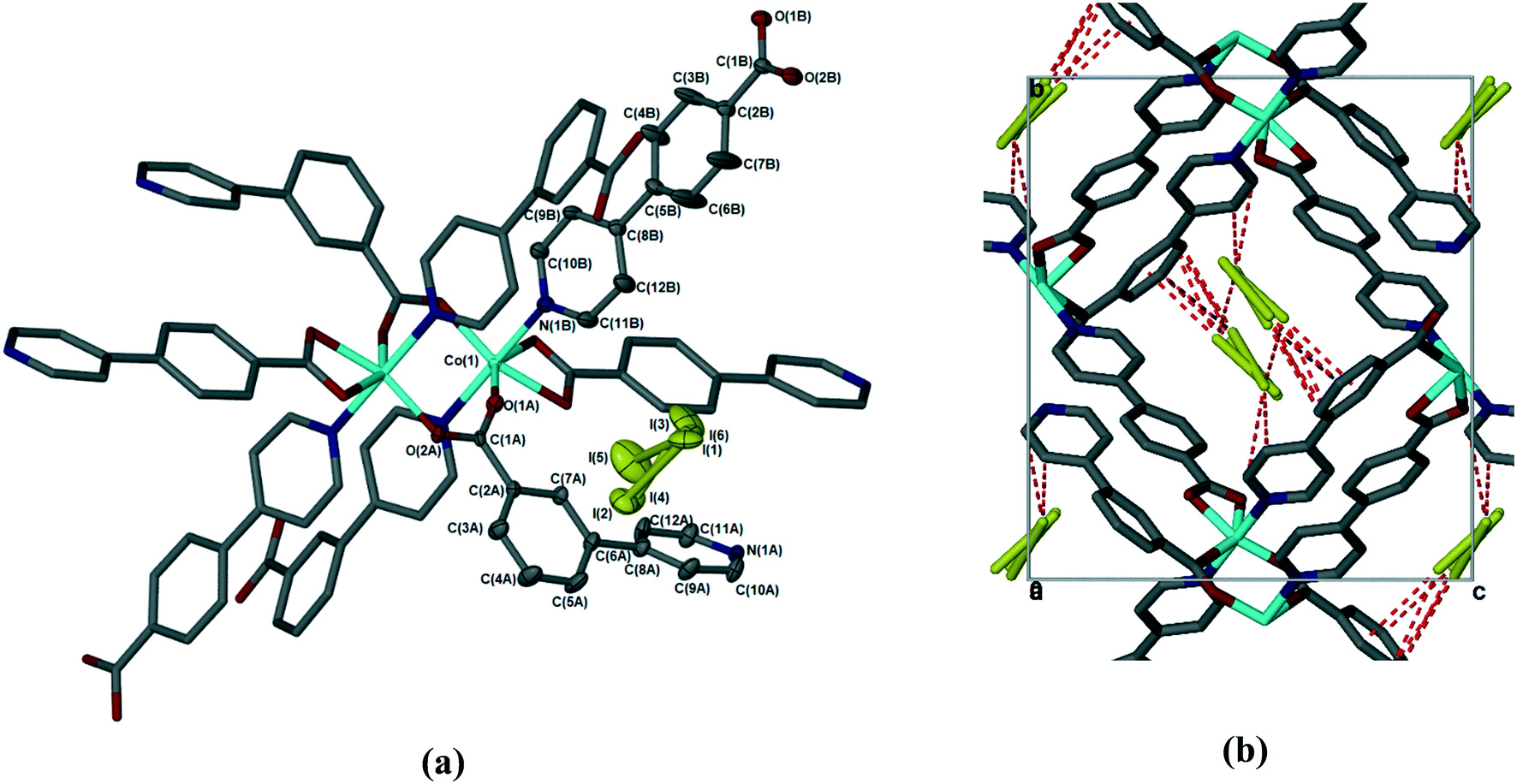
The sorption of iodine by 1d was sufficiently slow that the crystal used retained its single crystallinity and allowed us to obtain a fully elucidated structure, 1dI2 (Table 2 and Fig. 7). In common with all previously reported structures in which the MOF contains guest molecules, the framework is perfectly ordered, and is isostructural with the structures in which the MOF contains dimethylformamide, dichloromethane, chloroform, or chlorobenzene. To quantify the amount of iodine present in 1dI2, we used the SQUEEZE routine in Platon23 to estimate the void size and number of electrons present. For 1dI2 this indicated solvent-accessible voids of 644 Å3 and 418 e−/unit cell, which corresponds to ca. 2 iodine molecules per [Co(34pba)(44pba)] unit (in good agreement with the TGA data). We were able to locate several disordered iodine molecules occupying the same cavity; these were modelled over three sites with total occupancy adding to 0.8 iodine molecules per [Co(34pba)(44pba)] unit. We note that residual electron density in the same area indicates the possibility of further iodine being present but we were unable to model this in a chemically meaningful way. For the iodine molecules we could model we can identify a number of non-covalent interactions of the form I⋯π and I⋯I between the iodine molecules and the channel walls and between iodine molecules themselves. These are shown in Fig. 7 and detailed in Table 4.
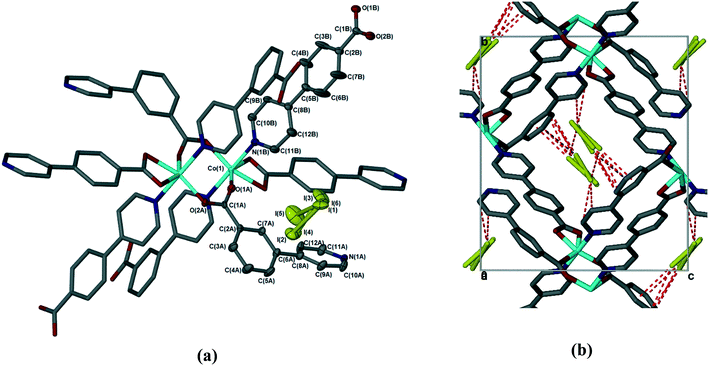 |
| | Fig. 7 (a) Coordination geometry and SBU in 1dI2. The asymmetric unit is shown with thermal ellipsoids at the 50% probability level. (b) Weak interactions (dotted lines) between the iodine guest molecule and MOF in 1dI2. Hydrogen atoms have been omitted for clarity. | |
Table 4 Non-covalent interactions in 1dI2
| Atoms |
Interatomic distance (Å) |
| I2–C4A |
3.566 |
| I2–C5A |
3.628 |
| I3–C9A |
3.604 |
| I3–C10A |
3.636 |
| I4–C4A |
3.533 |
| I4–C5A |
3.460 |
| I4–C6A |
3.662 |
| I5–C5A |
3.820 |
| I5–C6A |
3.625 |
| I3–I5 |
3.875 |
After the desorption in methanol, a single crystal was analyzed, to give the crystal structure 1dI2d (Table 2). 1dI2d reverts to the same structure as 1d, with identical disorder re-appearing in the pyridyl rings of the 34pba linker and in the benzoic acid rings of 44pba. Thus this material can be taken through successive desolvation, sorption, and desolvation cycles without significant changes.
We are now able to compare the similar structures obtained for the [Co(34pba)(44pba)] MOF 1, which include six inclusion compounds and two desolvated compounds. As already reported, the compounds are isostructural, with no significant trends emerging in looking at unit cell parameters, though the b-axis shows the greatest variation, from 15.3 Å (in 1d) to 17.8 Å (in 1DMF). Conformational analysis shows that the rings of both linkers are close to co-planar for inclusion compounds where guests have larger molecular volumes and are more twisted when smaller guests (e.g. CH2Cl2) are included. When no guest is included (in 1d or 1dI2d) the rings have even greater flexibility. This is particularly pronounced for the pyridyl ring of 34pba in which the major component of the disordered ring is rotated by ca. 70°. We noted that the other disordered ring (on the 44pba) aligns with its major component in the same orientation as in 1dCH2Cl2 and its minor component aligning with the other solvated compounds. These features are illustrated in Fig. 8. The flexibility of the MOF is confirmed by an analysis of the void space available in each instance (Table S5†), which increases with solvent molecular volume. To confirm the robustness of the MOF an individual crystal was taken through several cycles of desorption and sorption, which showed consistent changes in unit cell parameters (Table S6†).
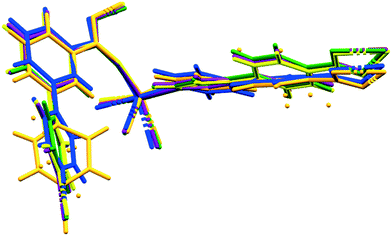 |
| | Fig. 8 Molecular overlays of the asymmetric unit for 1d (orange), 1dCH2Cl2 (blue), 1dCHCl3 (green), 1dClBenz (yellow), 1dI2 (purple). | |
3.4 Kinetics of desorption for 1dCH2Cl2 and 1dCH2Br2 and 1dI2
While 1d and 2d show similar sorption trends for halogenated VOCs in terms of adsorption capacity and desorption temperature, (Fig. S2†), we noted that the TGA traces for halogenated VOCs of aliphatic compounds show a single desorption step. On the other hand, halogenated VOCs containing aromatic ring showed a two-step desorption process. This may be attributed to the aromatic stacking interactions as seen the 1dClBenz structure (Fig. 3) which cause the guests to be more strongly retained and result in a larger temperature range for desorption.
TGA can also be used to determine the activation energy (Ea) of guest desorption processes, and we have applied this here to the desorption of the related guests dichloromethane and dibromomethane from 1dCH2X2, as well as to the desorption of iodine from 1dI2. A series of isothermal TG curves were obtained for each compound at several temperatures just below the desorption temperature observed in Fig. S2.† Each desorption curve was then fitted against a range of plausible desorption mechanisms,24 and a rate constant (k) was determined at each temperature (Fig. S5†). An Arrhenius plot then gave the value of the activation energy for each desorption process. The conversion vs. time curves are shown in Fig. S6,† and the activation energies determined are given in Table 5. The kinetics of desorption for CH2Cl2, CH2Br2, and I2 all showed a best fit to the 3D diffusion model, which indicates that the guest molecules interact with the wall of the adsorbent in a spherical zone. The Ea of desorption of CH2Cl2 and CH2Br2 are approximately equal while that for iodine is higher, probably owing to the increased non-covalent interactions which iodine is capable of making with the channel walls.
Table 5 Activation energy for the isothermal desorption of CH2Cl2, I2, and CH2Br2 from 1d
| Compound |
E
a (kJ mol−1) |
R
2 Coeff |
|
1
dCH
2
Cl
2
|
70 |
0.96 |
|
1
dCH
2
Br
2
|
60 |
0.97 |
|
1
dI
2
|
77 |
0.90 |
3.5 Sorption of carbon dioxide and hydrogen gases in 1d and 2d
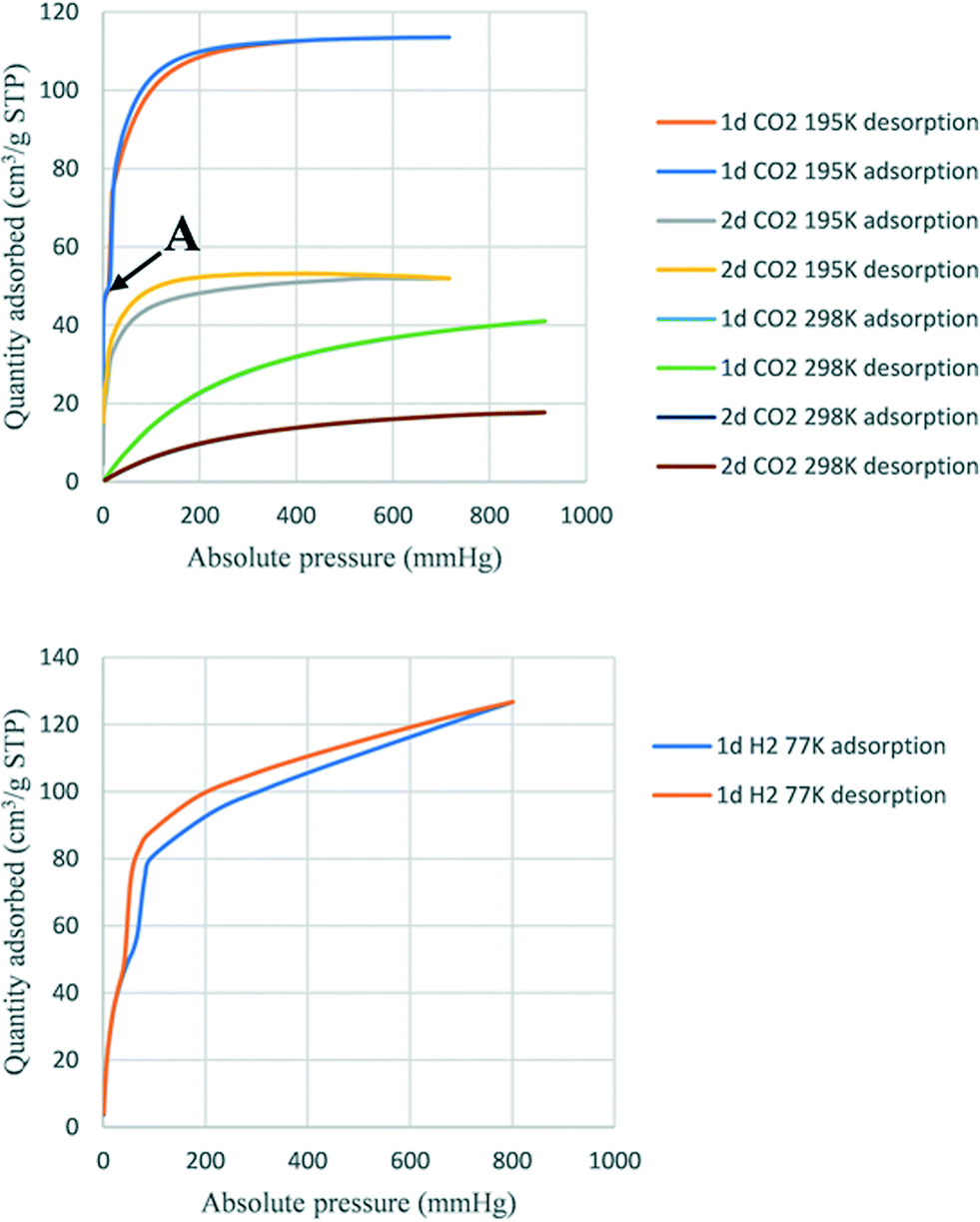
Activated 1d and 2d were investigated for the sorption of CO2 and H2 at different temperatures. The heat of adsorption of CO2 was also determined. Fig. 9 shows the adsorption traces for selected runs using both 1d and 2d. At the temperatures studied, the sorption behaviour was a type-Ib isotherm. In this model of sorption, the adsorbent is characterized by small external surfaces having a pore size with distributions over a broader range and the presence of wider mesopores with possibility of narrow micropores (≤2.5 nm).25 The steep uptake observed at low p/p0 is due to enhanced adsorbent–gas interactions in narrow micropores. Table 6 records the higher adsorption capacity at 195 K and 676 mmHg for both samples. 1d showed more than a double 114 cm3 (STP) g−1 (2.35 molecules per ASU) compared to that of 2d with 52 cm3 (STP) g−1 (1.11 molecules per ASU). Little hysteresis was observed in both isostructural frameworks at the lower temperature (195 K) owing to the lower mobility of gas molecules. Stage A (Fig. 9 top) may indicate the presence of some mesopores in 1d which are absent in 2d. Furthermore, this stage can also explain the flexibility of the ligand to open the pores and allow additional gas uptake.26 The highest adsorption recorded at 298 K and 910 mmHg for 1d and 2d was 41.0 cm3 (STP) g−1 (0.84 molecules per ASU) and 17.7 cm3 (STP) g−1 (0.37 molecules per ASU), respectively. Sample 1d showed higher adsorption capacity than 2d at a similar temperature and pressure.
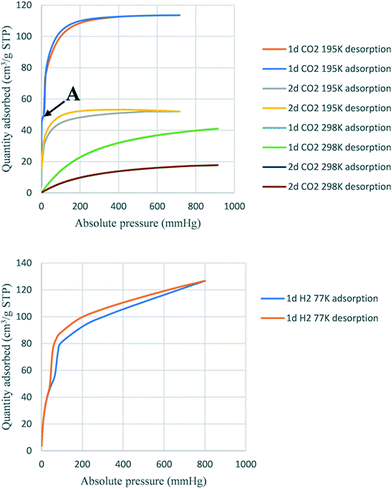 |
| | Fig. 9 Sorption of CO2 (top) and H2 (bottom). | |
Table 6 Sorption capacity for CO2
| No |
Sample |
Volume adsorbed (cm3 g−1) |
Pressure (mmHg) |
Temperature (K) |
Corresponding to (mmol g−1) |
| 1 |
2
d
|
52.0 |
676 |
195 |
2.40 |
|
1
d
|
114 |
5.06 |
| 2 |
2
d
|
22.4 |
784 |
273 |
1.00 |
|
1
d
|
47.6 |
2.10 |
| 3 |
2
d
|
21.7 |
803 |
278 |
1.00 |
|
1
d
|
47.0 |
2.10 |
| 4 |
2
d
|
19.7 |
910 |
288 |
0.90 |
|
1
d
|
44.6 |
2.00 |
| 5 |
2
d
|
18.8 |
900 |
293 |
0.84 |
|
1
d
|
43.1 |
1.92 |
| 6 |
2
d
|
17.7 |
910 |
298 |
0.80 |
|
1
d
|
41.0 |
1.80 |
The trend of higher adsorption capacity in 1d compared to 2d was observed for solvent vapours including the halogenated VOCs reported here and the volatile amines reported in ref. 18, and may be the result of a slightly smaller void space in 1d than 2d. However, this attribution of adsorption capacity related to the size of pores has been controversial in other reported structures.26 An alternative explanation is that the smaller size of a solvent-accessible void volume causes stronger interactions between the guest and the framework, depending on their respective structures and functionalization. The determination of isosteric heat of adsorption (Qst) of CO2 showed that the sample 1d adsorbed between 0.8 mmol and 1.38 mmol with heats of adsorption (Qst) between 29.8 kJ and 30.3 kJ, while sample 2d adsorbed between 0.1 mmol and 0.4 mmol CO2 (Qst 28.5 kJ and 28.9 kJ). These results suggest that there is a higher interaction in 1d than in 2d.27 The uptake for hydrogen gas in 1d was 120 cm3 (STP) g−1 at 800 mmHg at 77 K corresponding to 2.44 molecules of H2 per ASU. In contrast, 2d did not adsorb hydrogen under the same conditions.
4. Conclusion
The double-walled [Co(34pba)(44pba)]n·MOF (1d) was studied for its ability to take up vapours and gases from the desolvated, activated phase. This MOF has proved to be robust to several cycles of sorption and desorption, as evidenced by both single-crystal and powder diffraction. This is facilitated by the flexibility of the pyridylbenzoate linkers, in particular the ability of the rings to rotate and thus occupy additional space when the compound is desolvated. A similar effect was observed for the Zn(II) analogue (2d).
Inclusion of halogenated volatile organic compounds found that, for each series of VOCs studied, the uptake was generally higher for chlorinated over brominated and iodinated compounds. While only the chlorinated species CH2Cl2, CHCl3, and ClBenz afforded single crystal structures, TGA and PXRD analysis indicates that the type of interaction present in the Br- and I-derivatives are likely to be similar. On the other hand, the sorption of iodine into 1d could be followed in the powder form, and also afforded single crystals which are stabilized by I⋯π and I⋯I interactions. The desorption of iodine from 1dI2 gave 1dI2d whose structure was also fully elucidated, confirming that the desorption–sorption–desorption cycle proceeded without significant loss of crystallinity.
The isothermal kinetic desorption of I2, CH2Cl2, and CH2Br2 from 1d indicates that iodine requires more energy for the desorption in the range of the reported ones. The two adsorbents 1d and 2d were both capable of adsorption of carbon dioxide gas while only 1d adsorbed hydrogen. In general, across all types of vapour and gas sorption tested, the trend was for 1d to show stronger interactions and higher adsorption capacity. Further studies to determine the reasons for this observation are underway.
Author contributions
Conceptualization S. A. B.; data acquisition and analysis C. A. N.; assistance with crystallography and analysis S. A. B., C. L. O.; assistance with gas sorption N. C.; supervision S. A. B., C. L. O.; writing – original draft C. A. N., S. A. B.; all authors assisted with writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to have worked with Prof. Luigi Nassimbeni, and express their best wishes to him as he embarks on his 9th decade. Funding from the National Research Foundation of South Africa (grant number 111699) is gratefully acknowledged. C. A. N. thanks the University of Cape Town for a PhD fellowship.
References
- B. Wang, P. Wang, L.-H. Xie, R.-B. Lin, J. Lv, J.-R. Li and B. Chen, Nat. Commun., 2019, 10, 3861 CrossRef PubMed.
- N. Gcwensa, N. Chatterjee and C. L. Oliver, Inorg. Chem., 2019, 58, 2080 CrossRef CAS PubMed.
- S. Huh, Catalysts, 2019, 9, 34 CrossRef.
- X.-D. Yang, C. Chen, Y.-Z. Zhang, L.-X. Cai, B. Tan and J. Zhang, Dalton Trans., 2016, 45, 4522 RSC.
- D. N. Jiang, C. Huang, J. Zhu, P. Wang, Z. M. Liu and D. Fang, Coord. Chem. Rev., 2021, 444, 214064 CrossRef CAS.
- K. Hyojin and C. S. Hong, CrystEngComm, 2021, 23, 1377 RSC.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519 RSC.
- X. Yang and D. Yan, Chem. Commun., 2017, 53, 1801 RSC.
- M. Kobalz, J. Lincke, K. Kobalz, O. Erhard, J. Bergmann, D. Lässig, M. Lange, J. Möllmer, R. Gläser, R. Staudt and H. Krautscheid, Inorg. Chem., 2016, 55, 3030 CrossRef CAS PubMed.
- G. Mehlana, S. A. Bourne, G. Ramon and L. Öhrström, Cryst. Growth Des., 2013, 13, 633 CrossRef CAS.
- L. Mei, C. Wang, L. Wang, Y. Zhao, Z. Chai and W. Shi, Cryst. Growth Des., 2015, 15, 1395 CrossRef CAS.
- L. Wang, Y. Li, F. Yang, Q. Liu, J.-P. Ma and Y.-B. Dong, Inorg. Chem., 2014, 53, 9087 CrossRef CAS PubMed.
- Q.-Y. Yang, P. Lama, S. Sen, M. Lusi, K.-J. Chen, W.-Y. Gao, M. Shivanna, T. Pham, N. Hosono, S. Kusaka, J. J. Perry, S. Ma, B. Space, L. J. Barbour, S. Kitagawa and M. J. Zaworotko, Angew. Chem., Int. Ed., 2018, 57, 5684 CrossRef CAS PubMed.
- K. Davies, S. A. Bourne, L. Öhrström and C. L. Oliver, Dalton Trans., 2010, 39, 2869 RSC.
- G. Mehlana, S. A. Bourne and G. Ramon, CrystEngComm, 2014, 16, 8160 RSC.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244–245, 444 CrossRef CAS PubMed.
- A. Bacchi, S. A. Bourne, G. Cantoni, S. A. M. Cavallone, S. Mazza, G. Mehlana, P. Pelagatti and L. Rhighi, Cryst. Growth Des., 2015, 15, 1876 CrossRef CAS.
- C. A. Ndamyabera, S. C. Zacharias, C. L. Oliver and S. A. Bourne, Chemistry, 2019, 1, 111 CrossRef.
-
G. M. Sheldrick, SADABS, Version 2.05, University of Göttingen, Germany, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CAS.
- T. K. Trung, N. A. Ramsahye, P. Trens and N. Tanchoux, Microporous Mesoporous Mater., 2010, 134, 134 CrossRef CAS.
- N. Chatterjee and C. L. Oliver, Cryst. Growth Des., 2018, 18, 7570 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S8, Fig. S1–S6. CCDC 2100689–2100692. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01052b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Susan A.
Bourne
and
Susan A.
Bourne