
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluating the crystalline orbital hierarchy and high-pressure structure–property response of an extended-ligand platinum(II) bis(1,2-dioximato) complex†
Jonathan G.
Richardson
a,
Edward T.
Broadhurst
 a,
Helen
Benjamin
a,
Carole A.
Morrison
*a,
Stephen A.
Moggach
b and
Neil
Robertson
*a
a,
Helen
Benjamin
a,
Carole A.
Morrison
*a,
Stephen A.
Moggach
b and
Neil
Robertson
*a
aEaStCHEM School of Chemistry, University of Edinburgh, The King's buildings, David Brewster road, Edinburgh, EH9 3FJ, UK. E-mail: neil.robertson@ed.ac.uk
bCentre for Microscopy, Characterisation and Analysis, University of Western Australia, 35 Stirling Highway, Crawley, Perth, 6005, Western Australia, Australia
First published on 16th August 2021
Abstract
Herein the solid state structure and crystalline orbital hierarchy of a new extended-ligand platinum(II) bis(1,2-dioximato) complex is evaluated. The lack of direct stacking of Pt centres in the solid state, caused by the steric bulk on the exterior of the ligand, results in highly localised frontier bands, and thus minimal band gap compression upon the application of pressure. This is in contrast to its parent complex [Pt(bqd)2] which undergoes a semiconductor-to-metal transition by 1 GPa.
Over recent decades, interest has grown in molecular-based materials as conducting components in electronic devices. These aptly-named molecular conductors can be comprised of purely organic or organometallic components,1–3 with notable examples including tetrathiafulvalene (TTF) and Bechgaard charge transfer salts,4–7 dithiolene complexes,8–13 Magnus green salts and other analogous complexes,14–16 and bis(1,2-dioximato) complexes.17–20 While these materials may still be insulators at ambient conditions, conductivity can be enhanced under application of pressure; decreasing intermolecular separation results in enhanced orbital overlap which in turn results in band broadening and a narrowing of the electronic band gap. Evaluating how the behaviour of these complexes can be tuned by ligand design is necessary for identifying suitable candidate materials for applications such as field-effect transistors and charge-transport materials in solar cells.21–23
Previous work has evaluated and compared the structural and electronic responses to pressure of two members of the aforementioned bis(1,2-dioximato) family of platinum(II) complexes:24 platinum bis(1,2-benzoquinonedioximato), hereafter referred to as [Pt(bqd)2],25 and platinum bis(1,2-dimethylglyoximato), hereafter [Pt(dmg)2].26 Both have been shown to exhibit solid state packing that permits columnar stacking of the Pt metal centres, which results in short Pt⋯Pt distances (<3.3 Å) and direct orbital overlap. Upon application of pressure, anisotropic compression of the unit cells results in direct Pt⋯Pt contraction. The effect is more pronounced for [Pt(bqd)2], which can be attributed to the greater planarity of the ligand system. The nature of the band gaps and their response to pressure was also evaluated. The ambient-pressure band gap for [Pt(bqd)2] (0.52 eV) is notably smaller than for [Pt(dmg)2] (1.44 eV) and, due to the faster rate of Pt⋯Pt compression, closed much faster than Pt(dmg)2 under the application of pressure, such that the metallic state was reached by 1 GPa.24 This work also highlighted the importance of the role played by the ligands in the construction of the frontier orbitals, which had previously thought to be benign. Stronger interlayer ligand⋯ligand interactions increased the amount of dispersion in the conduction band, which facilitated a much faster band gap narrowing in these compounds than in, for example, the Magnus green salts, where much flatter ligand-based conduction bands were exhibited.27
As part of a wider study comparing the structural and property responses of pressure-responsive platinum-containing complexes, we have prepared a derivative complex of [Pt(bqd)2], in which two hydrogen atoms on each phenyl ring of the 1,2-dioximato ligand are replaced by n-butyl chains (Fig. 1, hereafter [Pt(bqd-dibutyl)2]). These modifications were undertaken to enhance the solubility of the complex and thus improve its processability to be applied as a thin film in transistors or other molecular-electronic devices. The effect of the ligand modification must be considered in its own right, however, as it may affect the ability of the Pt centres to stack in an idealised one-dimensional chain arrangement, and may alter the nature of the inter-ligand interactions. Herein we report the crystal structure of [Pt(bqd-dibutyl)2] and deduce how the molecular orbitals are recast as crystalline orbitals. We also report on the structural response and simulated electronic band gap response to pressure and compare with the parent complex [Pt(bqd)2].
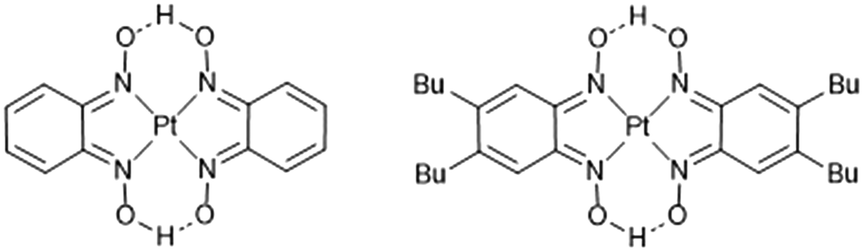 | ||
| Fig. 1 Schematic diagrams of [Pt(bqd)2] (left) and [Pt(bqd-dibutyl)2] (right). The label Bu refers to the n-butyl chains. | ||
Results and discussion
The synthesis of [Pt(bqd-dibutyl)2] is shown in Scheme 1. The butyl groups were introduced via Ag(I) assisted Suzuki coupling between butylboronic acid and 1,2-dibromo-4,5-dinitrobenzene, which proceeded in good yield (78%).28 An azide group was introduced via a nucleophilic aromatic substitution reaction, and the resulting ortho-nitro-azide was then cyclised to give the furoxan (56% – two steps). The dioxime ligand was then synthesised by reduction of the furoxan with diphenylhydrazine (65%). Treatment of a solution of the ligand (acetone) with an aqueous solution of K2[PtCl4] gave a dark brown precipitate, [Pt(bqd-dibutyl)2]. Unlike the parent complex [Pt(bqd)2], [Pt(bqd-dibutyl)2] was highly soluble in 1,2-dichlorobenzene at room temperature due to the inclusion of the long alkyl chains, allowing growth of single crystals via diffusion of hexane into a 1,2-dichlorobenzene solution.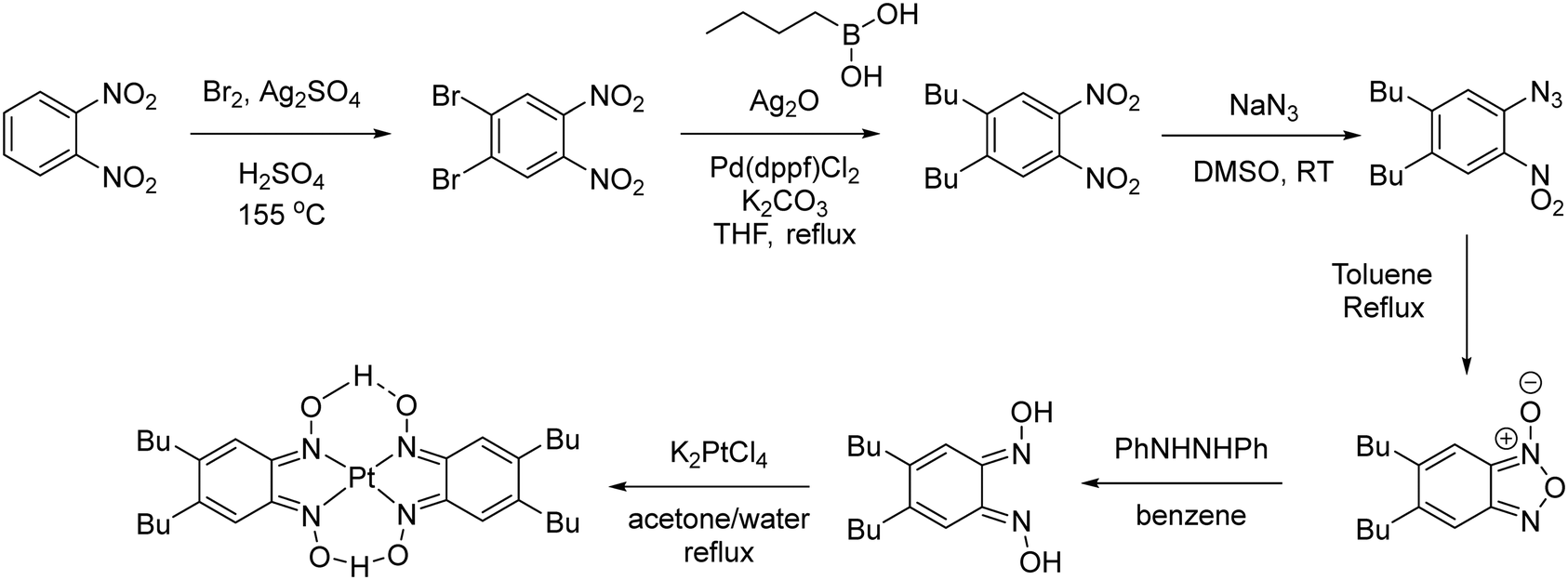 | ||
| Scheme 1 Synthesis of [Pt(bqd-dibutyl)2]. | ||
The structure of [Pt(bqd-dibutyl)2] was characterised by single-crystal X-ray diffraction (see Fig. 2 and Table 1), and found to crystallise in the orthorhombic space group Pna21 with four crystallographically-independent molecules per unit cell. It is readily apparent that the ligand modification has had a substantial impact on the nature of the crystal packing, with the columnar stacking of the Pt centres present in the parent [Pt(bqd)2] complex now completely disrupted.
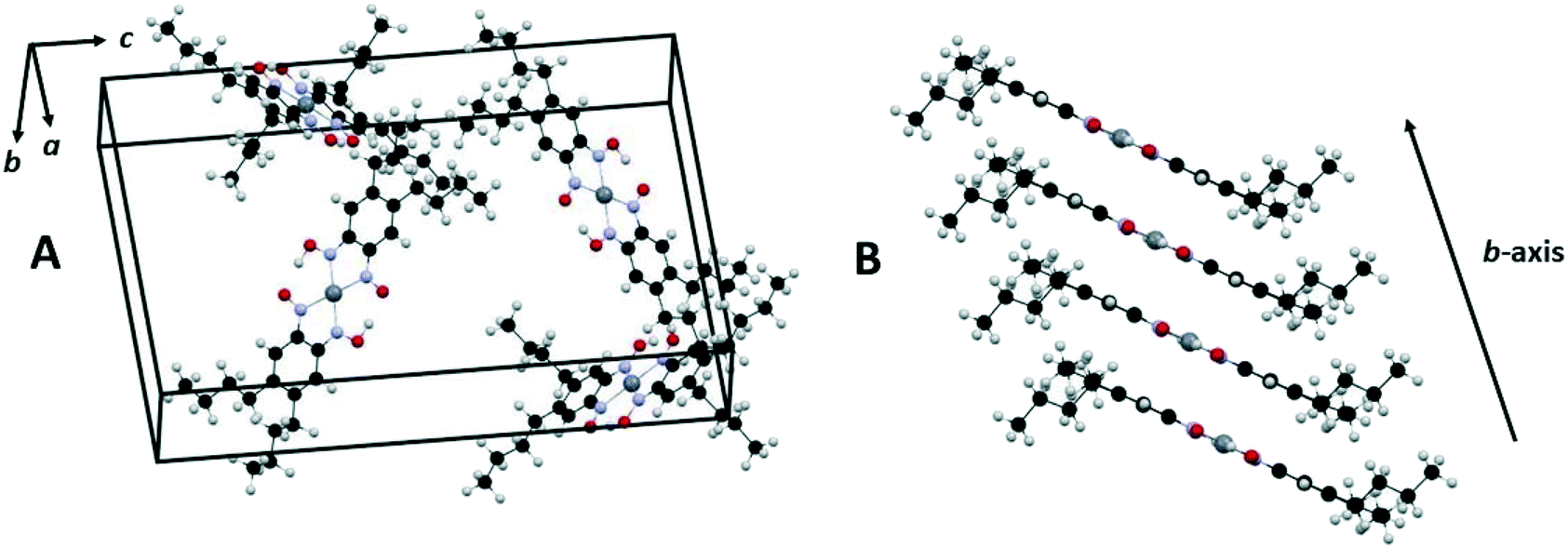 | ||
| Fig. 2 Unit cell of [Pt(bqd-dibutyl)2] illustrating the herringbone arrangement of molecules in the crystal lattice (A) and slipped stacking of [Pt(bqd-dibutyl)2] complexes along the b-axis direction (B), generated using Mercury.29 Pt, N, O, C and H atoms are coloured grey, blue, red, black and light grey respectively. | ||
| Formula | C28H42N4O4Pt |
| Crystal system | Orthorhombic |
| Space group | Pna21 |
| a (Å) | 20.7074(11) |
| b (Å) | 4.9052(2) |
| c (Å) | 28.1381(14) |
| Volume (Å3) | 2850.10(14) |
| Z | 4 |
| Wavelength (Å) | 0.71073 (Mo Kα) |
| R int | 0.042 |
| θ max (°) | 28.5 |
(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ/λ)max (Å−1) θ/λ)max (Å−1) |
0.672 |
| R[F2 > 2σ(F2)] | 0.044 |
| wR(F2) | 0.107 |
| S | 1.17 |
Instead, the structure adopts a herringbone arrangement, and the Pt⋯Pt separation, which lies along the b-axis direction, has expanded to 4.9051(2) Å (Fig. 2B), compared to 3.1764(2) Å for [Pt(bqd)2].24
The ordering of isolated molecular orbitals (MO), obtained from Gaussian09 calculations,30 and crystalline orbitals (CO), obtained from hybrid density functional theory (DFT) calculations using the CRYSTAL17 package,31 for [Pt(bqd-dibutyl)2] are presented in Fig. 3, alongside those for [Pt(bqd)2] for comparison.24 Further details of the computational methodology can be found in the ESI.†
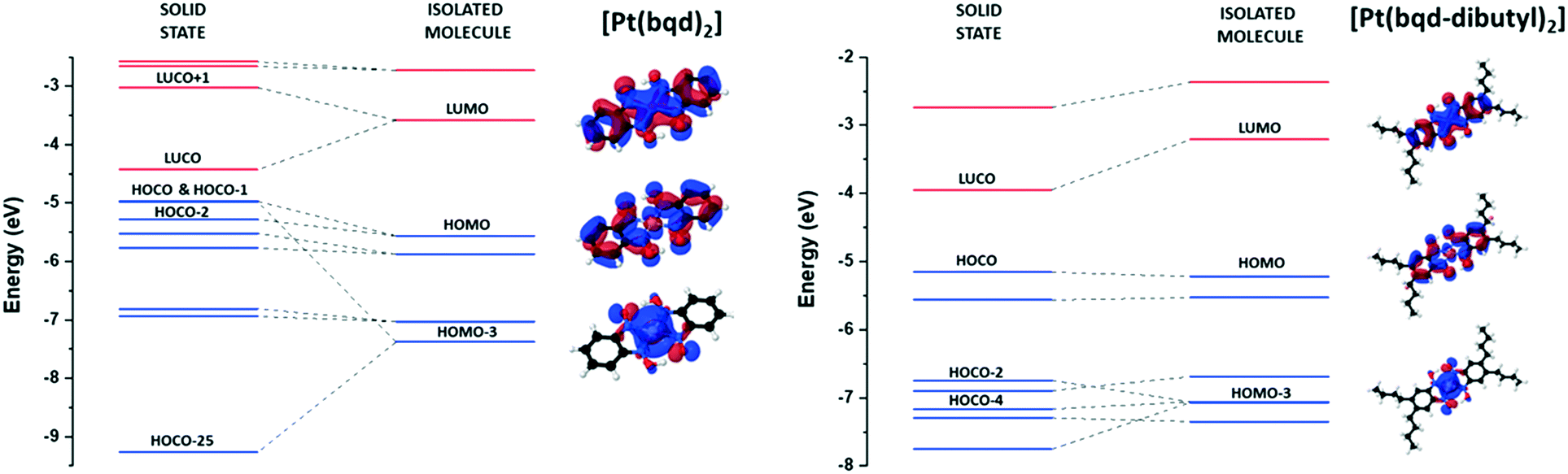 | ||
| Fig. 3 Construction of crystalline orbitals (COs) from molecular orbitals for [Pt(bqd)2], as previously published,24 and [Pt(bqd-dibutyl)2], as defined at the gamma point. Blue and red lines indicate occupied and unoccupied orbitals respectively. The COs from HOCO−7 to HOCO−24 inclusive for [Pt(bqd)2] have been removed for simplicity. | ||
Given that the modification was made to the extremity of the ligand, and thus the ligand backbone was untouched, the ordering of the molecular orbitals for [Pt(bqd-dibutyl)2] are very similar to those calculated for [Pt(bqd)2]. Three key molecular orbitals are highlighted in Fig. 3 to account for the differences in electronic behaviour between the two compounds, namely the LUMO, the HOMO and the HOMO−3.
Firstly, the LUMO sits at ca. −3.5 eV for both molecules, and mainly consists of π* interactions across the ligand backbone and the Pt centre (via the Pz orbital). As previously discussed for [Pt(bqd)2],24 in the crystal structure the LUMOs of neighbouring molecules interact along the stacking direction in a bonding and anti-bonding fashion, resulting in the formation of the LUCO (ca. −4.5 eV) and LUCO+1 (ca. −3.0 eV) bands, respectively. In [Pt(bqd-dibutyl)2], the absence of Pt⋯Pt interactions along the b-axis means this splitting is not observed, and therefore the LUCO (ca. −4.0 eV) essentially mirrors the LUMO. The HOMO for both complexes, present at ca. −5.3 eV, shows similar π* interactions to the LUMO, although the contributing orbital on the Pt centre has switched to the dxz/yz orbital. The crystal stacking in [Pt(bqd)2] causes the HOMO to split into the HOCO−1 (ca. −5.0 eV) and the HOCO−2 (ca. −5.3 eV), while again the absence of Pt⋯Pt interactions in [Pt(bqd-dibutyl)2] renders the HOCO as essentially a duplicate of the HOMO. Lastly, the HOMO−3 for both compounds is dominated by contribution from the Pt dz2 atomic orbital and resides at ca. −7.5 eV. As discussed previously,24 the crystal packing in [Pt(bqd)2] results in a very large energy splitting in the bonding and anti-bonding combinations of the HOMO−3 orbital between neighbouring molecules, to generate the HOCO−25 (below −9 eV) and HOCO (−5 eV) bands. In [Pt(bqd-dibutyl)2] the much weaker interaction between neighbouring HOMO−3 orbitals down the b-axis results in a much smaller energy splitting, and these MO contributions can be mapped onto the HOCO−2 and HOCO−4 bands. Taken together, these three orbital interactions, which in turn can be attributed solely to crystal packing effects, account for the narrow electronic band gap observed for [Pt(bqd)2] and the much wider gap for [Pt(bqd-dibutyl)2].
The full electronic band structure for the ambient pressure structure of [Pt(bqd-dibutyl)2], obtained from hybrid DFT calculations, is shown in Fig. 4. The bands are extremely flat (k-invariant), which confirms that the interactions between neighbouring molecules are weak. The band gap was calculated as 1.17 eV, considerably wider than the 0.52 eV obtained for [Pt(bqd)2] using the same level of theory.24 Noticeable, although weak, dispersion (k-variance) is observed for the LUCO, HOCO−2 and HOCO−4 bands along the Brillouin zone k-point path Y → S, which confirms intermolecular interactions, albeit fairly weak, must exist along the b-axis direction as a result of the molecular stacking. The limited and weak interactions are in keeping with our observation that [Pt(bqd-dibutyl)2] was found to have ambient-pressure resistance higher than could be measured in our equipment (>200 TΩ) even at elevated temperature up to the 353 K.
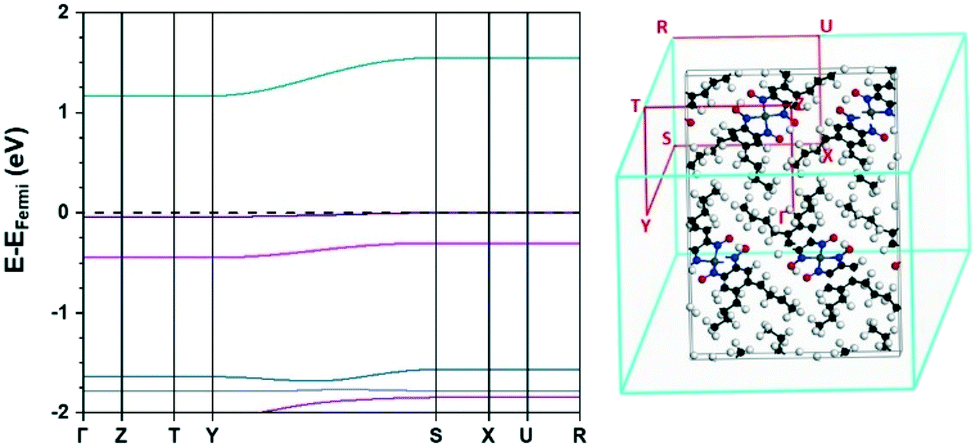 | ||
| Fig. 4 Electronic band structure (left) and Brillouin zone k-point path (right) of the ambient pressure structure of [Pt(bqd-dibutyl)2]. EFermi = −4.99 eV. | ||
The structural response of [Pt(bqd-dibutyl)2] to the application of pressure was investigated using single crystal X-ray diffraction; further details of the methodology and results obtained from these experiments can be found in the ESI.†
Structure solutions were obtained successfully up to 1.43 GPa, after which point no meaningful diffraction data could be collected; the compression curves for each of the unit cell parameters as a ratio of those obtained at ambient conditions is presented in Fig. 5. While only three non-ambient pressure points are reported, they still give a good sense of the structural response of [Pt(bqd-dibutyl)2] to pressure. The overall degree of anisotropic volumetric compression by 1.43 GPa (ca. 10%) is comparable to that observed for [Pt(bqd)2],24 with the majority contribution attributed to the b-axis direction which corresponds to the molecular stacking direction; the a-axis and c-axis directions are hindered by steric clashing of the n-butyl chains. Across the pressure series, the Pt⋯Pt stacking distance decreased from 4.9051(2) Å to 4.6778(3) Å. The experimental rate of compression can be compared to a simulated pressure series on [Pt(bqd-dibutyl)2] undertaken using the CASTEP simulation package,32 with the atomic positions and unit cell parameters optimised between ambient pressure and 1.50 GPa (also shown in Fig. 5). The calculated pressure series revealed a slightly reduced rate of volumetric compression, mostly stemming from the lower rates of compression along the a and c axis directions. However, using the smoother volumetric compression curve determined by the simulation, a bulk modulus of 7.5(8) GPa was determined (see ESI†), which was significantly lower (to 3σ) than that determined for [Pt(bqd)2] (11.7(7) GPa) from experimental data.24
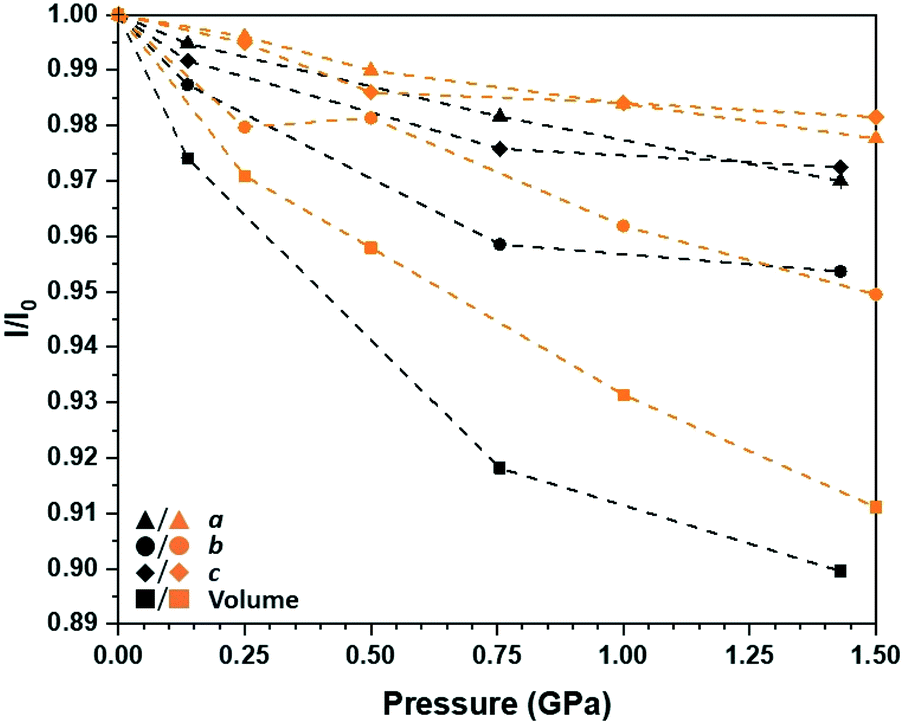 | ||
| Fig. 5 Compression curves for the unit cell parameters of [Pt(bqd-dibutyl)2] up to 1.50 GPa, as fraction of those recorded at ambient-pressure, determined from experiment (black) and a simulated pressure series using the CASTEP simulation package (orange). Error bars on the experimental data points are smaller than symbols used. | ||
The electronic band gap for the 1.43 GPa structure of [Pt(bqd-dibutyl)2] was calculated at 1.05 eV (see ESI†), a drop of slightly more than 0.1 eV across the pressure series. To provide some context, the calculated electronic band gap for [Pt(bqd)2] decreased from 0.5 eV to 0.0 eV by 1 GPa.24
To summarise, this investigation has highlighted how ligand modifications can have drastic effects on structure/property relationships, even when the modification is made far away from the electronically-delocalised metal–ligand centre. The addition of alkyl chains to assist the processability of the platinum bis(1,2-benzoquinonedioximato) system has completely changed the crystal packing arrangement, such that the Pt⋯Pt separation increases by ca. 1.7 Å. The resulting weakening of inter-layer interactions across the compressible b-axis direction acts to widen the electronic band gap, which could not be closed on application of pressure. This is in-stark contrast to the parent complex [Pt(bqd)2], which exhibits short Pt⋯Pt stacking and correspondingly stronger interlayer interactions facilitating a small ambient pressure band gap and an accessible conductive state upon application of 1 GPa external pressure.21 The alteration of the electronic structure was not apparent in the derivation of the molecular orbitals, which suggested that the two compounds should exhibit similar behaviour. This study therefore provides a precautionary note to material researchers on the effect of seemingly benign ligand modifications. Obtaining the desired solid-state packing is a crucial factor in material design to facilitate more rapid identification of candidate materials for desired electronic and optoelectronic applications. Furthermore, these findings emphasise the unusual character of the [Pt(II)bis(1,2-dioximato)] complex family in their tendency to show strong Pt⋯Pt interactions. This contrasts with the large majority of conducting molecular and organic materials that show weaker slipped plane-to-plane π-stacking. By adding the butyl groups to [Pt(bqd-dibutyl)2], any potential Pt⋯Pt interaction is prevented, and the behaviour reverts to the more common π-stacking.
Experimental data
Materials, synthesis and characterisation
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Leverhulme Trust (RPG-2016-152) for funding. J. R. thanks the University of Edinburgh for PhD stipend funding in the form of a Principal Career's Development Scholarship. We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1). This work has made use of the resources provided by the Edinburgh Compute and Data Facility (ECDF) (http://www.ecdf.ed.ac.uk/). The authors gratefully acknowledge the facilities, and the scientific and technical assistance of Microscopy Australian at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. We thank Sergejs Afanasjevs for conductivity measurements.References
- P. Batail, Chem. Rev., 2004, 104, 4887–4890 CrossRef CAS.
- H. Kobayashi, A. Kobayashi and H. Tajima, Chem. – Asian J., 2011, 6, 1688–1704 CrossRef CAS PubMed.
- L. Ouahab, Conducting and Magnetic Organometallic Molecular Materials, Springer-Verlag Berlin Heidelberg, Berlin, Heidelberg, 2009 Search PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95(3), 948–949 CrossRef CAS.
- P. Frere and P. J. Skabara, Chem. Soc. Rev., 2005, 34, 69–98 RSC.
- D. Jerome, A. Mazaud, M. Ribault and K. Bechgaard, J. Phys., Lett., 1980, 41, L95–L98 CrossRef.
- S. E. Brown, Phys. C, 2015, 514, 279–289 CrossRef CAS.
- B. Zhou, H. Yajima, Y. Idobata, A. Kobayashi, T. Kobayashi, E. Nishibori, H. Sawa and H. Kobayashi, Chem. Lett., 2012, 41, 154–156 CrossRef CAS.
- B. Zhou, S. Ogura, Q. Z. Liu, H. Kasai, E. Nishibori and A. Kobayashi, Chem. Lett., 2016, 45, 303–305 CrossRef CAS.
- H. Tanaka, Y. Okano, H. Kobayashi, W. Suzuki and A. Kobayashi, Science, 2001, 291, 285–287 CrossRef CAS PubMed.
- D. Belo, H. Alves, E. B. Lopes, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, A. Perez-Benitez, C. Rovira and J. Veciana, Chem. – Eur. J., 2001, 7, 511–519 CrossRef CAS.
- B. Zhou, S. Ishibashi, T. Ishii, T. Sekine, R. Takehara, K. Miyagawa, K. Kanoda, E. Nishibori and A. Kobayashi, Chem. Commun., 2019, 55, 3327–3330 RSC.
- R. Kato, H. Cui, T. Tsumuraya, T. Miyazaki and Y. Suzumura, J. Am. Chem. Soc., 2017, 139, 1770–1773 CrossRef CAS PubMed.
- M. Atoji, J. W. Richardson and R. E. Rundle, J. Am. Chem. Soc., 1957, 79, 3017–3020 CrossRef CAS.
- L. V. Interrante and F. P. Bundy, Inorg. Chem., 1971, 10, 1169–1174 CrossRef CAS.
- M. E. Cradwick, D. Hall and R. K. Phillips, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 480–484 CrossRef.
- J. W. Brill, M. Megnamisibelombe and M. Novotny, J. Chem. Phys., 1978, 68, 585–592 CrossRef CAS.
- I. Shirotani, A. Kawamura, K. Suzuki, W. Utsumi and T. Yagi, Bull. Chem. Soc. Jpn., 1991, 64, 1607–1612 CrossRef CAS.
- K. Takeda, I. Shirotani and K. Yakushi, Chem. Mater., 2000, 12, 912–916 CrossRef CAS.
- L. E. Godycki and R. E. Rundle, Acta Crystallogr., 1953, 6, 487–495 CrossRef CAS.
- A. Mizuno, H. Benjamin, Y. Shimizu, Y. Shuku, M. M. Matsushita, N. Robertson and K. Awaga, Adv. Funct. Mater., 2019, 1904181 CrossRef CAS.
- A. M. Whyte, Y. Shuku, G. S. Nichol, M. M. Matsushita, K. Awaga and N. Robertson, J. Mater. Chem., 2012, 22, 16967 RSC.
- Z. Yu, A. Hagfeldt and L. Sun, Coord. Chem. Rev., 2020, 406, 213143 CrossRef CAS.
- H. Benjamin, J. G. Richardson, S. A. Moggach, S. Afanasjevs, L. Warren, M. R. Warren, D. R. Allan, C. A. Morrison, K. V. Kamenev and N. Robertson, Phys. Chem. Chem. Phys., 2020, 22, 6677–6689 RSC.
- M. Megnamisibelombe, J. Solid State Chem., 1979, 27, 389–396 CrossRef CAS.
- E. Frasson, C. Panattoni and R. Zannetti, Acta Crystallogr., 1959, 12, 1027–1031 CrossRef CAS.
- J. G. Richardson, H. Benjamin, S. A. Moggach, L. R. Warren, M. R. Warren, D. R. Allan, L. K. Saunders, C. A. Morrison and N. Robertson, Phys. Chem. Chem. Phys., 2020, 22, 17668–17676 RSC.
- G. Zou, Y. Krishna Reddy and J. R. Falck, Tetrahedron Lett., 2001, 42, 7213–7215 CrossRef CAS.
- C. F. Macrae, L. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, H. X. Li, P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN09, Gaussian Inc., Wallingford, C.T., 2009 Search PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rerat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e:1360 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- L. Ma, Q.-S. Hu, D. Vitharana, C. Wu, C. M. S. Kwan and L. Pu, Macromolecules, 1997, 30(2), 204–218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic methodology and experimental details; computational methodology, orbital images and projected density of states. CCDC 2093361–2093364. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00892g |
| This journal is © The Royal Society of Chemistry 2021 |