
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-step solvothermal synthesis and growth mechanism of well-crystallized β-Ga2O3 nanoparticles in isopropanol
Kengo
Takezawa
a,
Jinfeng
Lu
b,
Chiya
Numako
 c and
Seiichi
Takami
*a
c and
Seiichi
Takami
*a
aDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: takami.seiichi@material.nagoya-u.ac.jp
bEP Application Department, EP Business Unit, JEOL Ltd., 3-1-2 Musashino, Akishima, Tokyo 196-8558, Japan
cDepartment of Chemistry, Graduate School of Science, Chiba University, 1-33 Yayoicho, Inage-ku, Chiba 263-8522, Japan
First published on 2nd September 2021
Abstract
Simple liquid-phase approaches for the synthesis of nanomaterials are attractive because their low costs, reduced nanoparticle aggregation, and compatibility with subsequent liquid processes widen the application scope of the resulting materials. This would be particularly interesting for β-Ga2O3 nanoparticles, which often suffer from aggregation issues during their synthesis processes. In this paper, we report a one-step synthesis of β-Ga2O3 nanoparticles in supercritical isopropanol. By simply heating Ga(NO3)3 in isopropanol at 400 °C for 24 hours, β-Ga2O3 nanoparticles with a size of ∼100 nm are obtained without requiring additional calcination in air. A structural characterization comprising X-ray diffraction, scanning electron microscopy, selected-area electron diffraction with transmission electron microscopy, and X-ray absorption fine structure measurements suggest that the synthesis process involves an initial conversion of Ga(NO3)3 to Ga(Oi-Pr)3 or a related species at 80 °C, which then transforms into γ-Ga2O3 nanoparticles upon increasing the temperature, to eventually produce β-Ga2O3 nanoparticles most likely via a dissolution and recrystallization process.
Introduction
Gallium oxide (Ga2O3) is a wide-bandgap semiconductor that possesses five polymorphs, namely, α-, β-, γ-, δ-, and ε-Ga2O3.1 Among them, β-Ga2O3 is the most stable phase below the melting point, and its properties and applications have been widely studied. Thus, the bandgap and breakdown field of β-Ga2O3 are known to be around 4.8 eV and 8 MV cm−1, respectively.2 This wide bandgap, along with its high thermal stability, renders β-Ga2O3 as a promising candidate for high-power electronic devices3,4 and solar-blind UV detectors,5–7 which only detect a specific wavelength range of 200–280 nm. With regard to its optical properties, β-Ga2O3 is almost transparent, and β-Ga2O3 nanoparticles have been demonstrated to exhibit photoluminescence upon doping.8–17 In addition to these applications, β-Ga2O3 has potential use in catalysis,18,19 solar cells,20 and gas sensors.21 β-Ga2O3 nanoparticles are also used as precursors in the synthesis of GaN nanoparticles via the nitration process.22,23Due to the wide application prospect of β-Ga2O3, its synthesis in the form of nanoparticles, nanorods, and thin-films has received intensive research attention. Thus, for the synthesis of β-Ga2O3 nanoparticles, a number of liquid processes based on the sol–gel method,8,12,19,24 hydrothermal synthesis,14,25,26 solvothermal synthesis,18,27–29 sonochemical method,30 and solution combustion31 were proposed. In most cases, the liquid processes involve the preparation of intermediate products such as GaOOH and γ-Ga2O3 nanoparticles, which are then converted to β-Ga2O3 nanoparticles by calcination at a temperature of 700 °C or higher in air. β-Ga2O3 nanoparticles with good luminescence, catalytic, and electrical properties are thereby obtained; however, calcination at elevated temperatures often causes nanoparticle aggregation. The calcination process also limits the in situ synthesis organic-modified nanoparticles. Therefore, the development of a one-step synthesis method that circumvents the aggregation issue to obtain highly crystalline β-Ga2O3 nanoparticles of a certain size is highly desirable. Herein, we propose a one-step synthesis of well-crystallized β-Ga2O3 nanoparticles via a solvothermal process that does not require additional calcination. This method consists of heating a dispersion liquid of Ga(NO3)3 in isopropanol at 400 °C for several hours to produce β-Ga2O3 nanoparticles with a size of ∼100 nm. We also discuss the growth process of the β-Ga2O3 nanoparticles by analyzing the products obtained at various reaction times.
Experimental
The one-step solvothermal synthesis of β-Ga2O3 was performed as follows. Gallium(III) nitrate n-hydrate (Ga(NO3)3, FUJIFILM Wako Chemicals, 072-02701, 60.2–68.5% was anhydrous according to an assay) was purchased and used as a precursor without further purification. Isopropanol was also purchased from FUJIFILM Wako Chemicals and used as a reaction medium. Ga(NO3)3 (0.0319 g) was put in isopropanol (2.00 mL) to prepare a 0.040 M dispersion liquid. It should be noted that Ga(NO3)3 did not dissolve in isopropanol at this concentration. The reactant liquid (2.0 mL) containing the undissolved Ga(NO3)3 was poured into a pressure-resistant Hastelloy C-276 reactor with an inner volume of 5.0 mL and capped tightly. The reactor was put in an electric furnace preheated to the desired reaction temperature, which was varied from 300 °C to 400 °C, above the critical temperature of isopropanol (235 °C).32After a certain reaction time, the reactor was removed from the electric furnace and immediately submerged in a water bath at room temperature to quench the reaction. The products were recovered from the reactor, and the solid products were separated by a combination of centrifugation (4 °C, 9600 × g, 15 min) and decantation. The solid products were purified by repeated centrifugation and decantation, using water and ethanol alternately. Finally, the products were freeze-dried. The products were obtained as white powders and analyzed as follows.
The crystal phase of the products was investigated by X-ray diffraction (XRD) analysis using a PANalytical Aeris diffractometer with Cu-Kα emission (λ = 1.5418 Å). The crystal phase fraction of the products was estimated using the PANalytical HighScore program (Ver. 4.6). The size and shape of the products were observed using a scanning electron microscope (SEM, JEOL JSM-IT-800). The crystal phase of each nanoparticle was confirmed by selected-area electron diffraction (SAED) measurements using a transmission electron microscope (TEM, JEOL JEM-ARM200F). To estimate the chemical states of the Ga species during the synthesis process, X-ray absorption spectra were measured around the Ga K-edge at BL-9A, Photon Factory, KEK, Japan. Data processing of the X-ray near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analysis was performed using REX2000 software (RIGAKU).33 Gallium isopropoxide [Ga(Oi-Pr)3] purchased from FUJIFILM Wako Chemicals (075-06071) was used as a reference material with known chemical state and crystal structure for the XAFS measurements.
Results and discussion
Fig. 1a shows the XRD profiles of the products obtained at 300 °C and different reaction times. At 10 min of reaction time, peaks attributable to the 113 and 044 reflections of the γ-Ga2O3 phase were observed, which is in agreement with previous studies describing the formation of γ-Ga2O3 nanoparticles by annealing Ga species in isopropanol at around 200 °C.18 Even at prolonged reaction times, the main component of the products was γ-Ga2O3; however, the appearance of peaks at around 30.1–31.7° and 35.2° at 360 and 1440 min of reaction time suggested the formation of the β-Ga2O3 phase. When the reaction was performed at 350 °C, the main product was also γ-Ga2O3 after 10 min of reaction (Fig. 1b). However, with increasing the reaction time, the β-Ga2O3 phase gradually appeared, to become the only observable phase at a reaction time of 720 min. At 400 °C (Fig. 1c), the product obtained at 10 min of reaction showed diffraction peaks attributable to the γ-Ga2O3 phase; however, at 30 min of reaction time, the main component was β-Ga2O3, and the γ-Ga2O3 phase gradually disappeared at longer reaction times. Under some synthetic conditions, unresolved peaks were observed. For example, the sample prepared at 300 °C and 60 min presented a peak at ∼28° which cannot be attributed to either β-Ga2O3 or γ-Ga2O3. These peaks may be assigned to metal oxides formed through the dissolution and subsequent solvothermal reaction of the constituent metals of the batch reactors during synthesis. These results suggest that the solvothermal treatment of Ga(NO3)3 in isopropanol first produced γ-Ga2O3, which gradually converted to β-Ga2O3 at 350 °C and 400 °C. | ||
| Fig. 1 Powder X-ray diffraction patterns of the products prepared at (a) 300 °C, (b) 350 °C, and (c) 400 °C at various reaction times. | ||
According to the XRD pattern, the fraction of β-phase in the products obtained at 400 °C and different reaction times was estimated using the PANalytical HighScore program, which performs a semiquantitative estimation based on the reference intensity ratio (RIR) method.34 In a binary system, the RIR method calculates the weight fraction of the components A and B (XA and XB, respectively) from the observed intensities of their strongest lines (IA and IB, respectively). The (I/Ic) values in eqn (1) are referenced from the crystal structure database file.
 | (1) |
The results are shown in Fig. 2. As can be extracted from the figure, the ratio of β-phase increased with increasing the reaction time. Notably, a quick crystal phase change from γ to β phase occurred within 30 min at 400 °C.
 | ||
| Fig. 2 Ratio of β-phase in the products prepared at 400 °C and various reaction times according to a semiquantitative analysis performed using the PANalytical HighScore program. | ||
Fig. 3 shows the SEM images of the samples prepared at 400 °C and different reaction times. The product obtained at 10 min of reaction time was smaller than 10 nm and did not exhibit an apparent crystal shape (Fig. 3a). Meanwhile, the product obtained at 30 min of reaction time was composed of small spherical nanoparticles and larger nanocrystals with distorted octahedral shapes (Fig. 3b). As can be seen in Fig. 3, the number of smaller nanoparticles gradually decreased with increasing the reaction time, whereas the size of the distorted octahedral nanoparticles gradually increased, reaching around 100 nm at 1440 min (Fig. 3d). Based on the XRD results (Fig. 1c), the nanoparticles shown in Fig. 3a and d were inferred as γ-Ga2O3 and β-Ga2O3 phases, respectively. To confirm this assumption and resolve the crystal structure of the smaller and larger nanoparticles produced at the intermediate reaction time (Fig. 3b), we performed TEM-SAED measurements. Fig. 4 shows the TEM images of the samples prepared at 400 °C with different reaction times with their SAED patterns. The SAED ring patterns of the products after 10 min of reaction (Fig. 4a inset) correspond to the 044 and 113 reflections of the γ-Ga2O3 phase. After 1440 min of reaction (Fig. 4c inset), the characteristic diffraction spots of β-Ga2O3 phases appear. These results agreed with the XRD and SEM measurements. Fig. 4b shows the TEM image of the sample prepared at 400 °C for 30 min. Consistent with Fig. 3b, the product was composed of smaller and larger nanoparticles. The SAED measurements revealed that the smaller nanoparticles (sized ∼10 nm) were composed of γ-Ga2O3 phase, whereas the larger nanoparticles (sized ∼100 nm) existed in the β-Ga2O3 phase.
 | ||
| Fig. 3 Scanning electron microscopy images of the Ga2O3 nanoparticles synthesized at 400 °C with the reaction times of (a) 10, (b) 30, (c) 120, and (d) 1440 min. | ||
 | ||
| Fig. 4 Transmission electron microscopy (TEM) images of the Ga2O3 nanoparticles synthesized at 400 °C with the reaction times of (a) 10, (b) 30, and (c) 1440 min. Insets: Selected-area electron diffraction pattern of the products shown in each TEM image. | ||
These results confirm that the crystalline phase transition from γ-Ga2O3 to β-Ga2O3 occurred within 1440 min at 400 °C. As discussed in the Introduction, the calcination of GaOOH, α-Ga2O3, or γ-Ga2O3 at 700 °C or above in air produces β-Ga2O3 particles,8,13,14,17–19,26,28,35,36 which is the most stable phase. The detailed mechanism of the phase transition from γ-Ga2O3 to β-Ga2O3 has been reported recently.37 Specifically, a synchrotron X-ray atomic pair distribution function analysis revealed that the local structure of γ-Ga2O3 started to change to β-Ga2O3 at around 300 °C, and then the bulk transformation occurred between 550 °C and 750 °C. However, our process proceeded at much lower temperatures, suggesting that the formation of the β-Ga2O3 phase occurred through a different process from that reported for the calcination in air. In our case, the production of γ-Ga2O3 and its transition to β-Ga2O3 occurred in isopropanol. Therefore, the effects of isopropanol on the phase transition should be considered. Typically, the synthesis of nanoparticles comprises nucleation, growth, and coarsening stages sequentially.38 In the final coarsening stage, i.e., the Ostwald ripening stage, smaller particles with higher free energy are dissolved in the solution and then recrystallized with lower free energy. This might also occur in the present solvothermal process; accordingly, the number of smaller γ-Ga2O3 nanoparticles would decrease to give rise to larger β-Ga2O3 nanoparticles. A similar crystal phase transition process was reported for the anatase-to-rutile phase transition via dissolution and precipitation.39 However, in the present study, the dissolution process of Ga species in isopropanol remains unclear. This could be explained in terms of the formation of Ga(Oi-Pr)3 or similar species that can be dissolved in isopropanol at the synthesis temperature. In this regard, the dissolution of Ga(Oi-Pr)3 and its dimer in isopropanol was previously described by Suslova et al.40 In our case, the initially produced γ-Ga2O3 nanoparticles might have slight solubility in isopropanol as Ga(Oi-Pr)3 or similar species, to be then recrystallized as β-Ga2O3 nanoparticles with apparent crystal shape.
To further investigate the growth of Ga2O3 nanoparticles in isopropanol, the local structure around Ga in the products, especially the chemical bonds between Ga and isopropanol molecules, was investigated by XAFS. During the hydrothermal synthesis of Ga2O3 nanoparticles, Ga3+ ions react with H2O to produce GaOOH.10,19,26 However, little is known about the solvothermal processes. The products obtained upon heating the precursor at 80 °C, 160 °C, 240 °C, and 270 °C for 10 min and at 300 °C for 60 min in isopropanol were analyzed by XAFS. To provide comparison, Ga(NO3)3 in isopropanol and Ga(Oi-Pr)3 were also analyzed. Fig. 5–7 show the XANES spectra, the k3χ (k) oscillation, and the derived radial structure function, respectively. The radial structure function of Ga(NO3)3 in isopropanol exhibited a peak at around 1.50 Å (Fig. 7a), which can be attributed to Ga–O bonds in Ga(NO3)3. After heating at 80 °C for 10 min, new peaks appeared at 1.48 and 2.70 Å (Fig. 7c). This spectrum was similar to that of Ga(Oi-Pr)3 (Fig. 7b), which exhibited peaks at 1.44 and 2.67 Å corresponding to Ga–O and Ga–C bonds in Ga(Oi-Pr)3, respectively. Therefore, the peaks at 1.48 Å and 2.70 Å in Fig. 7c can be also attributed to these chemical bonds. The corresponding EXAFS vibration in Fig. 6b and c also indicated that the product obtained upon heating Ga(NO3)3 80 °C for 10 min was similar to Ga(Oi-Pr)3. Taken together, these results suggest that Ga(NO3)3 in isopropanol was converted to Ga(Oi-Pr)3 or similar structures even at a low temperature of 80 °C.
 | ||
| Fig. 5 Ga K-edge X-ray absorption near-edge structure spectra of (a) Ga(NO3)3 in isopropanol, (b) Ga(Oi-Pr)3, and the samples obtained upon heating at (c) 80 °C, (d) 160 °C, (e) 240 °C, and (f) 270 °C for 10 min and at (g) 300 °C for 60 min. | ||
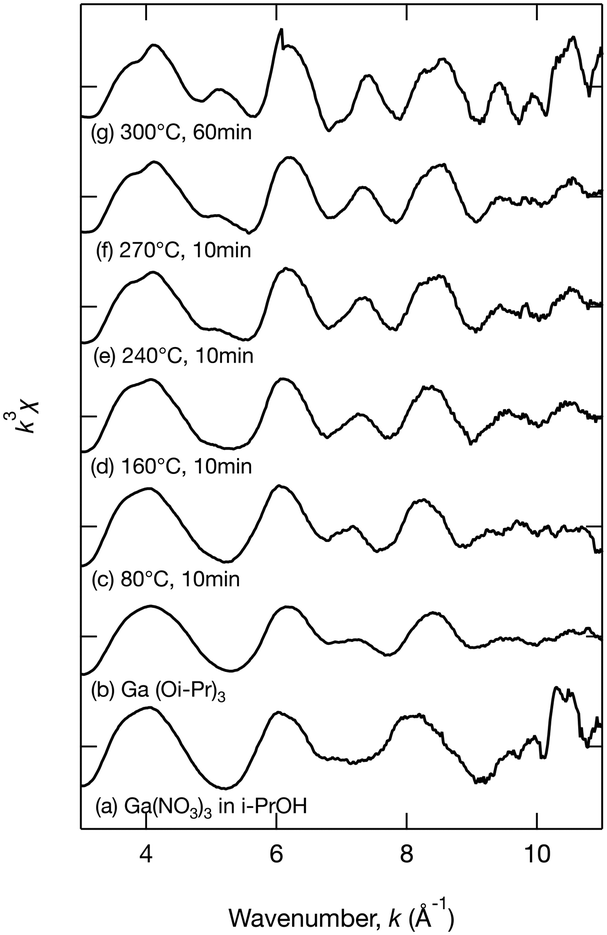 | ||
| Fig. 6 The k3χ (k) oscillation of the extended X-ray absorption fine structure analysis of (a) Ga(NO3)3 in isopropanol, (b) Ga(Oi-Pr)3, and the samples obtained upon heating at (c) 80 °C, (d) 160 °C, (e) 240 °C, and (f) 270 °C for 10 min and at (g) 300 °C for 60 min. | ||
 | ||
| Fig. 7 Radial structure function derived from the extended X-ray absorption fine structure analysis of (a) Ga(NO3)3 in isopropanol, (b) Ga(Oi-Pr)3, and the samples obtained upon heating at (c) 80 °C, (d) 160 °C, (e) 240 °C, and (f) 270 °C for 10 min and at (g) 300 °C for 60 min. | ||
As the reaction temperature increased, the EXAFS vibration spectra (Fig. 6) and the radial structure function (Fig. 7) became similar to that of the sample prepared at 300 °C for 60 min (Fig. 6g and 7g), which was confirmed to be γ-Ga2O3 by the abovementioned XRD analysis (Fig. 1a). The XANES spectra shown in Fig. 5 also indicated the gradual formation of periodic structures. These spectra confirmed that the products became more similar to γ-Ga2O3 upon increasing the reaction temperature. The peaks of the radial structure function of the sample obtained by heating at 300 °C for 60 min (Fig. 7g) were observed at 1.46 and 2.84 Å. The former peak can be assigned to Ga–O bonds, and the latter peak is attributable to Ga–O and Ga–Ga interactions in γ-Ga2O3. This suggests that the Ga(Oi-Pr)3 structures corresponding to Fig. 7c–f might gradually form polynuclear species having Ga–O–Ga bonds that resemble γ-Ga2O3 structures.
On the basis of these results, a plausible reaction pathway for the formation of β-Ga2O3 nanoparticles in isopropanol from Ga(NO3)3 can be proposed as summarized in Fig. 8. According to this mechanism, Ga(NO3)3 is converted to Ga(Oi-Pr)3 or similar structures in isopropanol at a temperature as low as 80 °C. As the reaction temperature increases, the intermediate species gradually forms periodic structures, producing γ-Ga2O3 nanoparticles. Further heating at 400 °C causes the crystal phase transition from γ-Ga2O3 to β-Ga2O3 within 24 h, possibly via a dissolution and recrystallization process.
 | ||
| Fig. 8 Plausible pathway for the formation of β-Ga2O3 nanoparticles from Ga(NO3)3 in isopropanol. | ||
Conclusions
A one-step method for the synthesis of β-Ga2O3 nanoparticles without requiring calcination in air has been demonstrated. XRD, SEM, and TEM measurements confirmed that the solvothermal treatment of Ga(NO3)3 in isopropanol at 400 °C successfully produced well-crystallized β-Ga2O3 nanoparticles with a size of ∼100 nm. XAFS measurements revealed that Ga(NO3)3 was converted to Ga(Oi-Pr)3 species at a temperature as low as 80 °C. These intermediate products were gradually converted to γ-Ga2O3 nanoparticles at higher temperatures. Then, the γ-Ga2O3 nanoparticles transformed into β-Ga2O3 through the Ostwald ripening process, and finally, β-Ga2O3 was the main constituent of the product obtained upon heating at 400 °C for 24 h.Author contributions
Kengo Takezawa: validation, formal analysis, investigation. Jinfeng Lu: investigation, resources, visualization. Chiya Numako: validation, formal analysis, investigation, resources, writing – review & editing. Seiichi Takami: conceptualization, methodology, formal analysis, resources, writing – original draft, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP17H06467 and JP20H02514. XAFS measurements were performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2019G650). We thank Dr. Yuta Yamamoto of the High-Voltage Electron Microscope Laboratory, Institute of Materials and Systems for Sustainability, Nagoya University, for taking the TEM measurements.References
- S. I. Stepanov, V. I. Nikolaev, V. E. Bougrov and A. E. Romanov, Rev. Adv. Mater. Sci., 2016, 44, 63–86 CAS.
- J. Zhang, J. Shi, D.-C. Qi, L. Chen and K. H. L. Zhang, APL Mater., 2020, 8, 020906 CrossRef CAS.
- M. Higashiwaki, K. Sasaki, A. Kuramata, T. Masui and S. Yamakoshi, Phys. Status Solidi A, 2014, 211, 21–26 CrossRef CAS.
- M. J. Tadjer, J. L. Lyons, N. Nepal, J. A. Freitas, A. D. Koehler and G. M. Foster, ECS J. Solid State Sci. Technol., 2019, 8, Q3187–Q3194 CrossRef.
- S. Oh, C.-K. Kim and J. Kim, ACS Photonics, 2017, 5, 1123–1128 CrossRef.
- J. Xu, W. Zheng and F. Huang, J. Mater. Chem. C, 2019, 7, 8753–8770 RSC.
- M. I. Pintor-Monroy, B. L. Murillo-Borjas and M. A. Quevedo-Lopez, ACS Appl. Electron. Mater., 2020, 2, 3358–3365 CrossRef CAS.
- W. Y. Shen, M. L. Pang, J. Lin and J. Fang, J. Electrochem. Soc., 2005, 152, H25 CrossRef CAS.
- S. Zhou, G. Feng, B. Wu, N. Jiang, S. Xu and J. Qiu, J. Phys. Chem. C, 2007, 111, 7335–7338 CrossRef CAS.
- G. Li, C. Peng, C. Li, P. Yang, Z. Hou, Y. Fan, Z. Cheng and J. Lin, Inorg. Chem., 2010, 49, 1449–1457 CrossRef CAS PubMed.
- T. Wang and P. V. Radovanovic, J. Phys. Chem. C, 2011, 115, 18473–18478 CrossRef CAS.
- R. Lorenzi, A. Paleari, N. V. Golubev, E. S. Ignat'eva, V. N. Sigaev, M. Niederberger and A. Lauria, J. Mater. Chem. C, 2014, 3, 41–45 RSC.
- X.-S. Wang, J.-Q. Situ, X.-Y. Ying, H. Chen, H. Pan, Y. Jin and Y.-Z. Du, Acta Biomater., 2015, 22, 164–172 CrossRef CAS PubMed.
- S. Ye, Y. Zhang, H. He, J. Qiu and G. Dong, J. Mater. Chem. C, 2015, 3, 2886–2896 RSC.
- X.-S. Wang, W.-S. Li, J.-Q. Situ, X.-Y. Ying, H. Chen, Y. Jin and Y.-Z. Du, RSC Adv., 2015, 5, 12886–12889 RSC.
- P. C. Stanish, P. Yin and P. V. Radovanovic, Chem. Mater., 2020, 32, 7516–7523 CrossRef CAS.
- F. Shi and H. Qiao, CrystEngComm, 2020, 22, 7794–7799 RSC.
- Y. Hou, J. Zhang, Z. Ding and L. Wu, Powder Technol., 2010, 203, 440–446 CrossRef CAS.
- S. Hong, C. K. Rhee and Y. Sohn, J. Alloys Compd., 2019, 774, 11–17 CrossRef CAS.
- M. García-Carrión, J. Ramírez-Castellanos, E. Nogales, B. Méndez, C. C. You, S. Karazhanov and E. S. Marstein, Mater. Lett., 2019, 261, 127088 CrossRef.
- M. Ogita, N. Saika, Y. Nakanishi and Y. Hatanaka, Appl. Surf. Sci., 1999, 142, 188–191 CrossRef CAS.
- W.-S. Jung, Mater. Lett., 2002, 57, 110–114 CrossRef CAS.
- Y. Li, Z. Xiong, D. Zhang, X. Xiu, D. Liu, S. Wang, X. Hua, Z. Xie, T. Tao, B. Liu, P. Chen, R. Zhang and Y. Zheng, Superlattices Microstruct., 2018, 117, 235–240 CrossRef CAS.
- E. B. Shaik, C. S. Kamal, K. Srinivasu, B. V. N. Kumar, P. K. Balla, R. D. K. Swamy and K. R. Rao, J. Mater. Sci.: Mater. Electron., 2020, 31, 6185–6191 CrossRef CAS.
- E. I. EL-Sayed, A. A. Al-Ghamdi, S. Al-Heniti, F. Al-Marzouki and F. El-Tantawy, Mater. Lett., 2011, 65, 317–321 CrossRef CAS.
- F. Shi and H. Qiao, CrystEngComm, 2020, 23, 492–498 RSC.
- G. Sinha and S. Chaudhuri, Mater. Chem. Phys., 2009, 114, 644–649 CrossRef CAS.
- L. Shi, J. Zhang, S. Wu, Y. Li, L. Jiang and Q. Cui, J. Am. Ceram. Soc., 2014, 97, 2607–2614 CrossRef CAS.
- D. S. Cook, R. J. Kashtiban, K. Krambrock, G. M. de Lima, H. O. Stumpf, L. R. S. Lara, J. D. Ardisson and R. I. Walton, Materials, 2019, 12, 838 CrossRef CAS PubMed.
- M. I. Dar, S. Sampath and S. A. Shivashankar, RSC Adv., 2014, 4, 49360–49366 RSC.
- T. Biljan, A. Gajović and Z. Meić, J. Lumin., 2008, 128, 377–382 CrossRef CAS.
- M. Gude and A. S. Teja, J. Chem. Eng. Data, 1995, 40, 1025–1036 CrossRef CAS.
- T. Taguchi, T. Ozawa and H. Yashiro, Phys. Scr., T, 2005, 115, 205–206 CrossRef.
- F. H. Chung, J. Appl. Crystallogr., 1974, 7, 519–525 CrossRef.
- Y. Quan, S. Liu, K. Huang, D. Fang, X. Zhang and H. Hou, Trans. Nonferrous Met. Soc. China, 2010, 20, 1458–1462 CrossRef CAS.
- H. J. Bae, T. H. Yoo, Y. Yoon, I. G. Lee, J. P. Kim, B. J. Cho and W. S. Hwang, Nanomaterials, 2018, 8, 594 CrossRef PubMed.
- P. Castro-Fernández, M. V. Blanco, R. Verel, E. Willinger, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2020, 124, 20578–20588 CrossRef.
- J. A. Marqusee and J. Ross, J. Chem. Phys., 1983, 79, 373–378 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, Nanoscale, 2014, 6, 11574–11632 RSC.
- E. V. Suslova, N. Y. Turova, Z. A. Starikova and A. V. Kepman, Russ. J. Coord. Chem., 2016, 42, 19–26 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |