DOI:
10.1039/D1CE00719J
(Paper)
CrystEngComm, 2021,
23, 5200-5207
From [B6O13]8− to [GaB5O13]8− to [Ga{B5O9(OH)}{BO(OH)2}]2−: synthesis, structure and nonlinear optical properties of new metal borates†
Received
1st June 2021
, Accepted 22nd June 2021
First published on 22nd June 2021
Abstract
Three new metal borates, namely K0.5[B6O10]·H2O·1.5H3O (1), [GaB5O10]·H2en (2) and NaCs[Ga{B5O9(OH)}{BO(OH)2}] (3), have been synthesized under solvothermal conditions. Compound 1 features an intricate 3D framework composed of [B6O13]8− clusters, resulting in a 6-connected achiral pcu net. After introducing the organic gallium, one of the BO4 groups of the [B6O13]8− cluster is substituted by a GaO4 polyhedron to form the acentric [GaB5O13]8− cluster in 2. It also displays a high second-harmonic generation (SHG) response about approximately 2.6 times that of KH2PO4 (KDP), which is higher than those of alumino-borates (ABOs) containing the acentric [AlB5O13]8− cluster (0.3–2.1 times that of KDP). Dipole moment calculations have been carried out to confirm these results. In the process of trying to synthesize a new compound with a denser structure than 2, we have unexpectedly obtained 3 crystallized in the centrosymmetric (CS) space group, which shows a 1D structure constructed from the [Ga{B5O9(OH)}{BO(OH)2}]2− cluster. This kind of low-dimensional structure is rare in gallo-borates (GBOs) as well as ABOs.
Introduction
Borates are an important class of nonlinear optical (NLO) crystals mainly because they can easily crystallize in the non-centrosymmetric (NCS) space group and such structures are conducive to obtaining a large second-harmonic generation (SHG) response.1–13 Alumino-borates (ABOs) with diverse structures can be obtained by introducing Al atoms (belonging to the same group as B atoms) into borates. Compared with the B atom, Al has more abundant coordination modes, such as an AlO4 tetrahedron, O2AlO3 triangular bipyramid, OAlO4 tetragonal pyramid and AlO6 octahedron.14–16 In 1973, Lehmann and Teske introduced Al atoms into a borate system for the first time, but did not perform detailed characterization.17 After that, Xu et al. reported several cases of ABOs, but due to the lack of high-quality crystals, the precise structure of the compound has not been determined.18–21 Lin et al. also successfully synthesized a series of PKU-n (n = 1, 2, 5, 6, 8) ABOs with an open framework by a boric acid flux synthesis method, and their structures were mostly determined by powder X-ray diffraction.22–26 Since 2009, our group has been committed to the synthesis of ABOs with special configurations oriented by organic amines or transition metal complexes.8,27,28 We introduce Al atoms into the borate framework based on the following considerations: (1) expanding the structural dimension of borates. The borate synthesized by a hydro(solvo)thermal method often exhibits a low-dimensional structure, and the introduction of Al is an effective way to form a multi-dimensional borate; (2) a twisted structure can be formed between Al and O atoms, thus forming a metal polyhedral configuration with a chiral center. The synergistic combination between acentric oxoboron clusters and chiral polyhedra (such as AlO4, GeO4 or GaO4 tetrahedra) is beneficial for increasing the probability of products crystallized in the NCS space group; (3) enriching the fundamental building blocks (FBBs) of borate, laying the foundation for the synthesis of more borate with unique FBBs and excellent properties. According to the above ideas, we have synthesized a new inorganic–organic hybrid solid with NLO properties, namely [Zn(dap)2][AlB5O10] (dap = 1,3-diaminopropane) under solvothermal conditions based on the structural features of 3D open framework inorganic solids and 2D metal–organic coordination polymers for the first time.29 In addition, we also use the “vacancy-oriented synthesis strategy” to construct multicomponent composite polyoxometalates containing new {B9O18(OH)3} and {B22O42} clusters.30 The existing borates can only form {B9O13(OH)4}, {B9O12(OH)6}, {B9O19}, {B9O17(OH)2} and {B9O16(OH)3} clusters in previous studies.
The Ga atom, like Al, belongs to the same main group elements and also has several coordination modes (i.e., a GaO4 tetrahedron, an O2GaO3 triangular bipyramid, and a GaO6 octahedron).31–33 In our previous work, we found that the NLO properties of dense structures are better.34,35 Compared with the Al atom, Ga has a larger atomic radius and more easily forms a dense structure, but the crystal structure of gallo-borates (GBOs) is more difficult to obtain than that of ABOs. In the synthesis of GBOs, we choose organic gallium compounds (i.e., gallium acetylacetonate and gallium isopropoxide) instead of traditional inorganic gallium sources (i.e., gallium oxide, gallium chloride and gallium nitrate) mainly based on the following points: (1) gallium acetylacetonate is easily dissolved in organic solvents (i.e., ethylenediamine and 1,3-propanediamine); (2) gallium acetylacetonate may undergo a slowly hydrolysis reaction to obtain a GaO4 tetrahedron with a chiral Ga center in the crystallization process; (3) the synergistic combination between chiral GaO4 tetrahedra and acentric oxoboron clusters increase the likelihood of producing new acentric GBOs with a dense structure, offering us the opportunity for choosing the desired NLO materials from these candidates.
In this work, in the process of trying to synthesize a dense structure crystallized in the NCS space group, we have obtained the following three new metal borates, namely K0.5[B6O10]·H2O·1.5H3O (1), [GaB5O10]·H2en (2) and NaCs[Ga{B5O9(OH)}{BO(OH)2}] (3). Compound 1 shows an achiral 3D framework with [B6O13]8− clusters. After replacing BO4 with chiral GaO4 polyhedra, the compound is transformed into a NCS structure and new compound 2 presents good NLO activity (2.6 times that of KDP), which is better than previously reported ABOs (0.3–2.1 times that of KDP). Therefore, the introduction of a chiral polyhedron with a larger atomic radius may be an effective way to synthesize better NLO crystals. In order to obtain a denser structure, we try to fill the void of 2 with alkali (earth) metal ions and unexpectedly obtained a GBO with rare 1D chains (compound 3). The synthesis, structure and comparison of the above compounds will be discussed in detail.
Experimental section
Materials and measurements
All chemical reagents were commercially available and used without further purification. Commercial high purity KH2PO4 (KDP, 99.99%) and LiNbO3 (99.99%) were purchased from Aladdin Co., Ltd., China. Fourier transform infrared (FTIR) spectra were recorded on a Nicolet iS10 FTIR spectrometer using a mixture of KBr and a sample wafer in the range of 4000–400 cm−1. Powder X-ray diffraction studies were performed on a Bruker D8 Advance X-ray diffractometer. Optical diffuse reflectance spectra in the range of 200–800 nm were obtained via a Lamda-950 UV/vis-NIR spectrophotometer at room temperature. Thermogravimetric analyses (TGA) were conducted on a METTLER TGA/SDTA 851e thermal analyser under a constant flow of oxygen from room temperature to 1000 °C at a heating rate of 10 °C min−1. SHG measurements were performed with a Nd:YAG solid-state laser (λ = 1064 nm) as the incident light source. Commercial high purity KDP and LiNbO3 were used as the reference for SHG measurements. Ion-exchange reactions were performed by stirring the samples of as-synthesized compound 2 (50 mg) in 5 M LiCl, NaCl, KCl, MgCl2, CaCl2, and BaCl2 solutions (10 mL) at room temperature for 12 h. The ion-exchanged products were recovered by filtration, washed with excess water, and dried in air. The Li, Na, K, Mg, Ca and Ba contents in the ion-exchanged products were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 725 ICP-OES).
Synthesis of K0.5[B6O10]·H2O·1.5H3O (1)
A mixture of H3BO3 (0.370 g, 6 mmol) and potassium tert-butoxide (0.112 g, 1 mmol) was added into pyridine (1.5 mL) and ethylenediamine (2 mL). After stirring for 15 min, the resulting mixture was sealed in a 25 mL Teflon-lined stainless steel autoclave, heated at 210 °C for 8 days, and then slowly cooled to room temperature. The colorless block crystals were obtained (yield 80% based on H3BO3). Elemental anal. (%) calcd. for B12H13KO25: K 6.70, found: K 6.33.
Synthesis of [GaB5O10]·H2en (2)
A mixture of compound 1 (0.104 g, 0.5 mmol) and gallium acetylacetonate (0.367 g, 1 mmol) was added into pyridine (1 mL) and ethylenediamine (1.5 mL). After stirring for 1 h, the resulting mixture was sealed in a 20 mL Teflon-lined stainless steel autoclave, heated at 210 °C for 8 days, and then slowly cooled to room temperature. The colorless block crystals were obtained (yield 50% based on 1).
Synthesis of NaCs[Ga{B5O9(OH)}{BO(OH)2}] (3)
A mixture of compound 2 (0.104 g, 0.3 mmol), sodium tert-butoxide (0.096 g, 1 mmol) and CsOH·H2O (0.168 g, 1 mmol) was added into pyridine (1 mL) and ethylenediamine (1.5 mL). After stirring for 1 h, the resulting mixture was sealed in a 20 mL Teflon-lined stainless steel autoclave, heated at 210 °C for 8 days, and then slowly cooled to room temperature. The colorless block crystals were obtained (yield 48% based on 2).
Crystallographic data collection and refinement
The single crystal X-ray diffraction data for compounds 1–3 were collected at 296 K on a Bruker SMART CCD diffractometer equipped with graphite monochromatized Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by the full-matrix least-squares method on F2 using the SHELXL-2018 programs.36 All non-hydrogen atoms were refined with anisotropic displacement parameters. The final structures were checked for possible missing symmetry with the PLATON program.37 The detail crystallographic data and selected bond distances for compounds 1–3 are summarized in Tables 1 and S1–S3,† respectively.
Table 1 Crystallographic data and structure refinements for compounds 1–3
| Compound |
1
|
2
|
3
|
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|.
wR2 = {∑w[(Fo)2 − (Fc)2]2/∑[w(Fo)2]2}1/2.
|
| Formula |
B12H13KO25 |
C2H10B5GaN2O10 |
B6H3CsGaNaO13 |
|
M (mol−1) |
581.92 |
345.89 |
501.50 |
|
T (K) |
296(2) |
296(2) |
296(2) |
| Crystal system |
Cubic |
Monoclinic |
Triclinic |
| Space group |
Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) no. 205 no. 205 |
Pc no. 7 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) no. 2 no. 2 |
|
a (Å) |
12.243(3) |
6.686(5) |
7.027(2) |
|
b (Å) |
12.243(3) |
8.082(6) |
8.039(2) |
|
c (Å) |
12.243(3) |
10.485(8) |
11.442(4) |
|
α (°) |
90 |
90 |
108.567(10) |
|
β (°) |
90 |
97.192(18) |
93.633(11) |
|
γ (°) |
90 |
90 |
90.442(11) |
|
V (Å3) |
1835.2(13) |
562.2(7) |
611.2(3) |
|
Z
|
4 |
2 |
2 |
|
F (000) |
1168 |
344 |
468 |
|
D
c (g cm−3) |
2.106 |
2.043 |
2.725 |
|
θ (°) |
2.88–31.61 |
3.19–25.05 |
2.67–29.28 |
|
R
int
|
0.0498 |
0.0516 |
0.0557 |
| Independent reflns |
1006 |
1004 |
3278 |
| Refined parameters |
74 |
183 |
203 |
| Flack parameters |
— |
0.03(4) |
— |
|
GoF
|
1.067 |
1.046 |
1.027 |
|
R
1
[I > 2σ(I)] |
0.0502 |
0.0554 |
0.0439 |
| wR2b [I > 2σ(I)] |
0.1350 |
0.1212 |
0.0898 |
Results and discussion
Synthesis of the compounds
From a synthetic point of view, the title compounds 1–3 are related to the synthesis conditions. All the compounds were synthesized under solvothermal conditions (pyridine and ethylenediamine) at 210 °C for 8 days. In 1, the compound containing [B6O13]8− clusters was synthesized with H3BO3 and potassium salt, resulting in a 3D framework with the centrosymmetric (CS) space group. Based on the idea of “the synergistic combination between oxoboron clusters and chiral polyhedra (such as GaO4 tetrahedra) to obtain the product with the NCS space group and high-dimensional structure”, we tried to add organogallium to compound 1, hoping to obtain a 3D GBO which crystallizes in the NCS space group. Fortunately, one of the BO4 groups of the [B6O13]8− cluster was substituted by GaO4 to form the acentric [GaB5O13]8− cluster. So, compound 2 which crystallizes in the NCS space group is constructed from the [GaB5O13]8− cluster and exhibits a 6-connected (6-c) 3D framework with (412·63) pcu topology, just similar to 1. Considering that the non-framework volume of 2 occupies 37.4% of the whole unit cell volume, a variety of alkali and alkaline earth metals with different sizes and charges of ions were introduced in order to obtain a dense structure, which may further change their NLO properties. Unexpectedly, compound 3 is constructed from the [Ga{B5O9(OH)}{BO(OH)2}]2− cluster and Na+ and Cs+ cations, and exhibits a rare 1D structure in GBOs with the CS space group. Attempts to produce compounds at higher temperatures using stronger bases or other alkali and alkaline earth metals were fruitless. It seems that the template effect is improved after adding the alkali or alkaline earth metal salt, which is not conducive to the formation of high-dimensional NCS borates under this condition.
Crystal structure of 1
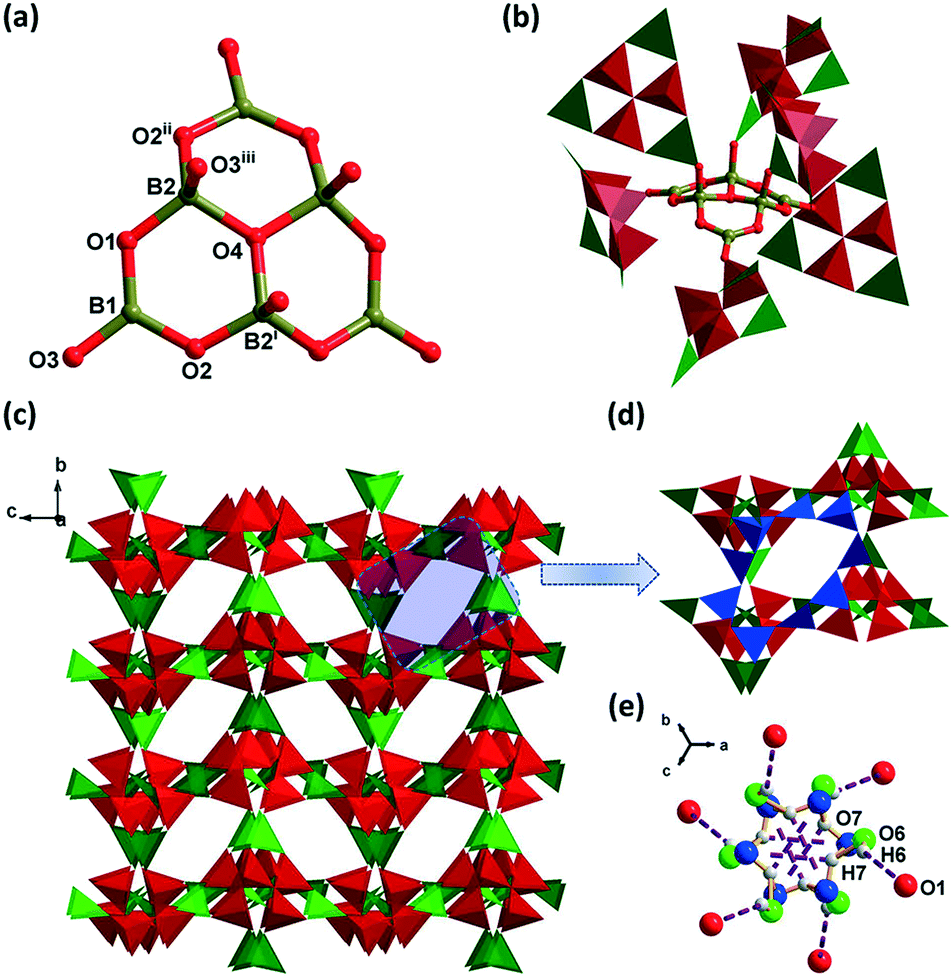
Compound 1 is the precursor of 2. Single-crystal XRD analysis shows that 1 crystallizes in the centrosymmetric (CS) cubic space group Pa. The asymmetric unit of compound 1 consists of two B atoms, 10/3 O atoms, 1/6 K+, 1/3 H2O, and 1/2 H3O+. BVS calculations show that K and B are consistent with the expected oxidation states.38,39 Three BO4 units [B–O(av.), 1.471 Å] are joined together through corner-sharing of the μ3-O4 atom and three BO4 units are connected with neighboring three BO3 units [B–O(av.), 1.366 Å] to form a compact cluster of [B6O13]8− ({B6}-1), with shorthand notation of 6:[3Δ + 3T)] (Fig. 1a). In {B6}-1 clusters, the μ3-O4–B bonds are unique because their bond distances (1.530 Å) are longer than those of the μ2-O–B bonds (1.358–1.457 Å) in BO3 and BO4. Each {B6}-1 is connected to six other {B6}-1 by corner-sharing O3 atoms to form a 3D framework with misplaced 9-membered ring (9-MR) windows (Fig. 1b–d). The overall 3D framework can be rationalized as a pcu topology with the Schläfli symbol of (412·63) and the cubic cavity with a diameter of 8.60 Å is constructed from eight {B6}-1 interconnected by O atoms (Fig. S1a and b†). The larger K+ ion in 1 (ionic radius: K+/1.38 Å, Na+/1.02 Å) makes the diameter of the cubic cavity longer (8.60 Å) than that in our previously reported NH4NaB6O10 (7.35 Å).40 Further, the structure is stabilized by the water cluster formed by O6–H6⋯O1 (2.460 Å, 3.265 Å, 158°) and O7–H7⋯O7 (1.800 Å, 2.266 Å, 117°) hydrogen bonds among the water molecule, hydronium and {B6}-1 (Fig. 1e, Table S4†). The channels of 1 are also filled with guest ions/molecules of K+, water molecules and hydroniums (Fig. S1c†).
 |
| | Fig. 1 (a) The structure of {B6}-1 of compound 1, symmetry codes: (i) y, z, x; (ii) z, x, y; (iii) y, −z + 1/2, x − 1/2. (b) The linkage of {B6}-1. (c) The 3D framework viewed along the a axis. (d) The misplaced 9-MR windows highlighted with blue color. (e) The water cluster of 1. | |
Crystal structure of 2
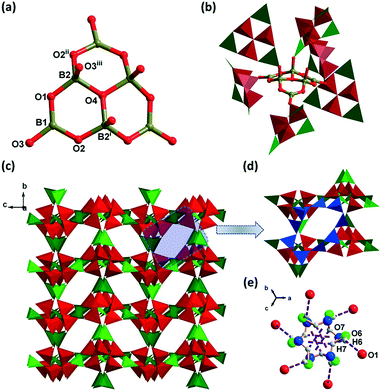
Considering that compound 1 crystallizes in the CS space group with a 3D framework, we tried to introduce a chiral GaO4 polyhedron, and use its chiral induction and transfer characteristics to construct compounds crystallized in the NCS space group. As expected, one of the BO4 groups of the [B6O13]8− cluster was substituted by GaO4, and then one BO3 triangle is transplanted to link two BO4 groups by corner-sharing, forming the acentric [GaB5O13]8− cluster in compound 2 (Fig. 2a and d). The new compound 2 crystallizes in the NCS monoclinic space group Pc and the [GaB5O13]8− cluster can be considered as the transfiguration of the centrosymmetric [B6O13]8− cluster. Each [GaB5O13]8− is connected to six others by corner-sharing O1, O6 and O7 atoms to form a 3D framework (Fig. 2b and f), and the 3D framework can also be regarded as having a pcu topology with the Schläfli symbol of (412·63), just similar to 1. As for the [GaB5O13]8− cluster, a GaO4 polyhedron is particularly important for the structural expansion of compound 2, which extends the 1D [B5O10]5− chain to a 3D framework by Ga–O–B bonds (Fig. 2c). In addition to the fact that 1 and 2 crystallize in different space groups, their structures are also somewhat different. In 2, a 3D-intersecting channel system contains a 10-, 9- and 9-MR along the a, b and c axes, respectively (Fig. 2e, f and S2†), and the template H2en (en = ethylenediamine) cations interact with the 10-MR channels through several N–H⋯O (1.810–2.390 Å, 2.693–3.275 Å, 151–175°) hydrogen bonds (Table S5†). Meanwhile only the misplaced 9-MR windows can be observed in 1 along the a, b and c axes, and the shapes of the windows are also different (Fig. 1d). Besides, unlike compound 1, there is no cavity structure in 2. Therefore, the introduction of a chiral GaO4 polyhedron changed the space group and the crystal structure of the compound.
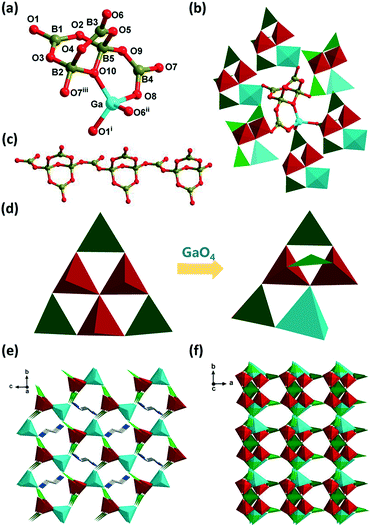 |
| | Fig. 2 (a) The structure of [GaB5O13]8− of compound 2, symmetry codes: (i) x, 1 − y, −0.5 + z; (ii) x, −y, −0.5 + z; (iii) 1 + x, y, z. (b) The linkage of the [GaB5O13]8− cluster. (c) The 1D chain formed by the oxoboron cluster. (d) Transformation from [B6O13]8− to [GaB5O13]8− by replacing a BO4 with a GaO4 group. (e) The 10-MR channels of the 3D framework located by the H2en cations along the a axis. The H atoms are omitted for clarity. (f) View of the 9-MR channels along the c axis. Color code: BO4: red, BO3: green, GaO4: turquoise, similarly hereinafter. | |
Table 2 shows the structure of known ABOs and GBOs containing [AlB5O10] and [GaB5O10], respectively.8,41–45 Clearly, although the FBBs of the compound are [MB5O10] (M = Al, Ga), they crystallize in a variety of space groups with different windows and different topological structures. The non-framework volumes and SHG responses are also different, which is related to the differences of templates and chiral polyhedra (AlO4, GaO4) in compounds. Among them, compound 2 has the smallest non-framework volume and the highest SHG responses. Compared with our previous structure of (H2EDAP)[GaB5O10]·H2O (2a, EDAP = N-ethyl-diaminopropane),42 they have some differences in structure and properties. Firstly, compound 2a crystallizes in the orthorhombic Pna21 space group, while 2 crystallizes in the monoclinic Pc space group. Different space groups may lead to different asymmetric units and stacking structures. Secondly, for the B5O10 cluster in 2a, the central B atom adopts a tetrahedral geometry and is connected with two B2O5 groups, where the B atoms adopt a triangular geometry. The GaO4 polyhedron further connects with one of the BO3 of the B5O10 cluster via the Ga–O–B bond (Fig. S3b†). Meanwhile in 2, the B4O9 unit containing two BO3 triangles (B1 and B3) and two BO4 tetrahedra (B2 and B5) is linked by a BO3 triangle (B4) via a μ2-O9 atom to form a [B4O9(BO2)] unit, this unit further chelates a chiral tetrahedral Ga center to form an acentric [GaB5O13]8− cluster through μ2-O8 and μ3-O10 atoms (Fig. S3a†). Thirdly, 2a and 2 reveal two distinct shapes of the 8-MR and 9-MR channels because of different structure-directing agents (SDAs) (Fig. S3c and d†). As for the properties, due to different SDAs, en in 2 is significantly shorter than EDAP in 2a, resulting in a smaller non-framework volume (2: 37.4%; 2a: 51.4%) and a higher SHG response (2: 2.6 × KDP; 2a: 0.5 × KDP). Compared with our other previous structure of [Al(B4O9)(BO)]·H2en (2b),8 the asymmetric unit and stacking structure of 2 and 2b are similar. However, due to the different radii of the chiral center metal ions, the bond length of the Ga–O in 2 is longer than that of the Al–O bond in 2b (2, Ga–O: 1.795–1.834 Å; 2b, Al–O: 1.714–1.770 Å), and the pore dimension along the a axis in 2 is also larger than 2b (2, 5.6 × 8.6 Å; 2b, 2.3 × 5.6 Å). In addition, in terms of the NLO properties, the SHG response of 2 is also higher than 2b (2: 2.6 × KDP; 2b: 1.9 × KDP), indicating that the dense structure more easily forms materials with better NLO properties.
Table 2 The structure of known [AlB5O10] and [GaB5O10]
| Formula |
Space group |
n-MR |
Topology/net type |
Non-framework volume (%) |
SHG responses (× KDP) |
Ref. |
|
Not mentioned in the references, but calculated in this work.
|
| [CH3NH3]1.5[CH3CH2CH2NH3]0.5[H2O]5[AlB5O10] |
P63 |
8, 9, 24 |
Zeolite CAN |
63.4 |
1.1 |
41
|
| (H3APEA)6[AlB5O10]9·12H2O |
P21 |
8, 11, 12 |
{4·65}2{66} |
56.5 |
0.3 |
42
|
| (HDETA)2(H2DETA)2[AlB5O10]3 |
Pna21 |
11 |
lon
|
58.5 |
1.5 |
| (H2EDAP)[GaB5O10]·H2O (2a) |
Pna21 |
8, 11 |
dia
|
51.4 |
0.5 |
| Al[B5O10]·H2dab·2H2O |
P21/c |
8, 11, 12 |
CrB4 |
54.6a |
— |
8
|
| [Al(B4O9)(BO)]·H2en (2b) |
Pc
|
9, 10 |
pcu
|
38.7a |
1.9 |
| [Al(B4O9)(BO)]·H2dap |
Cc
|
9, 10 |
pcu
|
44.9a |
2.1 |
| K2Al[B5O10]·4H2O |
C2221 |
8, 11 |
dia
|
49.1a |
2.0 |
| (NH4)2Al[B5O10]·4H2O |
C2221 |
8, 11 |
dia
|
51.9a |
2.0 |
| (H2TETA)[AlB5O10] |
Pna21 |
8, 11, 14 |
Sra
|
57.2 |
1.5 |
43
|
| (CH3NH3)2(H2O)2[AlB5O10] |
I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d 2d |
8, 10 |
dia
|
52.6 |
2.0 |
44
|
| (CH3NH3)2(H2O)[AlB5O10] |
C2 |
6, 8, 10, 11 |
dia
|
55.5 |
2.0 |
| [CH3NH3][(CH3CH2)2NH2][AlB5O10] |
Pbca
|
8, 11, 14 |
CrB4 |
56.4 |
— |
45
|
| [CH3CH2NH3][(CH3CH2)2NH2][AlB5O10] |
Pbca
|
8, 11, 14 |
CrB4 |
56.4 |
— |
| [CH3CH2NH3][(CH3)2NH2](H2O]0.5[AlB5O10] |
Pbca
|
8, 11, 14 |
CrB4 |
56.4 |
— |
| [CH3NH3][CH3CH2NH3][H2O]2[AlB5O10] |
Pna21 |
8, 14 |
Sra
|
54.7 |
0.5 |
| [GaB5O10]·H2en (2) |
Pc
|
9, 10 |
pcu
|
37.4 |
2.6 |
This work |
Crystal structure of 3
Compound 3 is transformed from compound 2 by introducing Na+ and Cs+. It crystallizes in the CS triclinic space group P. The asymmetric unit of 3 contains one [Ga{B5O9(OH)}{BO(OH)2}]2−, one Na+, and one Cs+, in which the [Ga{B5O9(OH)}{BO(OH)2}]2− cluster is formed by [B5O9(OH)]4− connected with [BO(OH)2]− through a GaO4 tetrahedron (Fig. 3a). Each GaO4 tetrahedron connects with three [B5O9(OH)]4− and one [BO(OH)2]− (Fig. 3b). The [B5O9(OH)]4−, GaO4, and [BO(OH)2]− construct two opposite-orientated single chains with 8-MR channels along the a axis with dimensions of 3.5 × 8.2 Å through the linkage of –GaO4–BO3–BO4–BO3–GaO4–BO3–BO4–BO3– (Fig. 3c and d). These two single chains can be considered as one inverted chain through an inversion center to obtain another chain. The double chains further extended to a 3D supramolecular framework by O1–H1⋯O12 (1.974 Å, 2.766 Å, 163°), O13–H13⋯O3 (1.945 Å, 2.761 Å, 176°), O12–H12⋯O11 (2.408 Å, 2.974 Å, 127°), and O12–H12⋯O9 (2.455 Å, 3.035 Å, 129°) hydrogen bonds with the distance between double chains of 8.0 Å (Fig. 3e and Table S6†).
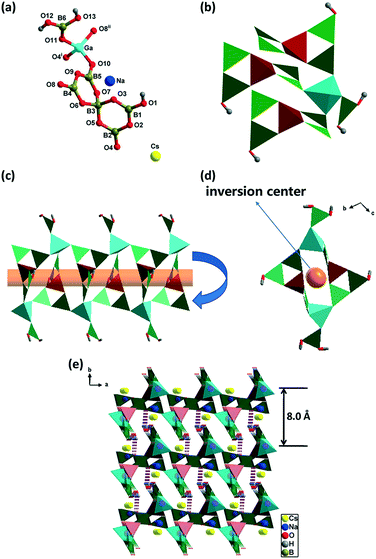 |
| | Fig. 3 (a) The asymmetric unit of 3, symmetry codes: (i) 1 − x, 1 − y, 1 − z; (ii) 1 + x, y, z. (b) The linkage of GaO4. (c) 1D structure constructed from two opposite-orientated monolayers. (d) Inversion operation along the a axis with 8-MR channels. (e) The 3D supramolecular structure along the c axis connected by several O–H⋯O hydrogen bonds (dashed lines) with the charge balancing Na+ and Cs+ ions. | |
In general, most ABOs/GBOs tend to form 2D or 3D structures via Al/Ga–O–B or Al–O–Al bonds (Table 3).31,32,42,46–49 So, a GBO with a 1D structure in 3 is rare. To the best of our knowledge, [Ga(en)2][B5O8(OH)2]·H2O (3a) is the only GBO as well as ABO with a 1D structure so far.49 The gallium(III) ion in 3a is octahedrally coordinated with four N atoms from two en molecules and two O atoms from the [B5O8(OH)2]3− clusters (Fig. S4a†). The two chelated en molecules prevent the connection of the gallium(III) ion with other oxoboron clusters in the form of GaO4, thus hindering the expansion of the structure of GBOs (Fig. S4b†). Further, two of the terminal oxygen atoms of [B5O8(OH)2]3− are protonated, that is another reason why 3a cannot form a high-dimensional structure.
Table 3 The structure of known GBOs
| Formula |
Space group |
GBO framework |
Ref. |
| [Ga2B7O14(OH)]·H21,6dah |
P |
2D |
31
|
| K2Ga2B7O14(OH)(en)0.5 |
P |
3D |
| (H2EDAP)[GaB5O10]·H2O |
Pna21 |
3D |
42
|
| Na4Ga3B4O12(OH) |
F3c |
3D |
46
|
| BaGa[B4O8(OH)]·H2O |
C2/c |
3D |
32
|
| BaGa(B4O8)(OH)·(H2O) |
P |
3D |
47
|
| Ba3Ga2[B3O6(OH)]2[B4O7(OH)2] |
Fdd2 |
2D |
48
|
| [Ga(en)2][B5O8(OH)2]·H2O (3a) |
P21/c |
1D |
49
|
| Rb2Ga(B5O10)(H2O)4 |
C2221 |
3D |
| NaCs[Ga{B5O9(OH)}{BO(OH)2}] (3) |
P |
1D |
This work |
Compound 3 does not contain chelated organic amines, but has protonated oxoboron clusters and alkali metal counterions, and also fails to form a high-dimensional structure. The possible reason for low-dimensional structure is that the synthesis temperature and alkalinity under this condition is not conducive to the dehydration and condensation of terminal hydroxyl groups of oxoboron clusters, so the alkali metal counterions are chosen to fill the chains of oxoboron clusters to balance the charge and block the expansion of the GBOs.
Compound characterization
The purity of compounds 1–3 are confirmed by powder XRD (Fig. S5†). The thermogravimetric (TG) curves and IR spectra of compounds 1–3 are discussed in the ESI† (Fig. S6 and S7). The UV/vis diffuse reflectance spectra of compounds 1–3 are shown in Fig. S8.† The band gaps obtained by extrapolating the linear part of the rising curve to zero for compounds 1–3 are 5.44, 4.91 and 5.14 eV, respectively, indicating that 1–3 are wide-band-gap semiconductors.8,31 Attempts to exchange the guest complexes of compound 2 with guest cations Li+/Na+/K+/Mg2+/Ca2+/Ba2+ were unsuccessful, mainly because the extensive H-bonds between the templates H2en and the frameworks play an important role in stabilizing the structures.
Nonlinear optical properties
Compound 2 crystallizes in the NCS space group, the SHG measurement shows that 2 displays a SHG efficiency about 2.6 times that of KDP in the same particle range of 150–212 μm (Fig. 4). In order to understand the origin of the SHG efficiency after GaO4/AlO4 groups have been introduced into the oxoboron cluster backbone, the relationship between the macroscopic SHG behavior and acentric distortions of the building units is studied. According to the anionic-group theory,50–53 the BO3 groups are responsible for the high SHG effect and the different orientation of the structure restricts their total NLO contribution. Therefore, we calculated the local dipole moments of BO3 triangles, distorted GaO4 and AlO4 polyhedra in the unit cells of 2 and 2b based on the bond-valence approach (Table 4).38,39 Overall, the dipole moments of the building units are totally cancelled along the b axis in 2 and 2b, while those are basically enhanced along the negative a and c axes. For 2b, the calculated dipole moments for the BO3 and AlO4 groups are 0.56–0.61 D and 1.39 D (D = Debye), respectively. The AlO4 groups are the main contribution of the SHG effect for 2b. When a larger GaO4 polyhedron is introduced into the crystal structure instead of smaller AlO4, the calculated dipole moments for the BO3 and GaO4 groups are 0.73–1.26 D and 1.00 D in 2, respectively. The component of the dipole moment of the unit cells on the negative a and c axes mainly comes from BO3. Therefore, the larger total dipole moments of the unit cell make 2 (∼2.6 times that of KDP, 6.04 D) show a relatively higher SHG response than 2b (∼1.9 times that of KDP, 2.93 D) as well as other ABOs containing the acentric [AlB5O13]8− cluster (0.3–2.1 times that of KDP). The results show that an effective way to obtain a denser structure is by introducing larger metal ions into borates, and a dense structure more easily forms a desired NLO material with a higher SHG response in the presence of a chiral polyhedron.
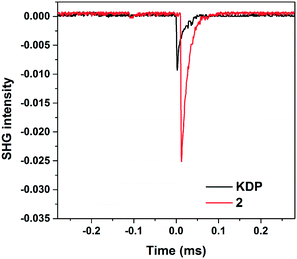 |
| | Fig. 4 Oscilloscope traces of SHG signals for compounds 2 and KDP in the same particle size range of 150–212 μm. | |
Table 4 The direction and magnitude (in Debye) of the polyhedral dipole moments for compounds 2 and 2b
| Species |
a
|
b
|
c
|
Magnitude |
| Compound 2 |
| B1O3 |
−0.92 × 2 |
±0.20 |
−0.83 × 2 |
1.26 |
| B3O3 |
−0.02 × 2 |
±0.45 |
−0.57 × 2 |
0.73 |
| B4O3 |
0.49 × 2 |
±0.28 |
−0.79 × 2 |
0.97 |
|
|
−0.90
|
0
|
−4.38
|
|
| GaO4 |
−0.75 × 2 |
±0.32 |
−0.58 × 2 |
1.00 |
|
|
−1.50
|
0
|
−1.16
|
|
|
Unit cell
|
−2.40
|
0
|
−5.54
|
6.04
|
| |
| Compound 2b |
| B1O3 |
−0.52 × 2 |
±0.21 |
−0.08 × 2 |
0.57 |
| B3O3 |
−0.15 × 2 |
±0.50 |
0.20 × 2 |
0.56 |
| B5O3 |
0.54 × 2 |
±0.26 |
−0.08 × 2 |
0.61 |
|
|
−0.26
|
0
|
0.08
|
|
| AlO4 |
−1.13 × 2 |
±0.15 |
−0.79 × 2 |
1.39 |
|
|
−2.26
|
0
|
−1.58
|
|
|
Unit cell
|
−2.52
|
0
|
−1.50
|
2.93
|
Although KDP is widely used as the reference to estimate the SHG response, the disadvantage of KDP is its ability to adsorb water, which may have a certain effect on the study of NLO properties. Therefore, LiNbO3 is more preferable as a sample for comparison than KDP. For the above reasons, we also characterized the SHG intensity of 2 at the commercial LiNbO3 level. Compound 2 displays a SHG response about approximately 0.4 times that of commercial LiNbO3 in the same particle range of 150–212 μm (Fig. S9†). Clearly, LiNbO3 exhibits a significantly larger SHG response than KDP. As far as we know, it has been reported that most of the SHG responses of compounds are obtained at the KDP or LiNbO3 level alone. The experimental results show that the SHG responses of LiNbO3 is about 6.5 times that of KDP (2.6/0.4 = 6.5), which is basically consistent with a previous study (8.7 times).54
Conclusions
In summary, two 3D framework borates K0.5[B6O10]·H2O·1.5H3O (1) and [GaB5O10]·H2en (2), and one 1D chain borate NaCs[Ga{B5O9(OH)}{BO(OH)2}] (3) have been synthesized under mild solvothermal conditions. The structures of 2 and 3 were constructed on the basis of 1. Compound 1 shows a 3D framework composed of [B6O13]8− clusters, while one of the BO4 groups of the [B6O13]8− cluster was substituted by GaO4, resulting in the acentric [GaB5O13]8− cluster in 2. The introduction of chiral GaO4 polyhedra may induce and transfer the chirality to the oxoboron framework, which is beneficial to the formation of borates crystallized in the NCS space group. In addition, the 1D structure constructed from [Ga{B5O9(OH)}{BO(OH)2}]2− clusters in 3 is rare in GBOs as well as ABOs. What needs to be emphasized is that the calculations reveal that the dipole moments along the negative a and c axes are enhanced in 2, which favor the relatively larger SHG response of 2 (∼2.6 times that of KDP) than ABOs containing the acentric [AlB5O13]8− cluster (0.3–2.1 times that of KDP). The larger atomic radius of 2 is more likely to form a dense structure, and thus the compound with better NLO properties can be obtained. We hope that this work is helpful to explore more new functional NLO materials with higher SHG responses.
Author contributions
Qi-Ming Qiu: Conceptualization, methodology, investigation, data curation, formal analysis, writing –original draft, preparation. Guo-Yu Yang: Writing – reviewing & editing, supervision, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the NSFC (No. 21571016, 21831001 and 91122028) and the NSFC for Distinguished Young Scholars (No. 20725101).
Notes and references
- J. Zhou, Y. Liu, H. Wu, H. Yu, Z. Lin, Z. Hu, J. Wang and Y. Wu, Angew. Chem., 2020, 132, 19168–19172 CrossRef.
- P. Ren, Y. Yang, M. Wen, H. Li, Z. Yang and S. Pan, Inorg. Chem., 2020, 59, 8396–8403 CrossRef CAS PubMed.
- M. Gai, Y. Wang and S. Pan, Chin. Sci. Bull., 2018, 63, 998–1011 CrossRef.
- Y. Song, G. Yang, P. Gong, M. Luo, Z. Lin and N. Ye, Inorg. Chem., 2020, 59, 7789–7794 CrossRef CAS PubMed.
- G. Zou and K. M. Ok, Chem. Sci., 2020, 11, 5404–5409 RSC.
- J. H. Huang, C. C. Jin, P. L. Xu, P. Gong, Z. Lin, J. W. Cheng and G. Y. Yang, Inorg. Chem., 2019, 58, 1755–1758 CrossRef CAS PubMed.
- B. B. Han, J. W. Cheng and G. Y. Yang, CrystEngComm, 2019, 21, 1262–1266 RSC.
- C. Rong, Z. Yu, Q. Wang, S. T. Zheng, C. Y. Pan, F. Deng and G. Y. Yang, Inorg. Chem., 2009, 48, 3650–3659 CrossRef CAS PubMed.
- Z. E. Lin, J. Zhang and G. Y. Yang, Inorg. Chem., 2003, 42, 1797–1799 CrossRef CAS.
- G. Zou and K. M. Ok, Chem. Sci., 2020, 11, 5404–5409 RSC.
- H. Lee and K. M. Ok, Bull. Korean Chem. Soc., 2020, 41, 139–142 CrossRef CAS.
- Q. M. Qiu, X. Y. Li, C. A. Chen, K. N. Sun and G. Y. Yang, J. Solid State Chem., 2021, 299, 122193 CrossRef CAS.
- Q. M. Qiu and G. Y. Yang, J. Solid State Chem., 2021, 301, 122303 CrossRef CAS.
- Q. Wei, S. J. Sun, J. Zhang and G. Y. Yang, Chem. – Eur. J., 2017, 23, 7614–7620 CrossRef CAS PubMed.
- D. Qin, T. J. Zhang, C. B. Ma and G. Y. Yang, Dalton Trans., 2020, 49, 3824–3829 RSC.
- C. A. Chen, R. Pan and G. Y. Yang, Dalton Trans., 2020, 49, 3750–3757 RSC.
- H. A. Lehmann and A. Teske, Z. Anorg. Allg. Chem., 1973, 400, 169–175 CrossRef CAS.
- J. H. Wang, S. H. Feng and R. R. Xu, J. Chem. Soc., Chem. Commun., 1989, 265–266 CAS.
- J. H. Yu, R. R. Xu, Y. H. Xu and Y. Yue, J. Solid State Chem., 1996, 122, 200–205 CrossRef CAS.
- X. S. Liu and R. R. Xu, J. Chem. Soc., Chem. Commun., 1989, 1837–1839 RSC.
- J. H. Yu, R. R. Xu, Q. B. Kan and Y. Xu, J. Mater. Chem., 1993, 3, 77–82 RSC.
- J. Ju, J. Lin, G. Li, T. Yang, H. Li, F. Liao, C. K. Loong and L. You, Angew. Chem., Int. Ed., 2003, 42, 5607–5610 CrossRef CAS PubMed.
- T. Yang, A. Bartoszewicz, J. Ju, J. Sun, Z. Liu, X. Zou, Y. Wang, G. Li, F. Liao, B. Martin-Matute and J. Lin, Angew. Chem., Int. Ed., 2011, 50, 12555–12558 CrossRef CAS PubMed.
- J. Ju, T. Yang, G. Li, F. Liao, Y. Wang, L. You and J. Lin, Chem. – Eur. J., 2004, 10, 3901–3906 CrossRef CAS PubMed.
- T. Yang, J. Ju, G. Li, F. Liao, X. Zou, F. Deng and J. Lin, Inorg. Chem., 2007, 46, 4772–4774 CrossRef CAS PubMed.
- W. Gao, Y. Wang, G. Li, F. Liao, L. You and J. Lin, Inorg. Chem., 2008, 47, 7080–7082 CrossRef CAS PubMed.
- J. Zhou, W. H. Fang, C. Rong and G. Y. Yang, Chem. – Eur. J., 2010, 16, 4852–4863 CrossRef CAS PubMed.
- L. Cheng and G. Y. Yang, Chem. Commun., 2014, 50, 344–346 RSC.
- L. Wei, Q. Wei, Z. E. Lin, Q. Meng, H. He, B. F. Yang and G. Y. Yang, Angew. Chem., Int. Ed., 2014, 53, 7188–7191 CrossRef CAS PubMed.
- Y. Chen, Z. W. Guo, X. X. Li, S. T. Zheng and G. Y. Yang, CCS Chem., 2021, 3, 1232–1241 CrossRef.
- C. A. Chen, R. Pan, T. J. Zhang, X. Y. Li and G. Y. Yang, Inorg. Chem., 2020, 59, 18366–18373 CrossRef CAS PubMed.
- Q. Wei, L. Li, L. Cheng, Q. Meng and G. Y. Yang, Dalton Trans., 2014, 43, 9427–9430 RSC.
- W. Gao, Y. Jing, J. Yang, Z. Zhou, D. Yang, J. Sun, J. Lin, R. Cong and T. Yang, Inorg. Chem., 2014, 53, 2364–2366 CrossRef CAS PubMed.
- Q. Wei, J. W. Cheng, C. He and G. Y. Yang, Inorg. Chem., 2014, 53, 11757–11763 CrossRef CAS PubMed.
- H. Q. Wu, H. He, B. F. Yang and G. Y. Yang, Inorg. Chem. Commun., 2013, 23, 77–79 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- J. H. Wang, J. W. Cheng, Q. Wei, H. He, B. F. Yang and G. Y. Yang, Eur. J. Inorg. Chem., 2014, 4079–4083 CrossRef CAS.
- G. J. Cao, Q. Wei, J. W. Cheng, L. Cheng and G. Y. Yang, Chem. Commun., 2016, 52, 1729–1732 RSC.
- L. Cheng and G. Y. Yang, Inorg. Chem., 2018, 57, 13505–13512 CrossRef CAS PubMed.
- Q. Meng, G. M. Wang, B. F. Yang, H. He and G. Y. Yang, Inorg. Chem. Commun., 2014, 40, 168–171 CrossRef CAS.
- G. J. Cao, J. Lin, J. Y. Wang, S. T. Zheng, W. H. Fang and G. Y. Yang, Dalton Trans., 2010, 39, 8631–8636 RSC.
- G. J. Cao, J. Lin, W. H. Fang, S. T. Zheng and G. Y. Yang, Dalton Trans., 2011, 40, 2940–2946 RSC.
- S. J. Yu, X. Y. Gu, T. T. Deng, J. H. Huang, J. W. Cheng and G. Y. Yang, Inorg. Chem., 2017, 56, 12695–12698 CrossRef CAS PubMed.
- T. T. Deng, Y. E. Gao, L. X. Zhang, X. Y. Gu, H. R. Tian, Y. Liu, Y. L. Feng and J. W. Cheng, CrystEngComm, 2014, 16, 5689–5694 RSC.
- L. Cheng, Q. Wei, H. Q. Wu, L. J. Zhou and G. Y. Yang, Chem. – Eur. J., 2013, 19, 17662–17667 CrossRef CAS PubMed.
- T. Hu, C. L. Hu, F. Kong, J. G. Mao and T. C. W. Mak, Inorg. Chem., 2012, 51, 8810–8817 CrossRef CAS.
- N. Ye, Q. X. Chen, B. C. Wu and C. T. Chen, J. Appl. Phys., 1998, 84, 555–558 CrossRef CAS.
- C. T. Chen, Y. C. Wu and R. K. Li, Int. Rev. Phys. Chem., 1989, 5, 65–91 Search PubMed.
- Y. Liu, Y. Shen, S. Zhao and J. Luo, Coord. Chem. Rev., 2020, 407, 213152 CrossRef CAS.
- L. Kang, F. Liang, X. Jiang, Z. Lin and C. Chen, Acc. Chem. Res., 2020, 53, 209–217 CrossRef CAS PubMed.
- W. J. Alford and A. V. Smith, J. Opt. Soc. Am. B, 2001, 18, 524–533 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR, TGA, and UV-vis spectra and PXRD patterns. CCDC 1962950, 2077862 and 2077863 for compounds 1–3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00719j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Guo-Yu
Yang
and
Guo-Yu
Yang