
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable protein crystal size distribution via continuous slug-flow crystallization with spatially varying temperature
Nicholas J.
Mozdzierz
a,
Moo Sun
Hong
 a,
Yongkyu
Lee
ab,
Moritz H. P.
Benisch
ac,
Mo
Jiang
d,
Allan S.
Myerson
a and
Richard D.
Braatz
*a
a,
Yongkyu
Lee
ab,
Moritz H. P.
Benisch
ac,
Mo
Jiang
d,
Allan S.
Myerson
a and
Richard D.
Braatz
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: braatz@mit.edu
bSchool of Chemical & Biological Engineering, Seoul National University, Seoul, South Korea
cInstitute for Chemical & Bioengineering, ETH Zurich, Zurich, Switzerland
dDepartment of Chemical & Life Science Engineering, Virginia Commonwealth University, Richmond, VA, USA
First published on 29th July 2021
Abstract
Accompanied with the growth of the biopharmaceuticals market has been the interest in developing processes with increased control of product quality attributes at low manufacturing cost, with one of the approaches being through genuinely continuous manufacturing processes. Part of this interest is in new drug product formulations that extend shelf-life and improve the patient experience. Some of these drug product formulations require the production of protein crystals of controlled size distribution. This article describes a continuous tubular crystallizer in which the size distribution of the produced protein crystals is tuned by controlling the spatial temperature along the tube. Under appropriate buffer and pH conditions, the magnitude and dispersion of product protein crystals are reproducibly manipulated using a fully controlled temperature profile over a residence time of 25 to 30 minutes, and the formation of amorphous precipitates can be achieved under higher supersaturation conditions via the addition of a concentrated precipitant for drug products in which higher solubility is desired. The tunable continuous process for protein crystallization has the potential to become a low-cost platform technology for producing protein crystals for a variety of biologic drug product formulations.
1. Introduction
From an industrial perspective, an opportunity exists to develop scalable non-chromatographic protein separation methods that disrupt the traditional batch-wise paradigm and support continuous purification modes.1–3 Despite efforts to demonstrate the future viability of sequential ‘bind-and-elute’ chromatography, resin-based adsorption processes are costly and widely perceived within the biomanufacturing field to be a major process bottleneck.1,4–6 Technologies such as periodic counter-current chromatography (PCC), simulated moving bed chromatography (SMB), and multi-column counter-current solvent gradient purification (MCSGP) have been demonstrated to reduce this bottleneck.7 Each of these processes, however, employs more columns, valves, and pumps than sequential chromatography, substantially increasing both system complexity and capital equipment costs.2,8,9 Further, given that they require the same number of stages as sequential chromatography to achieve the same purification efficiency – but operate at higher throughputs using more resin – PCC, SMB, and MCSGP have operating expenses (OPEX) that scale linearly with respect to conventional techniques. In contrast, non-chromatographic protein purification methods such as precipitation, aqueous two-phase extraction (ATPE), and crystallization are relatively simple to execute, require a small initial capital investment, and have OPEX (primarily buffers/solvents) that scale sub-linearly with throughput.2,6,7 A single example of the application of each of these three techniques to continuous protein purification has been published.10–12 In addition to reducing facility footprints, precipitation, ATPE, and crystallization have the potential to dramatically increase equipment utilization, allowing the biopharmaceutical sector to realize higher productivities and improved operational flexibility.2,3,7 These methods could also support the robust control of short product residence times, allowing for the rapid recovery of labile proteins and standardization of critical quality attributes across each lot.3,7 Finally, improved purification efficiency has been gleaned by coupling these techniques with ‘clean’ expression hosts (e.g., Komagataella phaffii), as well as their amenability to scale-out by parallelization.13,14Despite their potential cost-effectiveness and scalability, however, the intrinsic difficulty of optimizing and controlling protein crystallization has prevented their broad adoption as preparative purification techniques.7,15 Specifically, a generalized set of heuristics governing the myriad physical, chemical, and biochemical factors that can impact protein crystal nucleation and growth has yet to be realized.15–17 Consequently, while a wide range of proteins has been crystallized at the μL scale for structure determination using various combinatorial screening approaches (e.g., hanging and sitting drop vapor diffusion, free interface diffusion, and dialysis), recombinant insulin is the only biopharmaceutical reported to be purified by crystallization at the industrial scale.7,18 Studies surrounding the batch crystallization of enzymes (e.g. hen egg white lysozyme (HEWL), lipase) and monoclonal antibody fragments from both homo- and heterogeneous mixtures at volumes ranging from 100 mL to 1 L represent promising demonstrations of this technique as a method for ‘at-scale’ purification, but fall short of proving industrial applicability.15
In addition, protein crystallization from nearly pure solution is suitable for producing crystals in drug product formulation/delivery.19 Amorphous lyophilizates and aqueous solutions are commonly used for formulation/delivery but have low stability and high viscosity at high concentration. Crystals have higher stability and could lead to a better patient experience with consistent controlled properties. For injection, which is currently the primary mode of administration, high viscosity suspension requires a large-bore needle and a high amount of force to push the needle into the body, which is painful for the patient. Experimental results for monoclonal antibodies have demonstrated that injection of a crystalline suspension reduces the syringe force by about 50% for the same protein concentration compared to liquid formulations.15 The injection of crystals also enables the protein to be taken into the blood stream at a slower rate for a more sustained release. In all of these protein crystal-based drug product formulations, the rate of uptake of biotherapeutic protein molecules into the bloodstream depends critically on the size distribution of protein crystals.
A recent set of experiments has demonstrated tubular designs as viable technologies for protein crystallization.12,20 Using a low-cost setup designed around disposable plastic components and syringe pumps, crystallization of HEWL was reported from a purified solution at a rate of 0.72 g h−1. Such studies have not demonstrated robust feedback process control. In this article, we leverage optimization/control theory – building off from prior art surrounding the tubular crystallization of small molecules and active pharmaceutical ingredients – and develop a flexible flow-through system for the continuous crystallization of therapeutically relevant proteins under feedback control.
Here, a fully automated system designed to operate under segmented slug-flow conditions and capable of on-line control of the cooling process is applied to the continuous crystallization of the model protein HEWL. In addition to temperature, pH and buffered precipitant solution are used to control the supersaturation of HEWL at various defined points along the length of the crystallizer. These parameters are carefully combined to permit tuning the particle size distribution (PSD) generated by the counter current heat exchanger (CCHEX) platform under controlled and seeded inlet conditions. The system further demonstrated that this control is possible over a residence time as short as 25–30 minutes (2–4× shorter than similar recent reports). Powder X-ray diffraction (PXRD) and cross-polarized transmitted light microscopy are used to qualitatively score the relative ratio of amorphous-to-crystalline HEWL generated under each set of conditions tested. The images acquired using cross-polarized light is analyzed using the custom crystal image analysis algorithm designed for adjusting contrast and segmenting the overlaid crystal image.
2. Experimental method
2.1. Materials
Hen egg white lysozyme (HEWL) was used as the model protein for all experiments as described. Buffer preparation involved sodium acetate (NaOAc), hydrochloric acid (HCl), HPLC-grade distilled deionized water, and disposable 0.2 μm vacuum filtration systems. A 1 M solution of sodium hydroxide (NaOH) was used to flush the CCHEX crystallizer at the beginning and end of each day of experiments.Seed solutions for all experiments were prepared by first suspending HEWL in a refrigerated solution of 2% w/v sodium chloride (NaCl) and 100 mM NaOAc (pH 4.0) to a concentration of 70 g L−1. The suspension was then placed in a water batch held at 30 °C and stirred for 3.5 hours. The resulting hazy solution was subsequently vacuum filtered, placed back in the water bath, and allowed to cool spontaneously overnight. Before first use, the seed solution was heated to the appropriate temperature as indicated in Table 1.
| Exp. | Rep. | Seed bath temperature (°C) | Temperatures (°C) | |||
|---|---|---|---|---|---|---|
| Pre-HEX | HEX 1 | HEX 2 | HEX 3 | |||
| 1 | 1 | 32.6 | 5 | 5 | 5 | 23 |
| 1 | 2 | 32.35 | 5 | 5 | 5 | 23 |
| 2 | 1 | 28.65 | 14 | 20 | 11 | 7 |
| 2 | 2 | 28.45 | 14 | 20 | 11 | 7 |
| 3 | 1 | 23.8 | 17 | 15 | 11 | 8 |
| 3 | 2 | 23.9 | 17 | 15 | 11 | 8 |
| 4 | 1 | 42 | 5 | 5 | 5 | 5 |
2.2. Experimental apparatus
The experimental apparatus in Fig. 1 consists of peristaltic pumps, heat exchangers (HEXs), heating and cooling baths, and controlled-temperature water baths.21,22 All but one of the components are integrated in a computer used for collecting data and performing real-time control calculations. The precipitant pump containing NaCl aqueous solution was not integrated into the larger control system, as the in-line precipitant mixing was manually set at the beginning of the experiment and was not changed during each experiment. The precipitant solution was stored in a magnetically stirred, jacketed, 1 L, glass, two-necked round-bottom flask plumbed in series with the cold coolant reservoir and seed/feedstock solution Allihn condenser.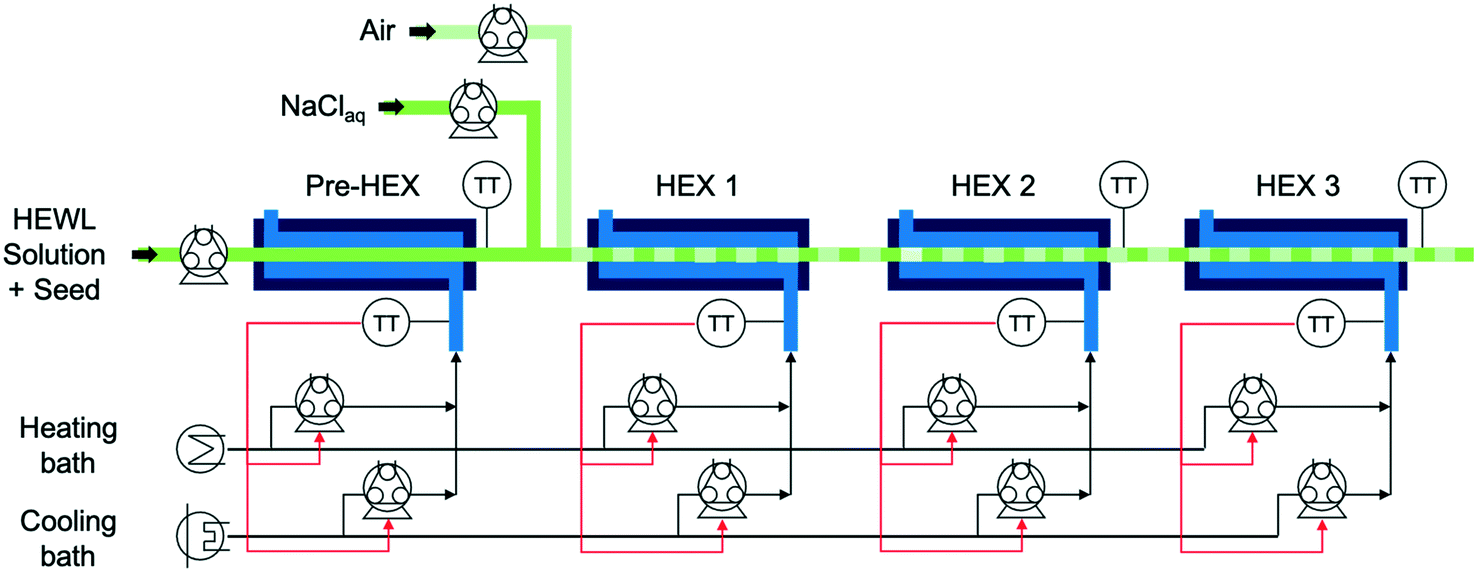 | ||
| Fig. 1 A process flow diagram of the continuous crystallizer with in-line precipitant mixing capabilities, where HEWL is hen egg white lysozyme, HEX refers to a heat exchanger, and TT refers to a temperature transmitter. | ||
The temperature-controlled HEWL solution enters the crystallizer from the round-bottom flask at the left in Fig. 1 and traverses a short segment of insulated tubing before transiting through the preliminary heat exchanger (pre-HEX) and mixing with a slipstream of concentrated, chilled, and buffered NaCl. The mixture of HEWL and precipitant then traverse under an indirect ultrasonication probe and mixes with filtered air to form stable liquid slugs. The slug flow is hydrodynamically stable for a very large range of gas and liquid flow rates for the tubing diameter and fluid properties in the experiments in this study, as observed experimentally which is consistent with theoretical expressions as detailed in a recent book chapter.23 The slugs then move through HEXs 1, 2, and 3 prior to collection for imaging and PXRD analysis at the outlet. For all experiments that did not employ concentrated precipitant addition, the air and liquid flow rates into the slugging tee were both 7 mL min−1. The air and liquid flow rates into the slugging tee during experiments that involved concentrated precipitant addition were 7 and 9.61 mL min−1 (HEWL solution 7 mL min−1 and precipitant solution 2.61 mL min−1), respectively. For each HEX, the shell-side flow rate from the peristaltic pumps is given by the proportional-integral (PI) controllers to achieve the set point temperature.
2.3. Design of experiments
Each of the four sets of experimental conditions tested was designed to expose HEWL transiting the continuous system to markedly different supersaturation conditions while holding constant the residence time of the slugs (Table 1). All temperature and the concentration of precipitant set points were determined with the aid of empirical models fitted to solubility data.24 Specifically, experiment 1 was designed to ‘crash cool’ the HEWL solution immediately upon entering the pre-HEX module by exposure to a maximum instantaneous supersaturation (σmax, where σ = C/Csat − 1, C is the HEWL concentration, and Csat is the solubility) equal to ∼18. Experiment 2 is designed to reduce σmax to ∼6.4 and promote dissolution in HEX 1 following primary nucleation in the pre-HEX. Experiment 3 was designed to expose slugs of dissolved and crystalline HEWL to a shallow temperature – and by extension, supersaturation – gradient with σmax of ∼4.3 in an effort to favor the growth of seed crystals relative to the nucleation of new particles. Finally, experiment 4 represents a situation in which σmax was varied aggressively (∼220) by using a combination of concentrated precipitant (i.e., NaCl) addition and crash cooling. Each of experiments 1–3 was performed in duplicate. This study explores a wide range of supersaturations to demonstrate the ability of the system to access a very large experimental design space, and to assess the effects on the product crystals. Further, relative to experiments 1–3, experiment 4 was designed to exploit the generalized phase behavior of protein solutions, which predicts that extreme values of σmax will preferentially induce the formation of amorphous precipitates over well-ordered crystals.25 In this way, disordered aggregates of HEWL are generated intentionally as a control to aid in distinguishing crystalline from non-crystalline samples by both cross-polarized microscopy and PXRD.2.4. Automated imaging analysis for particle size distribution
For each experiment, a single pulse of HEWL (Table 1) was fed into the continuous crystallizer. Pulses of air (each 10 s long) bracketed the HEWL to aid in identifying crystal-containing slugs at the outlet. All slugs of HEWL were collected as they exited the crystallizer in a single sterile 50 mL conical tube. Four 45 μL droplets of the collected slurry were immediately transferred to an air-dusted microscope slide and protected from evaporation with cover glasses. A set of position matched images of each droplet was then acquired using a microscope fitted with a digital camera, a 10× trinocular eyetube, and a 4×/0.10 HI PLAN objective. The resolution of this optical setup was 1 μm per pixel. Light intensity, aperture, and condenser settings were kept constant across all images and all experiments.Each of the four cross-polarized micrographs corresponding to a given experiment was analyzed using the watershed algorithm with markers and boundaries described in Fig. 2. First of all, the original color image is mapped to grayscale for adjusting the contrast. The adjusted image is dilated with structuring element neighborhood where pixels are connected along the horizontal or vertical direction for protecting tiny size crystals from erosion. The boundaries of segmented objects are calculated in pixels and separate the threshold of regions for the watershed method. The foreground markers in the object are obtained by a closing followed by erosion and are superimposed on the grayscale image with the boundaries of regions. The magnitudes of markers are modified to regional minima of the objective region and scaled to different integer values. The flooding process is performed from the marker (the regional minima), and the borderline is constructed between the extended regions of different labeled markers. Finally, the area and length of crystals are estimated and used to acquire the particle size distribution.
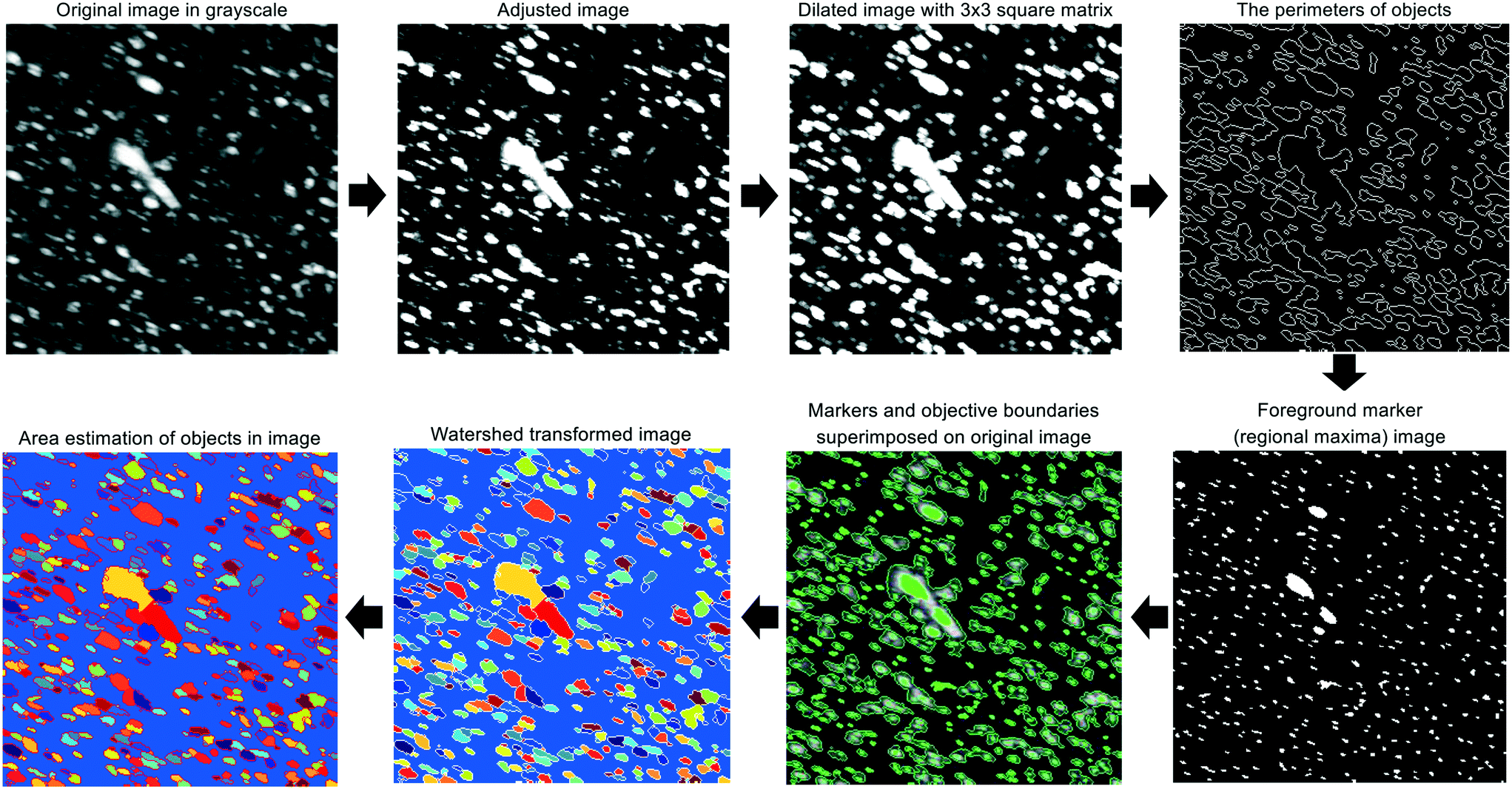 | ||
| Fig. 2 Graphical summary of the image processing algorithm which is a modified version of marker-controlled watershed segmentation for sorting out overlaid crystals. | ||
2.5. Powder X-ray diffraction
Approximately 10 minutes after collection (the time required to perform all imaging described in section 2.3), the remaining slurry from a given experiment was divided into 12 to 15 1.0 mL aliquots and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 22 °C for 2 minutes. The resulting supernatant was subsequently aspirated off and discarded. A second identical centrifugation step was employed in the case when bulk liquid remained after aspiration. Pellets were then stored under ambient conditions for ∼4 hours prior to analysis by PXRD. The obtained crystals were crushed using a mortar and pestle in order to maximize the number of visible crystal faces.26
000g and 22 °C for 2 minutes. The resulting supernatant was subsequently aspirated off and discarded. A second identical centrifugation step was employed in the case when bulk liquid remained after aspiration. Pellets were then stored under ambient conditions for ∼4 hours prior to analysis by PXRD. The obtained crystals were crushed using a mortar and pestle in order to maximize the number of visible crystal faces.26
PXRD was performed using a PANalytical X'Pert PRO diffractometer. The instrument was configured as described in a previous study26 and operated at a tension of 45 kV and an anode current of 40 mA. All scans were conducted under the following programmable settings to maximize resolution at low angles: 3.507–13.5° 2θ range; 0.0167113° step; 455.295 s step time; 0.004661° s−1 scan speed; and 1 rps spinner stage rotation speed. Each scan analyzed the equivalent of at least 8 pellets pressed onto a zero-background sample tray. A blank sample tray diffractogram was acquired under these same settings.
A negative control diffractogram of the 2% w/v NaCl and 100 mM NaOAc (pH 4.0) buffer used to prepare all HEWL solutions was obtained using the same instrument and hardware configuration noted above. 75 mL of the buffer was first boiled on a hot plate under stirring for 2 hours to evaporate most of the bulk liquid. The remaining slurry was then dried in a vacuum oven overnight to yield a powder of crystalline NaCl and NaOAc in a mass ratio identical to that in the original buffer.
3. Results and discussion
3.1. Protein crystal populations
Microscope images were acquired for each experiment using cross-polarized light (Fig. 3). Particles of HEWL that appear white are birefringent and are very likely to be crystalline.27 Particles that appear dark are amorphous precipitates of HEWL or crystals possessing a cubic (i.e., isotropic) lattice instead of the desired tetragonal structure. The inlet seed and outlet particles for experiments 1 to 3 are overwhelming anisotropic crystals (Fig. 3). Experiment 4, which is at extremely high supersaturation σmax, yielded minimal anisotropic crystals (Fig. 4). The production of different solid states during particle formation operating under different magnitude supersaturation is commonly observed for small molecules,28 and can certainly occur for protein molecules which have many more degrees of freedom. | ||
| Fig. 3 Representative micrographs from HEWL DoE experiments 1–3. The upper image in each set is representative of the seed PSD used in the associated experiment. The lower image in each set is representative of the outlet PSD generated by the associated experiment. All images were acquired under cross-polarized lighting conditions using identical microscope aperture, condenser, and magnification (40×) settings. The scale bars are 250 μm in all images. (a.1) Seed of experiment 1. (a.2) Outlet of experiment 1. (b.1) Seed of experiment 2. (b.2) Outlet of experiment 2. (c.1) Seed of experiment 3. (c.2) Outlet of experiment 3. | ||
 | ||
| Fig. 4 Representative micrographs from experiment 4. The left image in each set was acquired under cross-polarized lighting conditions, while the right image was captured using plane polarized light. The crystal sample as not repositioned between image acquisitions. Identical microscope aperture, condenser, and magnification (40×) settings were used for all four images. The scale bars are 250 μm in all images. (a) Seed. (b) Outlet. | ||
With the image analysis procedure described in section 2.3, the micrographs of experiments 1, 2, and 3 were used to measure the PSDs of the seed and generated crystals in Fig. 5 (experiment 4 was excluded from the analysis since the particles have a different solid state and so would have different solubility and crystallization kinetics). The crystallization conditions in experiments 1 and 2 yielded markedly different final PSDs than those in experiment 3. The leftward shift of the experimental cumulative distribution functions (ECDFs) for experiments 1 and 2 from relatively heavy-tailed seed populations to substantially more monodisperse final distributions of small crystals (L < 30 μm) suggests that, at an initial σmax ≥ 5, nucleation of HEWL crystals was strongly favored over the growth of existing particles. In contrast, the product PSD is much more similar to the seed PSD in experiment 3, with a similar level of broadness. The resulting PSDs are consistent with the temperatures used in the experiments (Table 1). The temperature in experiment 1 was decreased to its lowest value at the crystallizer inlet and kept at a low value, which would promote nucleation, until increasing the temperature at the end to promote growth. The large number of nuclei generated upstream in experiment 1 would limit the size in which the crystals can grow downstream. The temperature in experiment 2 also decreased to a low value at the crystallizer inlet, then was increased which would result in dissolution, and then the temperature was decreased to a very low value, and then kept it low until the outlet. The large number of nuclei generated upstream would then largely be dissolved in the dissolution part of the crystallizer, but then a burst of nuclei would be generated again, which would then grow. The temperature in experiment 3 was monotonically decreased in small steps (Table 1), which resulted in much less nuclei formation. Taken collectively, Fig. 5 demonstrates that, even over residence times as short as 25–30 minutes, the continuous protein crystallization system can be used to tune the characteristics of the protein crystal populations.
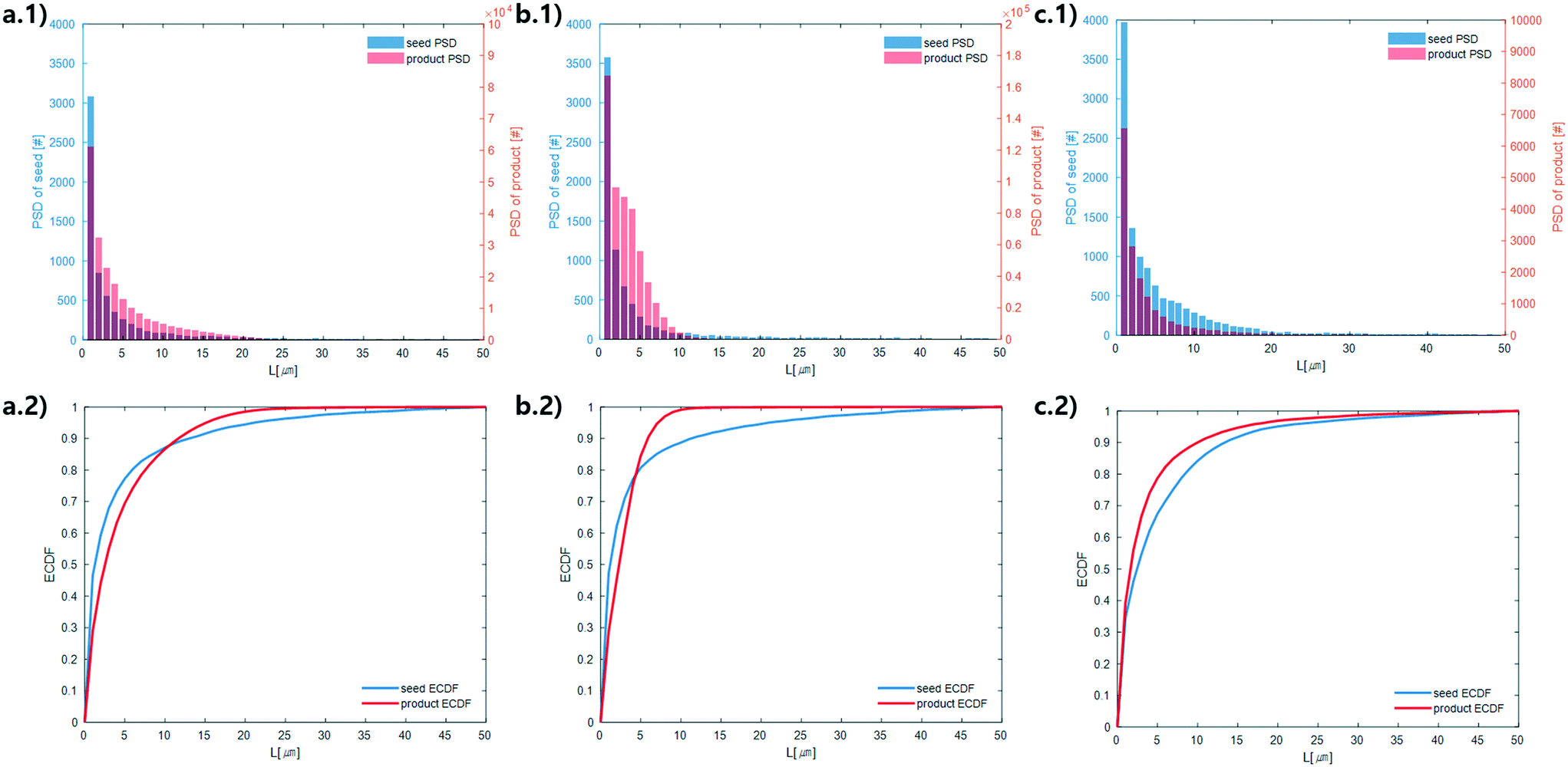 | ||
| Fig. 5 Measured particle size distribution (upper) and cumulative distribution function (lower) of the seed (blue) and product (orange) crystals. (a.1) and (a.2), (b.1) and (b.2), and (c.1) and (c.2) are for experiments 1, 2, and 3 (summation of replicates), respectively. For all images, the horizontal axes are crystal length (μm). The vertical axes are different for the seed and product crystals. As stated in section 2.3, only particles that exhibited high-intensity constructive interference under cross-polarized light (Fig. 3) were considered crystals to calculate these PSDs. | ||
Continuously differentiable analytical expressions for these ECDFs were fitted using piecewise cubic Hermite interpolating polynomials and used to calculate the summary statistics in Table 2via
 | (1) |
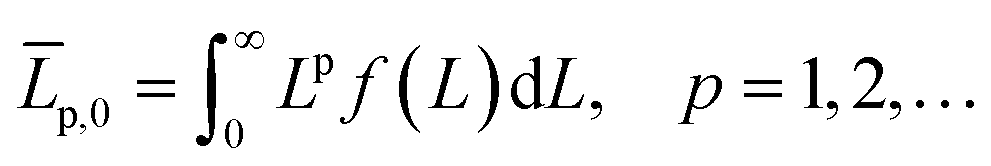 | (2) |
![[L with combining overline]](https://www.rsc.org/images/entities/i_char_004c_0305.gif) p,0 are the weighted mean crystal sizes.29
(1,0), (2,0), and (3,0) are the mean crystal length (μm), surface area (μm2), and volume (μm3) in given experiments (summation of replicates), respectively. All mean size statistics were calculated using moments of the analytical derivatives of the ECDFs reported in Fig. 5. All NT values were determined directly from the PSDs reported in the same figures
p,0 are the weighted mean crystal sizes.29
(1,0), (2,0), and (3,0) are the mean crystal length (μm), surface area (μm2), and volume (μm3) in given experiments (summation of replicates), respectively. All mean size statistics were calculated using moments of the analytical derivatives of the ECDFs reported in Fig. 5. All NT values were determined directly from the PSDs reported in the same figures
| Moment | Experiment 1 | Experiment 2 | Experiment 3 | |||
|---|---|---|---|---|---|---|
| Seed | Product | Seed | Product | Seed | Product | |
| N T | 6.7 × 103 | 2.1 × 105 | 7.6 × 103 | 5.8 × 105 | 1.2 × 104 | 1.7 × 104 |
|
(1,0)
|
5.7 | 5.0 | 5.4 | 3.3 | 6.8 | 4.9 |
|
(2,0)
|
1.3 × 102 | 5.5 × 101 | 1.3 × 102 | 1.9 × 101 | 1.6 × 102 | 9.3 × 101 |
|
(3,0)
|
5.7 × 103 | 1.1 × 103 | 5.4 × 103 | 3.1 × 102 | 7.4 × 103 | 3.9 × 103 |
The above observations from Fig. 5 are seen in the summary statistics in Table 2. Given that all PSDs were acquired from the consistent total volumes of well-mixed slurries, the fact that  for experiments 1 and 2 and ∼1 for experiment 3 indicates that experiments 1 and 2 produced a much larger number of small crystals than experiment 3. Furthermore, nucleation was so strongly favored in experiment 1 that the process resulted in a drastic decrease in the mean crystal volume (3,0) (<0.2×) between the inlet and outlet of the system.
for experiments 1 and 2 and ∼1 for experiment 3 indicates that experiments 1 and 2 produced a much larger number of small crystals than experiment 3. Furthermore, nucleation was so strongly favored in experiment 1 that the process resulted in a drastic decrease in the mean crystal volume (3,0) (<0.2×) between the inlet and outlet of the system.
3.2. Structural characterization of HEWL
PXRD was employed as an orthogonal method to assess the qualitative crystallinity of all samples generated and confirm the results of cross-polarized microscopy. Fig. 6 shows the low angle diffractograms for each of experiments 1–4 from top to bottom. Where appropriate, diffractograms for replicate experiments are plotted on common axes. Additionally, an idealized powder diffractogram (bottom panel; purple line) for tetragonal HEWL served as a reference. All samples collected at the outlet of the crystallizer during experiments 1–3 exhibit relatively defined diffraction peaks that match the reference peak positions to within ±0.5° 2θ, suggesting that a substantial fraction of the particles formed in each run was crystalline tetragonal HEWL. This small offset in 2θ can likely be attributed to the higher resolution and signal-to-noise ratio of single-crystal to powder XRD.30 Differences in the number of structure-bound water molecules between samples could also convolute the traces. The systematic translation in 2θ evident between each pair of traces for experiments 1–3 is the result of small (O(mm)) differences in the positioning of each sample within the focusing circle of the diffractometer.31 While the PXRD data unequivocally corroborate the formation of crystals in experiments 1–3, all six diffractograms exhibit a broad parabolic baseline (‘halo’) that indicates the presence of some amorphous or short-range ordered nanocrystalline phases of matter.32 Although techniques for estimating the relative ratio of amorphous to crystalline material in PXRD traces exist (e.g., the Rietveld method), they are generally regarded as being difficult to implement and subject to large uncertainties.33 A comparison of the top three panels of Fig. 6 to the black trace (exp. 4; confirmed an amorphous precipitate) in the bottom panel of the figure, however, bolsters the claim that experiments 1–3 yielded HEWL particles exhibiting significant crystalline character. HEWL analyzed as received from the manufacturer also produced a purely amorphous diffractogram (Fig. 6; bottom panel; gray line), which confirms the ability of the end-to-end seed population preparation and crystallization process to generate long-range ordered protein particles from otherwise disordered precursor materials.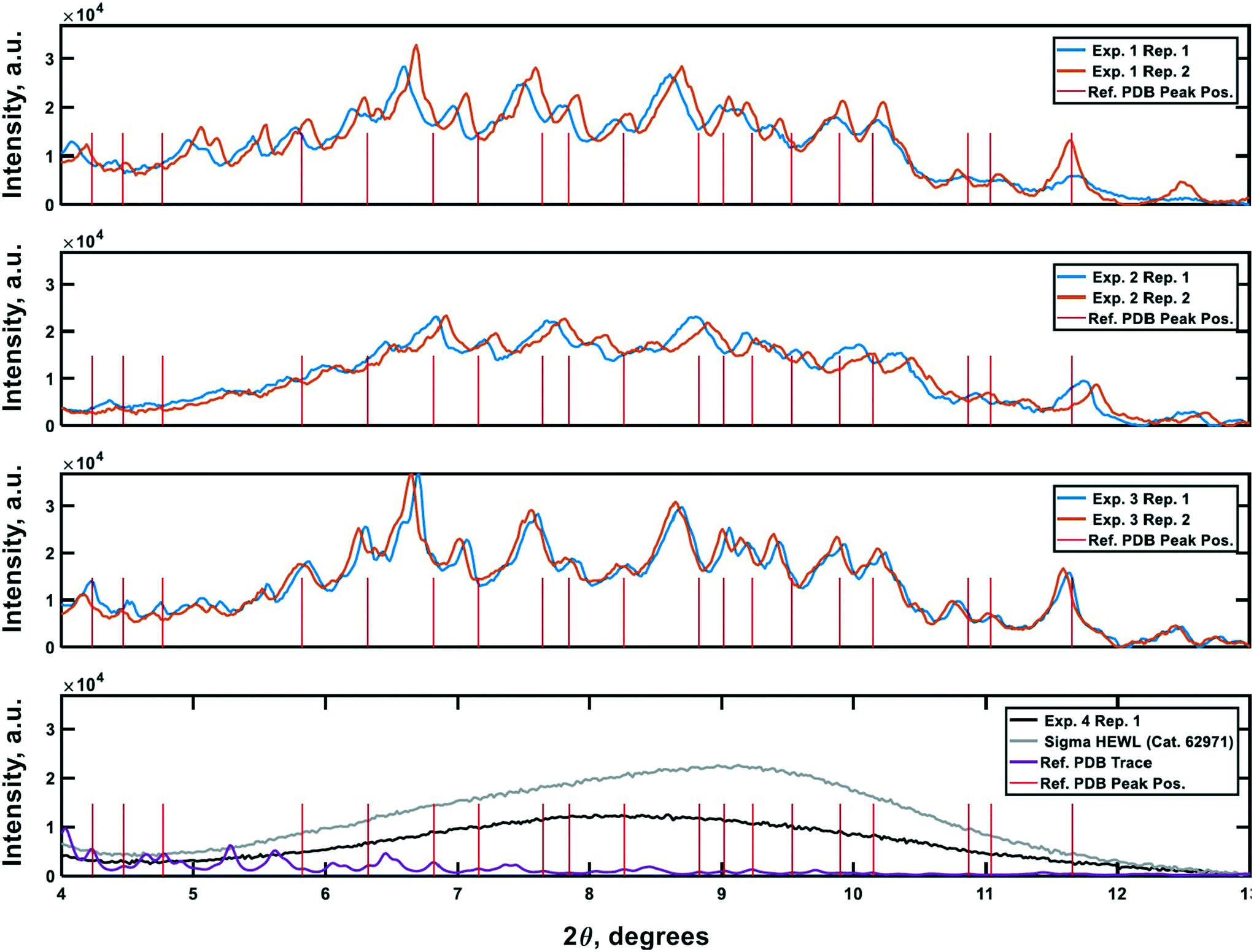 | ||
| Fig. 6 Powder X-ray diffraction traces characterizing the crystallinity of the samples generated during the experiments. Plots 1–3 (top to bottom) are the PXRD traces gathered for the materials generated in experiments 1–3, respectively. The 4th plot shows reference non-crystalline spectra for manufacturer-supplied HEWL, and HEWL intentionally precipitated out of solution using the CCHEX platform (black; exp. 4). The purple trace in the bottommost plot is an idealized PXRD diffractogram calculated using publicly available single-crystal XRD data banked in the RCSB Protein Data Bank (ID: 3wun). The vertical red lines in each plot correspond to the positions of a subset of the critical peaks in the idealized PXRD trace. The intensity of the lines is arbitrary. | ||
Lastly, negative control diffractograms for a blank sample tray and vacuum-dried HEWL dissolution buffer are presented in Fig. 7. The diffractogram for the blank sample tray exhibits a flat baseline for 2θ ≥ 3°. Similarly, the HEWL dissolution buffer diffractogram exhibits only a single peak within the range 9.5° ≤ 2θ ≤ 10°, which is characteristic of NaOAc. These controls indicate that neither the dissolution buffer nor the sample tray is expected to obscure the PXRD peaks of HEWL under the measurement conditions employed.34
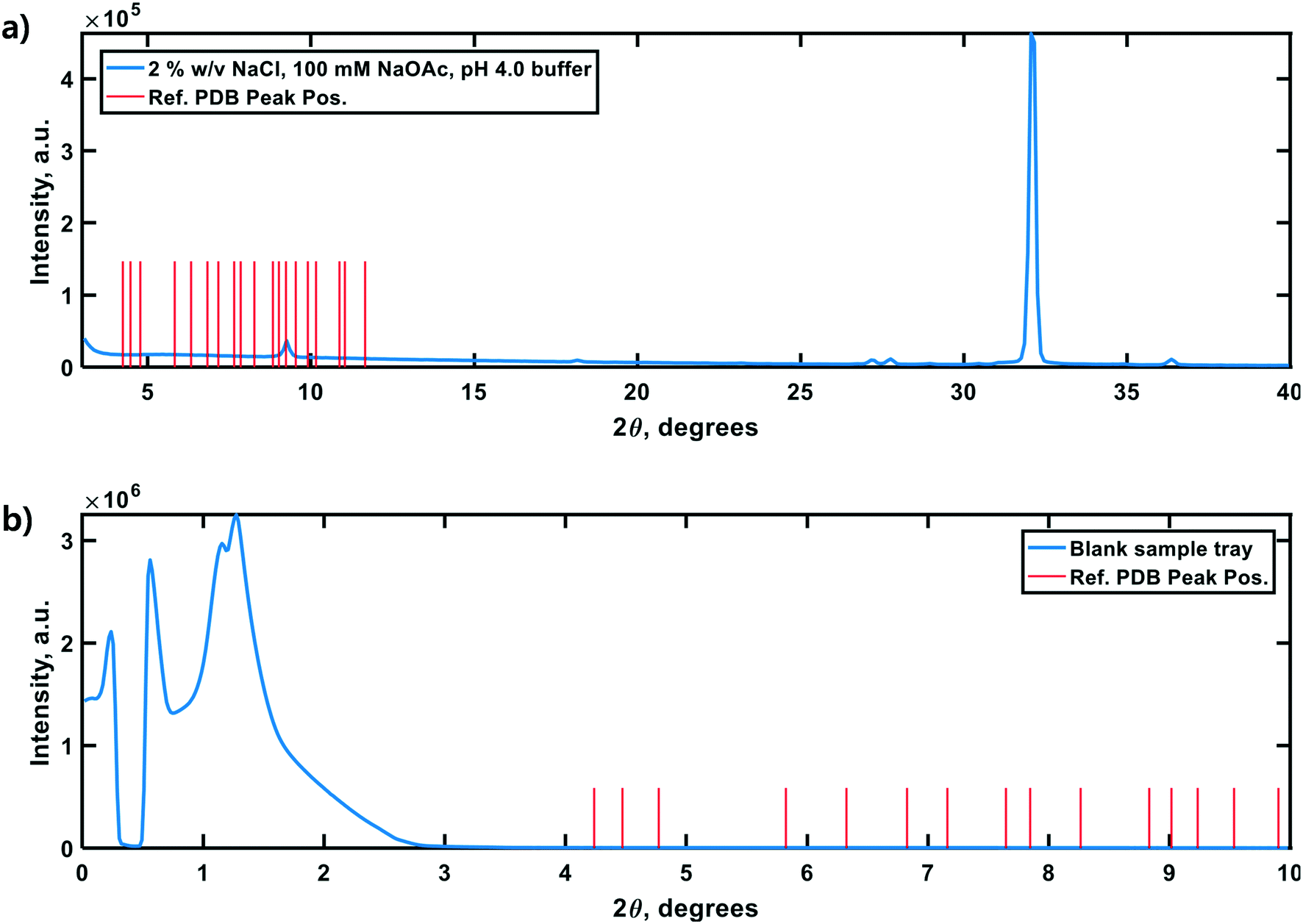 | ||
| Fig. 7 Control PXRD diffractograms of the HEWL dissolution buffer (a) and zero-background sample tray (b). The vertical red lines in each panel correspond to a subset of the critical peak positions in an idealized diffractogram calculated from single-crystal XRD data for tetragonal HEWL (PDBid: 3wun). (a) The doublet at ∼27.5° 2θ is characteristic of NaCl. The low intensity peak at ∼9° 2θ is characteristic of NaOAc (and is subject to shift above 10° 2θ due to variations in molecular hydration). (b) The high-intensity baseline below 3° 2θ is likely the result of direct beam scattering off of the sample tray itself at these extremely shallow angles. | ||
4. Conclusion
A continuous slug-flow crystallizer comprising reconfigurable, feedback-controlled, counter-current heat exchangers is applicable to mediating protein crystallization. Experiments using hen egg white lysozyme as the model protein showed that particle size distributions could be reproducibly manipulated using temperature gradients alone over a residence time of only 25–30 minutes. The formation of XRD-crystalline particles of HEWL was robust to maximum relative supersaturation gradients spanning two orders of magnitude, with σmax values ≥5 favoring the nucleation over the growth of existing crystals. In addition, the in-line mixing of the concentrated precipitant solution allowed σmax values as large as 220 to be achieved in concert with steep temperature gradients. Powder X-ray diffraction indicated that σmax of this magnitude overwhelmingly favored the formation of amorphous precipitates, which would have lower stability and higher solubility than crystals. The low cost and disposable nature of the slug-flow continuous crystallizer (∼$100 for the disposable tubing) and the ability to tune the particle size distribution suggest that this crystallization platform could be suitable in applications where the protein therapeutic is delivered in crystalline form, since the PSD directly affects the rate in which the protein would be absorbed by the body.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Bill & Melinda Gates Foundation [OPP1154682]. The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation. Financial support is acknowledged from the Engineering Development Research Center (EDRC) at Seoul National University for Yongkyu Lee. Leia Dwyer and Carlos Sieperman are acknowledged for PXRD training.References
- P. Gagnon, Technology trends in antibody purification, J. Chromatogr. A, 2012, 1221, 57–70 CrossRef CAS PubMed.
- A. Jungbauer, Continuous downstream processing of biopharmaceuticals, Trends Biotechnol., 2013, 31(8), 479–492 CrossRef CAS PubMed.
- K. B. Konstantinov and C. L. Cooney, White paper on continuous bioprocessing May 20–21 2014 continuous manufacturing symposium, J. Pharm. Sci., 2015, 104(3), 813–820 CrossRef CAS PubMed.
- B. Kelley, Very large scale monoclonal antibody purification: The case for conventional unit operations, Biotechnol. Prog., 2008, 23(5), 995–1008 Search PubMed.
- B. Kelley, Industrialization of mAb production technology: The bioprocessing industry at a crossroads, mAbs, 2009, 1(5), 443–452 CrossRef PubMed.
- M. S. Hong, K. A. Severson, M. Jiang, A. E. Lu, J. C. Love and R. D. Braatz, Challenges and opportunities in biopharmaceutical manufacturing control, Comput. Chem. Eng., 2018, 110, 106–114 CrossRef CAS.
- A. L. Zydney, Continuous downstream processing for high value biological products: A Review, Biotechnol. Bioeng., 2015, 113(3), 465–475 CrossRef PubMed.
- M. Bryntesson, M. Hall and K. Lacki, Chromatography method, U.S. Pat., 7901581, Mar 8, 2011 Search PubMed.
- V. Warikoo, R. Godawat, K. Brower, S. Jain, D. Cummings, E. Simons, T. Johnson, J. Walther, M. Yu, B. Wright, J. McLarty, K. P. Karey, C. Hwang, W. Zhou, F. Riske and K. Konstantinov, Integrated continuous production of recombinant therapeutic proteins, Biotechnol. Bioeng., 2012, 109(12), 3018–3029 CrossRef CAS PubMed.
- J. Eggersgluess, T. Wellsandt and J. Strube, Integration of aqueous two-phase extraction into downstream processing, Chem. Eng. Technol., 2014, 37(10), 1686–1696 CrossRef CAS.
- N. Hammerschmidt, A. Tscheliessnig, R. Sommer, B. Helk and A. Jungbauer, Economics of recombinant antibody production processes at various scales: Industry-standard compared to continuous precipitation, Biotechnol. J., 2014, 9(6), 766–775 CrossRef CAS PubMed.
- P. Neugebauer and J. G. Khinast, Continuous crystallization of proteins in a tubular plug-flow crystallizer, Cryst. Growth Des., 2015, 15(3), 1089–1095 CrossRef CAS PubMed.
- L. E. Crowell, A. E. Lu, K. R. Love, A. Stockdale, S. M. Timmick, D. Wu, Y. Wang, W. Doherty, A. Bonnyman, N. Vecchiarello, C. Goodwine, L. Bradbury, J. R. Brady, J. J. Clark, N. A. Colant, A. Cvetkovic, N. C. Dalvie, D. Liu, Y. Liu, C. A. Mascarenhas, C. B. Matthews, N. J. Mozdzierz, K. A. Shah, S.-L. Wu, W. S. Hancock, R. D. Braatz, S. M. Cramer and J. C. Love, On-demand manufacturing of clinical-quality biopharmaceuticals, Nat. Biotechnol., 2018, 36(10), 988–995 CrossRef CAS PubMed.
- A. E. Lu, J. A. Paulson, N. J. Mozdzierz, A. Stockdale, A. N. F. Versypt, K. R. Love, J. C. Love and R. D. Braatz, Control systems technology in the advanced manufacturing of biologic drugs, in Proc. of the IEEE Conference on Control Applications, 2015, pp. 1505–1515 Search PubMed.
- D. Hekmat, Large-scale crystallization of proteins for purification and formulation, Bioprocess Biosyst. Eng., 2015, 38(7), 1209–1231 CrossRef CAS PubMed.
- A. McPherson, Introduction to protein crystallization, Methods, 2004, 34(3), 254–265 CrossRef CAS PubMed.
- I. Russo Krauss, A. Merlino, A. Vergara and F. Sica, An overview of biological macromolecule crystallization, Int. J. Mol. Sci., 2013, 14(6), 11643–11691 CrossRef PubMed.
- J. C. Baker and B. M. Roberts, Preparation of stable insulin analog crystals, U.S. Pat., 5597893, Jan 28, 1997 Search PubMed.
- S. Mitragotri, P. A. Burke and R. Langer, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies, Nat. Rev. Drug Discovery, 2014, 13(9), 655–672 CrossRef CAS PubMed.
- D. Zhang, S. Xu, S. Du, J. Wang and J. Gong, Progress of pharmaceutical continuous crystallization, Engineering, 2017, 3(3), 354–364 CrossRef.
- N. J. Mozdzierz, Developing scalable and modular technologies for continuous biopharmaceutical production, Ph.D. thesis, Massachusetts Institute of Technology, Cambridge, MA, 2018 Search PubMed.
- N. J. Mozdzierz, Y. Lee, M. S. Hong, M. H. P. Benisch, M. L. Rasche, U. E. Tropp, M. Jiang, A. S. Myerson and R. D. Braatz, Mathematical modeling and experimental validation of continuous slug-flow tubular crystallization with ultrasonication-induced nucleation and spatially varying temperature, Chem. Eng. Res. Des., 2021, 169, 275–287 CrossRef CAS.
- J. C. Pirkle, M. L. Rasche, R. D. Braatz and M. Jiang, Slug-flow continuous crystallization: Fundamentals and process intensification, in The Handbook of Continuous Crystallization, The Royal Society of Chemistry, Croydon, UK, 2020, ch. 5, pp. 219–247 Search PubMed.
- E. L. Forsythe, R. A. Judge and M. L. Pusey, Tetragonal chicken egg white lysozyme solubility in sodium chloride solutions, J. Chem. Eng. Data, 1999, 44(3), 637–640 CrossRef CAS.
- N. E. Chayen, Methods for separating nucleation and growth in protein crystallisation, Prog. Biophys. Mol. Biol., 2005, 88(3), 329–337 CrossRef CAS PubMed.
- C. A. Pons Siepermann, S. Huang and A. S. Myerson, Nucleation inhibition of benzoic acid through solution complexation, Cryst. Growth Des., 2017, 17(5), 2646–2653 CrossRef CAS.
- R. T. Dombrowski, 1 - Microscopy techniques for analyzing the phase nature and morphology of biomaterials, in Characterization of Biomaterials, ed. M. Jaffe, W. Hammond, P. Tolias and T. Arinzeh, Woodhead Publishing, Waltham, MA, 2013, pp. 1–33 Search PubMed.
- A. Myerson, Handbook of Industrial Crystallization, Butterworth-Heinemann, Woburn, MA, 2002 Search PubMed.
- A. Ranodolph, Theory of Particulate Processes: Analysis and Techniques of Continuous Crystallization, Elsevier, New York, 2012 Search PubMed.
- P. Varlashkin, Approaches to quantification of amorphous content in crystalline drug substance by powder X-ray diffraction, Am. Pharm. Rev., 2011, 14(1), 22–28 CAS.
- S. Speakman, Basics of X-Ray Powder Diffraction: Training to Become an Independent User of the X-Ray SEF at the Center for Materials Science and Engineering at MIT, 2017, http://prism.mit.edu/xray/oldsite/Basics%20of%20X-Ray%20Powder%20Diffraction.pdf Search PubMed.
- S. Bates, G. Zografi, D. Engers, K. Morris, K. Crowley and A. Newman, Analysis of amorphous and nanocrystalline solids from their X-ray diffraction patterns, Pharm. Res., 2006, 23(10), 2333–2349 CrossRef CAS PubMed.
- S. Kemethmüller, A. Roosen, F. Goetz-Neunhoeffer and J. Neubauer, Quantitative analysis of crystalline and amorphous phases in glass–ceramic composites like LTCC by the Rietveld method, J. Am. Ceram. Soc., 2006, 89(8), 2632–2637 CrossRef.
- R. Kumar, S. Vyas, R. Kumar and A. Dixit, Development of sodium acetate trihydrate-ethylene glycol composite phase change materials with enhanced thermophysical properties for thermal comfort and therapeutic applications, Sci. Rep., 2017, 7(1), 1–11 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |