
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled titration-based ZnO formation†
Mark M. J.
van Rijt
 ,
Bernette M.
Oosterlaken
,
Heiner
Friedrich
and
Gijsbertus
de With
*
,
Bernette M.
Oosterlaken
,
Heiner
Friedrich
and
Gijsbertus
de With
*
Laboratory of Physical Chemistry, Centre for Multiscale Electron Microscopy, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P. O. Box 513, Eindhoven, 5600 MB, The Netherlands. E-mail: G.deWith@tue.nl
First published on 24th March 2021
Abstract
Hexamethylenetetramine (HMTA) is commonly used as a base releasing agent for the synthesis of ZnO under mild aqueous conditions. HMTA hydrolysis leads to gradual formation of a base during the reaction. Use of HMTA, however, does have limitations: HMTA hydrolysis yields both formaldehyde and ammonia, it provides no direct control over the ammonia addition rate or the total amount of ammonia added during the reaction, it results in a limited applicable pH range and it dictates the accessible reaction temperatures. To overcome these restrictions, this work presents a direct base titration strategy for ZnO synthesis in which a continuous base addition rate is maintained. Using this highly flexible strategy, wurtzite ZnO can be synthesized at a pH >5.5 using either KOH or ammonia as a base source at various addition rates and reaction pH values. In situ pH measurements suggest a similar reaction mechanism to HMTA-based synthesis, independent of the varied conditions. The type and concentration of the base used for titration affect the reaction product, with ammonia showing evidence of capping behaviour. Optimizing this strategy, we are able to influence and direct the crystal shape and significantly increase the product yield to 74% compared to the ∼13% obtained by the reference HMTA reaction.
Introduction
For the aqueous formation of zinc oxide (ZnO) at a mild and controlled pH, hexamethylenetetramine (HMTA) is typically used as a base releasing agent.1–4 Using this strategy, a variety of ZnO morphologies can be synthesized5 ranging from hexagonal twin pillar structures6,7 and ZnO arrays8–10 to more complicated morphologies11–13 and nanocomposites.14 At elevated reaction temperature, HMTA hydrolysis results in the gradual formation of ammonia (NH3) and formaldehyde (CH2O, eqn (1)) in the reaction medium. The produced ammonia is protonated by the present water, releasing hydroxide in the process (eqn (2)).15 The formed hydroxide is subsequently consumed as a base during the formation of ZnO (eqn (3)).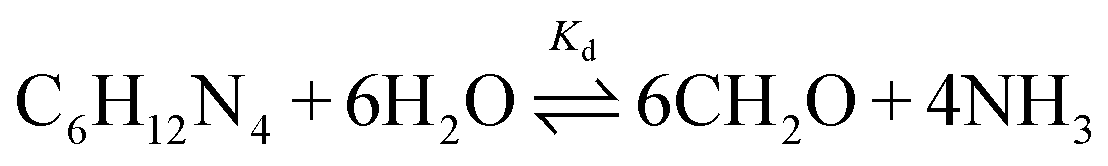 | (1) |
 | (2) |
| Zn2+ + 2OH− → ZnO(s) + H2O | (3) |
| kobs = (kw + kh[H]+)f+ | (4) |
HMTA hydrolysis is acid catalysed, resulting in a concomitant increase in the HMTA decomposition rate k with decreasing reaction pH (eqn (4)), with f+ representing the fraction of HMTA in protonated form.16 Combining this with a hydroxide consuming reaction, like the formation of ZnO, the system will move to an equilibrium pH where the formation of hydroxide by HMTA hydrolysis and ammonia protonation is balanced with the hydroxide consumption by the reaction, effectively making HMTA a pH buffer.17 Given that for a crystallization process the reaction rate is temperature dependent, the reaction pH and final HMTA decomposition rate will also be influenced by the reaction temperature. Although the decomposition of HMTA is further catalysed by the presence of strong acids,18 Ashfold et al.19 showed that the rate of HMTA decomposition is not catalysed or retarded by the formation of ZnO after a 2 h reaction time.
Besides HMTA being used as a base source and pH buffer, additional influences of HMTA on the ZnO reaction have been investigated.20 HMTA has been proposed to act as a capping or structure directing agent on the non-polar side facets of ZnO promoting growth along the c-axis21–23 and acting as a shell preventing the merging of nanorods.22,24 However, these additional HMTA influences are under debate. Notably, in situ XANES measurements showed no long-lived zinc–HMTA intermediates during the reaction, whereas IR measurements showed no adsorption of HMTA on ZnO.25,26 One generally overlooked aspect is the influence of the HMTA hydrolysis products: ammonia, formaldehyde and hydroxide. As hydrolysis of HMTA yields all these products simultaneously, studies on product specific effects become highly challenging.
To overcome the abovementioned limitations, a titration-based strategy may be employed which allows for the addition of any reactant at any rate, facilitating the study of the influence of individual components. Furthermore, titration is not limited to a narrow equilibrium pH window during reaction (I) and by the reaction temperature (II), and does not need reactant refreshing steps over long reaction times (III)27–29 or a reaction condition dependent base addition rate (IV), disadvantages inherent to the use of HMTA. Titration-based strategies have been used previously for the formation of biomimetic magnetite,30,31 silica32 and to a limited extent ZnO,26 but not in the context of imitating existing reaction conditions such as those in the HMTA-based ZnO synthesis.
Therefore, this work aims to design an HMTA-inspired base-titration strategy for the formation of ZnO in water under mild pH conditions. By investigating the contribution of HMTA decomposition products, we find that among its hydrolysis products, mainly ammonia affects the ZnO reaction product. It is further found that the decomposition rate of HMTA is higher during the initial formation of ZnO than that under native conditions. Finally, using the flexibility of the titration strategy, we show that the ZnO crystal shape and size is sensitive to the reaction conditions, including the initial pH and the added base concentration. We finally demonstrate a protocol for the formation of highly faceted twin-pillared ZnO crystals with a high yield.
Results
Hydrolysis of HMTA and experimental design
Previous work on the formation of ZnO using HMTA showed a constant pH of 5.8 during most of the reaction.33 Hence, the hydrolysis of 25 mM HMTA in water at a constant pH of 5.8 was taken as a model system.33 For experimental details, see ESI† section 1. The HMTA hydrolysis rate was measured by titrating 0.25 M acetic acid into the reaction vessel to maintain a constant pH (Fig. S1†), as discussed in detail in the ESI† section 2. These measurements showed a constant but pH dependent HMTA hydrolysis rate kobs of 0.013 h−1 (pH 6.0), 0.018 h−1 (pH 5.8) and 0.023 h−1 (pH 5.7), which matches excellently with the results from Ashfold et al.19 (Fig. 1a). | ||
| Fig. 1 Observed hydrolysis rates (a) of HMTA at pH 6.0, 5.8 and 5.7 (black) compared to results from Ashfold et al.19 (results from Ashfold have been corrected with a factor of two to compenstate for the difference HMTA starting concentration, see ESI† section 2.2). Measured formation of hydroxide ions (OH−) by HMTA hydrolysis (b) at pH 5.8 (green) and the calculated amount of formed hydroxide as a function of time (b), with the corresponding titration protocol, by using 73.35 mM ammonia or KOH (red) or 3.1 mM KOH (black), see inset. | ||
To mimic the base formation rate during HMTA hydrolysis with a preprogrammed run in our titration setup, the minimal titration rate and increment steps of 0.01 mL min−1 of the equipment should be considered. Taking this limitation into account, two titration strategies were designed, both presuming that ZnO formation does not influence the hydrolysis of HMTA, as indicated by Ashfold et al.19 The first is based on the addition of ammonia (both with and without the simultaneous addition of formaldehyde) or KOH without taking the dissociation constant of ammonia Kd into account. Given the near linear hydrolysis rate of HMTA, we calculated that 73.35 mM ammonia or KOH has to be added at a constant rate of 0.02 mL min−1 or 146.70 mM ammonia and 221.55 mM formaldehyde, both at a constant rate of 0.01 mL min−1 (Fig. 1b, red). The second titration strategy is based on the addition of KOH while taking the Kd of ammonia into account.15,34 For this, a progressively decreasing addition rate was calculated for 3.1 mM KOH (Fig. 1b, black). Both strategies showed an increase in hydroxide ions which closely matched with the previously identified decomposition of HMTA (25 mM) at a pH of 5.8 (Fig. 1b).
Influence of reaction components
Upon initiating an HMTA–ZnO reference reaction by heating, in situ pH measurements show a gradual pH decrease from 6.8 to 5.8 (Fig. 2). After this initial pH drop, the pH remains stable until 120 min reaction time. At this time, a second pH drop of 0.1 is observed, which our previous work has shown to indicate a rapid transition from a layered basic zinc acetate (LBZA) rich phase to a predominantly wurtzite ZnO phase (LBZA–ZnO transition).33 Following the LBZA–ZnO transition, the pH gradually recovered to 5.8. At the end of the reaction, a white precipitate is observed for all reactions but to a varying degree, both in dispersion and on the flask. After purification of the dispersed reaction products, a yield of 12.6% was obtained. Repeating the HMTA reaction to exclude the influence of titration itself (water, 0.2 mL min−1), no significant influence on the kinetics, final product or yield was observed (Fig. S2a–c†). PXRD and SEM analyses confirm the formation of wurtzite ZnO (Fig. S2d–f†).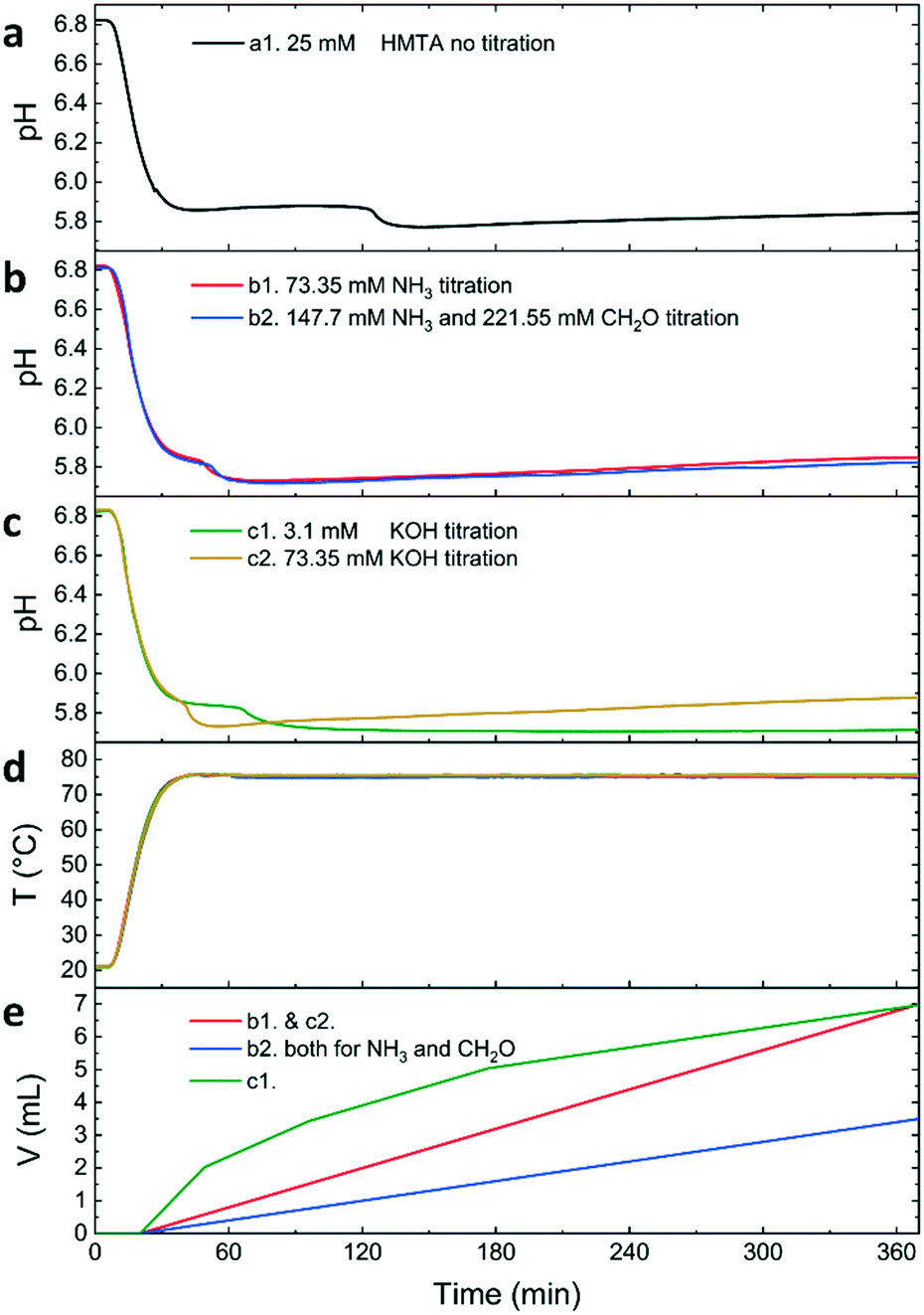 | ||
| Fig. 2 HMTA- and titration-based synthesis of ZnO from zinc acetate showing the pH evolution of the HMTA control experiment (a), the pH evolution of the ammonia-based titration experiments (b), the pH evolution of the potassium hydroxide-based titration experiments (c), and the temperature T (d) and titration volume V (e) as a function of time. | ||
When titrating with 73.35 mM ammonia directly (Fig. 2b, d and e), the pH profile showed the same features as the reference HMTA reaction with one notable difference: the LBZA–ZnO transition is observed at 51 min instead of 120 min reaction time. This indicates a significant acceleration of the reaction kinetics when using direct base-titration. Simultaneous titration of formaldehyde showed no influence on the pH evolution during the reaction. After purification, PXRD confirmed the formation of wurtzite ZnO, independent of simultaneous titration of formaldehyde (Fig. 3). As all PXRD reflections are sharp, we conclude that the materials formed are crystalline. The product yield was 3.5% when titrating with ammonia and 3.7% when titrating with ammonia and formaldehyde.
 | ||
| Fig. 3 PXRD data of ZnO formed by HMTA- and titration-mediated synthesis. The PXRD data are normalized on the highest intensity signal and, the broad PXRD signal visible at about 20° is due to the substrate. All further observed spacings match with wurtzite ZnO. | ||
The effects of direct hydroxide addition by KOH titration were investigated using the two protocols described above: 1) taking the Kd of ammonia into account by gradually adding 3.1 mM KOH and 2) mimicking the ammonia titration protocol using 73.35 mM KOH. Again, both strategies showed the typical pH features (Fig. 2c–e) observed when using HMTA.33 The LBZA–ZnO transition was observed at 64 min reaction time for 3.1 mM and 21 min reaction time for the 73.35 mM KOH titration strategy. Furthermore, when using 3.1 mM KOH, the pH remained at 5.7 after the LBZA–ZnO transition, resulting in a relatively low final pH compared to all other reactions. This suggests that the added amount of hydroxide is significantly lower for this experiment than for all other investigated reactions. Indeed, after purification, the yield was 0.4% for 3.1 mM and 4.3% for the 73.35 mM KOH titration strategy. Due to the limited yield, only the 73.35 mM KOH product could be analysed by PXRD, which confirmed the formation of wurtzite ZnO (Fig. 3).
Comparing the experimentally obtained reaction yields to theoretically calculated values (ESI† section 3), it is found that the result for the 3.1 mM KOH strategy matches well with the expected yield (0.43%) when taking the ammonia Kd into account. The 73.35 mM KOH and ammonia strategies showed a notably higher yield close to ∼4%. Given the close match between the 73.35 mM KOH and ammonia yields, we presume that almost all ammonia dissociates into ammonium during the reaction. Therefore, the result using 73.35 mM KOH is expected to be close to the result obtained using ammonia. This is likely caused by the formation of Zn–ammonium complexes,28 possibly in combination with the capping of LBZA and ZnO by ammonium.
Using SEM, the crystal size and shape were studied (Fig. 4 and S3†). An approximately constant crystal aspect ratio is observed for all conditions. The crystal habit of wurtzite ZnO results in either a rod-like or twin-pillared hexagonally faceted shape.28 The ZnO crystals resulting from the 25 mM HMTA reaction generally show this hexagonal shape. Size measurements revealed a diameter of 400 ± 150 nm, where ± indicates the sample standard deviation (Fig. 4a). Although all four HMTA-inspired titration strategies show hexagonally shaped ZnO crystals, the hexagonal faceting is generally less well expressed as compared to crystals of similar size synthesized by using HMTA. Measuring the crystal size revealed a diameter of 250 ± 40 nm when titrating with ammonia (Fig. 4c) and 260 ± 70 nm when titrating with ammonia and formaldehyde (Fig. 4d). This shows that formaldehyde has no notable influence on the crystal shape or size. Titrating with KOH results in a diameter of 270 ± 110 nm for 3.1 mM (Fig. 4e) and 110 ± 30 nm for the 73.35 mM titration strategy (Fig. 4f). This indicates that the above titration-based strategies result in the formation of smaller ZnO crystals compared to the use of HMTA, especially when using KOH as the base. Additional size measurements from the PXRD data, by the use of the Scherrer equation, did not yield reliable results because the crystals exceed the maximum measurable crystal size imposed by instrumental limitations.35
 | ||
| Fig. 4 SEM images of ZnO formed using 25 mM HMTA (a) and 8.33 mM HMTA (b) as a reference. Titration based-ZnO using 73.35 mM ammonia titration (c), 146.7 mM ammonia with simultaneous 221.55 mM formaldehyde titration (d) and 3.1 mM (e) and 73.35 mM (f) KOH titration. All scale bars equal 1 μm. Size analysis is shown in Fig. S3.† | ||
Overall, these results show that by using an HMTA-inspired direct base-titration strategy, wurtzite ZnO can be synthesized at a mild and controlled pH, and is formed independent of the added base (ammonia or KOH) and that the addition of formaldehyde does not seem to influence the reaction. A similar pH evolution is observed for all the titration strategies compared to the reference HMTA reaction, suggesting that the underlying ZnO formation mechanism remains unchanged, although with increased reaction kinetics. The HMTA-inspired titration-based ZnO crystals are smaller, show a loss in crystal habit definition and have a smaller yield than those from the reference HMTA reaction.
Increased base addition rates
Given the promising results using the HMTA-inspired direct base-titration strategy, the flexibility of the approach was used to optimize the formation of ZnO. As the initial step, the base addition was increased by using a 1 M ammonia or KOH stock solution while maintaining a titration rate of 0.02 mL min−1. After 1 h reaction time, no base was added in the case that the pH was 6.0 or higher to minimize pH-induced ZnO etching.33Using either a 1 M KOH or ammonia solution, all typical pH features were observed (Fig. 5a and b). The LBZA–ZnO transition was again clearly observed in both cases at 41 min and 44 min reaction time, respectively. After the LBZA–ZnO transition, the pH gradually increased (from 5.8) to 6.0 at 140 min for KOH and 169 min for ammonia. Upon reaching a pH of 6.0, the base addition rate gradually decreased to 0 mL min−1, indicating that the reaction was complete. This was observed between 180 and 240 min reaction time. In total, 2.70 mL of 1 M KOH and 3.27 mL of 1 M ammonia were required for the reaction with no base added after a 4 h reaction time (Fig. 5c). This new titration strategy resulted in a significantly increased yield of 31.8% using KOH and 41.3% using ammonia. PXRD confirmed the formation of pure ZnO (Fig. 6). For 1 M KOH, SEM analysis showed the formation of crystals with a diameter of 140 ± 30 nm (Fig. 7b and S3†) with some clear evidence of hexagonal faceting. For 1 M NH3 titration, crystals with a diameter of 240 ± 90 nm and clear hexagonal faceting were observed (Fig. 7c and S3†), however, rods tend to form clusters instead of the expected twin-pillared structure.
 | ||
| Fig. 5 Titration-based synthesis of ZnO from zinc acetate using 1 M KOH or 1 M ammonia solutions showing the time evolution of pH (a) magnified between 30 and 50 min reaction time (inset), the temperature T (b) and the titration volume V (c). The dotted line at 1 h reaction time indicates the change in the titration protocol to a target pH of 6. The LBZA–ZnO transitions (a) are indicated using coloured arrows. * indicates that the base has been gradually added prior to heating, see Fig. S4.† | ||
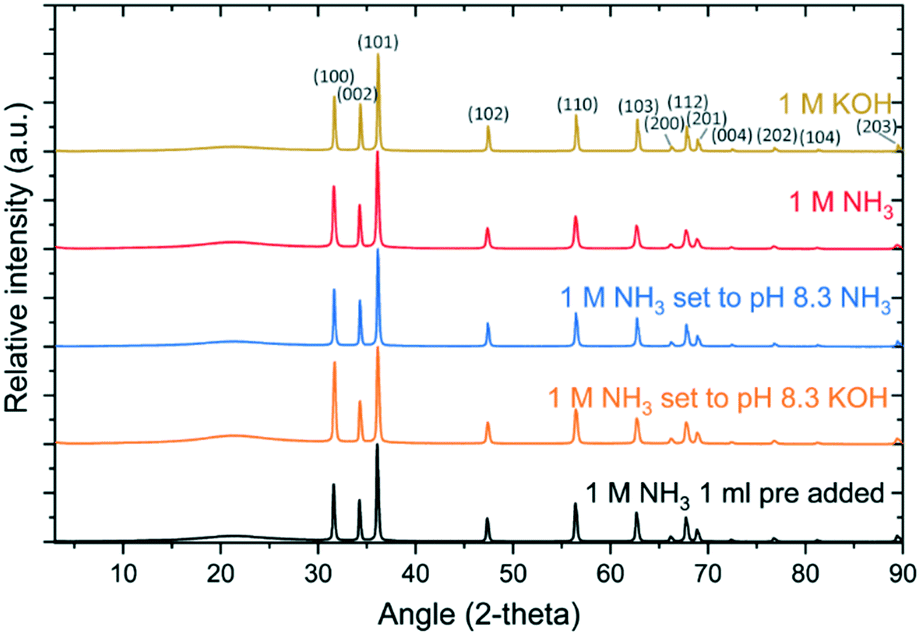 | ||
| Fig. 6 PXRD data of ZnO formed by direct base titration using 1 M NH3 or KOH. The PXRD data are normalized on the highest intensity signal and the broad pXRD signal visible at about 20° is due to the substrate. All further observed spacings match with wurtzite ZnO. | ||
 | ||
| Fig. 7 SEM images of ZnO formed with 25 mM HMTA as a reference (a, duplicate of Fig. 2a), titrating with 1 M KOH at native starting pH (b) and 1 M ammonia at native starting pH (c), starting at a pH of 8.3 set using ammonia (d), starting at a pH of 8.3 set using KOH (e) and titrating with 1 mL of 1 M ammonia before initiating the reaction (f). The PXRD data are normalized on the highest intensity signal and the broad pXRD signal visible at about 20° is due to the substrates. All scale bars equal 1 μm. | ||
Influence of pH and promoting twin-pillar ZnO formation
Given the narrow pH range of 6.8–5.7 under standard conditions, the influence of pH was further investigated. We used 1) a lower starting pH of 5.3 combined with a low target pH of 5.5 and 2) studied the influence of an initially higher pH of ∼8.3 combined with a similar reaction protocol to that used at standard pH. In all cases, 1 M ammonia was gradually titrated into the system.Starting the reaction at pH 5.3, the pH gradually increased until reaching 5.5 (Fig. 5a and b), after which the addition of the base was stopped, resulting in a constant pH of 5.5 (Fig. 5c). During and after the reaction, no turbidity was observed, nor could any product be obtained by post purification. This indicates that no LBZA or ZnO is formed under these conditions, suggesting that the typical reaction pH of 5.7–5.8 is close to the minimal pH required for the formation of ZnO.
To probe the higher pH range, the starting pH was set to 8.3 by either titrating 1 M ammonia (4.7 mL, Fig. 5) or KOH (4.8 mL, Fig. S4†) to the reaction mixture (Fig. S5†). Upon reaching pH 6.9, this instantly led to the precipitation of LBZA using either bases, independent of the base addition rate (Fig. S6†). Upon reaching pH 8.3 by titrating with ammonia or KOH, a stable pH was obtained.
Upon initiating the reaction by heating (Fig. 5a and b and S4†), in situ pH measurements show an immediate drop in pH. In the case where ammonia was used to set the initial pH, an accelerated decrease in the pH is observed 21 minutes after starting the reaction, suggesting a very early LBZA–ZnO transition. Around 32 min, a minimum pH of 6.6 is reached, followed by a gradual increase in pH up to 6.8 at 60 min due to the constant titration of ammonia. At 60 min, ammonia titration stops resulting in a slight drop in pH to 6.7 after which the pH remains stable for the remainder of the reaction. When adjusting the pH using KOH, a comparable pH evolution is observed with one notable difference. A small rise in pH is observed at 22 min, followed by a second pH decrease at 42 min (Fig. S4a†). These two pH observations make it unclear when the transition from LBZA to ZnO exactly occurs. After purification, an even higher yield of 74.5 or 68.2% was obtained using ammonia or KOH, respectively, to set the initial pH. PXRD analysis showed the formation of wurtzite ZnO for both bases (Fig. 6). SEM images show the formation of twin-pillared ZnO (Fig. 7d and e) with the amount of ammonia used during the start of the reaction strongly affecting their morphology. The use of KOH resulted in ZnO crystals with a diameter of 220 ± 90 nm with a less defined crystal habit, particularly visible on the side facets (Fig. 7e and S3†). Using ammonia to set the pH resulted in crystals with a diameter of 390 ± 110 nm and a complete loss of crystal habit, i.e., multifaceted ZnO crystals (Fig. 7d and S3†).
Using the high starting pH of 8.3, predominantly twin-pillared ZnO crystals were observed whereas clusters of ZnO are predominantly formed using the milder native reaction pH. LBZA is formed upon increasing the starting pH and LBZA is believed to act as a nucleation template during the formation of ZnO in dispersion.6,33 Therefore, to promote the formation of twin-pillared ZnO at a mild reaction pH, a titration reaction was designed starting at pH 6.9, after adding 1 mL of ammonia at RT to promote the initial formation of LBZA (Fig. 5, and S5†). This resulted in a yield of 34% wurtzite ZnO (Fig. 6). Indeed, after purification, SEM analysis (Fig. 7f and S3†) showed the presence of predominantly straight twin-pillared ZnO crystals with a diameter of 200 ± 50 nm.
In short, it was observed that titrating an increased concentration of the base during the reaction results in the formation of wurtzite ZnO crystals that are similar in size and shape compared to the use of HMTA, while maintaining a similar pH evolution and range. Simultaneously, the yield increased significantly to ∼35%, when aiming at a pH of 6. Decreasing the reaction pH does not result in LBZA or ZnO formation; however, upon increasing the starting pH to 8.3, the ZnO yield is increased to ∼70%. Crystals formed at elevated pH show the loss of habit, especially when adding relatively large amounts of ammonia. The formation of a twin-pillared ZnO structure over clustered pillars can be promoted by simply adding a base before initiating the reaction.
Discussion
Yield
As mentioned, the reference HMTA reaction resulted in a yield of ∼13%. This value exceeds both the yields observed for the HMTA-inspired ammonia- and KOH-based titration strategies (∼4%) and the expected maximum yield, based on the predicted decomposition rate of HMTA (10%, ESI† section 3). To obtain an HMTA reference with a lower yield, the initial amount of HMTA was decreased from 25 mM to 8.33 mM. However, this still resulted in a yield of 7.2%, significantly exceeding the yield of the titration reaction and the expected maximum yield (∼3.3%, Fig. S2†). Ashfold et al.19 showed that the decomposition rate of HMTA is not affected by the formation of ZnO after a reaction time longer than 2 h and these authors are on occasion cited as to prove that the ZnO formation reaction does not affect the HMTA decomposition rate. However, using their data for the first 1.5 h, we calculated a close to 9-fold faster decomposition rate in the presence of the ZnO forming reaction (ESI† section 3). This higher initial decomposition rate of HMTA during the formation of ZnO explains the higher yield for the reference HMTA reaction compared to the 73.35 mM base titration reactions. This emphasizes the unpredictability of the reference HMTA reaction and makes the direct imitation of HMTA decomposition highly challenging.When titrating with 1 M of base, an increase in yield with reaction pH could be observed. Titrating ammonia to an equilibrium pH of 6.0 resulted in a yield of 34.0 or 41.3% with or without an initial addition of base, respectively. For the high pH reactions with a final pH of 6.6 and 6.4, a yield of 74.5 or 68.2% was obtained, respectively. This suggests that the equilibrium between soluble Zn species and ZnO shifts with increasing reaction pH, allowing for a larger ZnO yield when terminating the reaction at a higher final pH. This also shows that significantly more base is added using these 1 M base titration strategies than when using HMTA.
Kinetics
Comparing the LBZA–ZnO transition time between experiments, we note that by titrating with 73.35 mM KOH, this transition is observed slightly earlier at 40 min compared to 51 min using 73.35 mM ammonia. In general, faster reaction kinetics are observed with an increase in the concentration of the base added (Fig. 3 and 5). By increasing the added KOH concentration from 3.1 mM to 73.35 mM, the time at which the LBZA–ZnO transition occurs decreases from 64 min to 40 min. However, further increasing the KOH concentration to 1 M does not further accelerate the occurrence of the LBZA–ZnO transition. Moreover, using 1 M ammonia, the addition of 1 mL base before initiating the reaction results in a delay of the LBZA–ZnO transition from 44 min to 50 min reaction time (Fig. 5). Given that the LBZA–ZnO transition is presumably caused by LBZA crystals exceeding a critical size,33 this suggests that the LBZA–ZnO transition time depends on small variations in the (initial) reaction conditions influencing the LBZA growth rate and/or stability. This point is further supported by the transition time of ∼120 min when using HMTA, close to an hour longer than all titration reactions including those with a slower base addition. However, based on the present data, it cannot be excluded that the presence of HMTA itself has a retarding effect on the kinetics.Particle size, shape and capping
The shape of the ZnO crystals seems to be strongly influenced by the added concentration and type of the base. Titration of KOH results in the formation of relatively poorly defined crystals with some hexagonal features, independent of the titration concentration of KOH (3.1 mM, 73.35 mM or 1 M). The size of the crystals decreases with increasing amounts of the base from 270 ± 110 (at 3.1 mM) to 140 ± 30 (at 1 M). This implies that the increased presence of the base in the system increases the number of nuclei resulting in smaller particles.36When using ammonia, additional changes in the crystal habit of the reaction product are observed. Titration with a relatively low amount of ammonia (73.35 mM, Fig. 4c) results in crystals with poor hexagonal faceting, while the titration with a higher amount (1 M, Fig. 7c) results in highly defined hexagonal crystals. This suggests, especially when taking the KOH observations into account, that the ZnO crystal habit definition increases with the amount of ammonia in the system, as further supported by the HMTA observations. This strategy adds an intermediate amount of ammonia compared to the two ammonia titration strategies and results in a less clear hexagonal habit compared to the use of 1 M ammonia titration (Fig. 4a and 7a). The influence on the crystal shape due to the presence of ammonia is a strong indicator of capping behaviour. Additional evidence for the capping of ammonia is provided from the reactions starting at an elevated pH of 8.3. In contrast to using KOH, using ammonia to set the initial pH results in the formation of multifaceted ZnO structures. Strikingly, Amin et al.37 observed similar multifaceted ZnO crystals for HMTA-based ZnO but at a very low starting pH of 1.8. This large difference in the starting pH excludes the pH effect. However, both systems are expected to contain a large amount of ammonia before the onset of ZnO formation, either due to the initial correction of pH starting at 8.3 or by rapid HMTA decomposition when the pH is increased from 1.8 to >5.5 to conditions under which ZnO can be formed.
The observation of capping behaviour by ammonia is not surprising. Adsorption of ammonia on ZnO is a known behaviour38,39 and exploited for the use of ZnO as ammonia sensors.40–42 With an increasing concentration of ammonia, both the (001) c-plane and the (210) m-plane become more pronounced, suggesting that both planes are capped (Fig. S7†). Wurtzite ZnO has an innate preferential growth in the [001] direction in water.43 Hence, observation of preferred growth suggests that capping along the m-plane is either comparable to the capping of the c-plane or more pronounced. The c-plane is composed of alternating layers of Zn+ or O2− resulting in a positively or negatively charged polar surface. The m-plane has no net surface charge and is non-polar. Although the surface chemistry is different, the adsorption of ammonia in the gas phase has been experimentally confirmed on both the c-plane44 and the m-plane,45 supporting this hypothesis.
Besides the effect of HMTA on crystal faceting, there is an additional factor that influences both the size and shape of the ZnO crystals, namely the early formation of LBZA during the reaction. As discussed earlier, LBZA promotes the formation of twin-pillared ZnO crystals by acting as a nucleation template.6 Thus, if more LBZA is formed at the start of the reaction, ZnO nucleation will become more favourable compared to growth, thereby decreasing the ZnO particle size. Indeed, in this work, early formation of LBZA decreased the ZnO diameter from 240 ± 90 nm to 200 ± 50 nm.
Direct HMTA influences
Compared to the HMTA-inspired titration strategies, the reference HMTA reaction results in larger and more clearly faceted ZnO particles, a higher reaction yield and retarded reaction kinetics. This makes it arguable whether HMTA has a direct influence on the reaction, besides its gradual formation of ammonia. Unfortunately, given the influence of the ZnO formation reaction on the decomposition rate of HMTA, it can also be argued that all these effects are caused by variations in the base formation rate. Therefore, based on the present data, no firm conclusion can be made whether HMTA directly influences the ZnO formation process. However, it should be noted that by increasing and optimizing the ammonia addition rate, ZnO crystals can be formed similar to those formed when using HMTA. This shows that, although HMTA may directly influence the formation of ZnO, the same results can be achieved by optimizing ammonia titration conditions.Conclusion
Wurtzite ZnO was synthesized at mild pH in water using a direct base titration strategy which offers excellent control over the base addition rate. Using HMTA inspired titration rates, we found that ZnO is formed independent of the added base, either ammonia or KOH. The evolution of pH with time indicates no change in the ZnO formation mechanism compared to that of HMTA, but only changed reaction kinetics. In contrast to statements in the literature, the formation of ZnO was found to influence the decomposition rate of HMTA. Formaldehyde, a side product of HMTA decomposition, shows no influence on the reaction.Use of KOH as the base results in relatively small crystals independent of the added amount of the base. Using increasing amounts of ammonia instead, the shape and degree of faceting closely matches the typically observed hexagonal ZnO twin-pillars, which implies that ammonia caps the formed ZnO crystals.
Reducing the reaction pH to ≤5.5 prevented any reaction from occurring, showing that a minimal pH value is required for the reaction. In contrast, increasing the starting pH to 8.3 resulted in ZnO formation with a loss in crystal habit. Here, the use of ammonia to set the starting pH resulted in multifaceted ZnO crystals. The typical twin pillared ZnO crystal shape was predominantly observed when LBZA was formed before initiating the reaction. This supports the hypothesis that LBZA acts as a nucleation template for the formation of twin pillared ZnO crystals in dispersion. Given the observed influences of ammonia on the formation of ZnO in this work, it can be argued that it is ammonia and not HMTA that further directs the formation of the ZnO crystals. Due to the direct control over the base addition rates, ZnO yields exceeding 74% could be achieved. This yield increases with the target pH.
Given the direct control over base addition and the entailing flexibility provided by the titration-based ZnO synthesis strategy, we believe that it is a promising tool for mineralization in general. For ZnO in particular, we expect it to have further uses for heterogenous ZnO nucleation and growth, due to the high control over the growth conditions, and for the formation of ZnO at reduced temperatures thanks to the temperature independent addition of the base.
Funding sources
This project received funding from a TopPunt grant (Bi-Hy, 718.016.003) of the Netherlands Organization for Scientific Research (NWO).Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Fujita, K. Murata, T. Nakazawa and I. Kayama, J. Ceram., 1984, 92, 227–230 CAS.
- W. L. Feng, B. C. Wang, P. Huang, X. D. Wang, J. Yu and C. W. Wang, Mater. Sci. Semicond. Process., 2016, 41, 462–469 CrossRef CAS.
- S. Xu and Z. L. Wang, Nano Res., 2011, 4, 1013–1098 CrossRef CAS.
- V. Gerbreders, M. Krasovska, E. Sledevskis, A. Gerbreders, I. Mihailova, E. Tamanis and A. Ogurcovs, CrystEngComm, 2020, 22, 1346–1358 RSC.
- S. Maiti, S. Pal and K. K. Chattopadhyay, CrystEngComm, 2015, 17, 9264–9295 RSC.
- E. S. Jang, J. H. Won, Y. W. Kim, Z. Cheng and J. H. Choy, CrystEngComm, 2011, 13, 546–552 RSC.
- Z. Q. Hou, Y. X. Wang, L. H. Shen, H. Guo, G. X. Wang, Y. Li, S. F. Zhou, Q. Q. Zhang and Q. Jiang, Nanoscale Res. Lett., 2012, 7, 507 CrossRef PubMed.
- L. Vayssieres, Adv. Mater., 2003, 15, 464–466 CrossRef CAS.
- S. Xu, Y. Wei, M. Kirkham, J. Liu, W. Mai, D. Davidovic, R. L. Snyder and Z. L. Wang, J. Am. Chem. Soc., 2008, 130, 14958–14959 CrossRef CAS PubMed.
- Y. Rajesh, S. K. Padhi and M. G. Krishna, RSC Adv., 2020, 10, 25721–25729 RSC.
- T. R. Zhang, W. J. Dong, M. Keeter-Brewer, S. Konar, R. N. Njabon and Z. R. Tian, J. Am. Chem. Soc., 2006, 128, 10960–10968 CrossRef CAS PubMed.
- Z. R. Tian, J. A. Voigt, J. Liu, B. McKenzie, M. J. McDermott, M. A. Rodriguez, H. Konishi and H. Xu, Nat. Mater., 2003, 2, 821–826 CrossRef CAS PubMed.
- Y. Miao, H. J. Zhang, S. Yuan, Z. Jiao and X. D. Zhu, J. Colloid Interface Sci., 2016, 462, 9–18 CrossRef CAS PubMed.
- Y. C. Liang and W. C. Zhao, CrystEngComm, 2020, 22, 7575–7589 RSC.
- R. G. Bates and G. D. Pinching, J. Res. Natl. Bur. Stand., 1949, 42, 419–430 CrossRef CAS.
- J. G. Strom and H. W. Jun, J. Pharm. Sci., 1980, 69, 1261–1263 CrossRef CAS PubMed.
- K. Govender, D. S. Boyle, P. B. Kenway and P. O'Brien, J. Mater. Chem., 2004, 14, 2575–2591 RSC.
- H. Tada, J. Am. Chem. Soc., 1960, 82, 255–263 CrossRef CAS.
- M. N. R. Ashfold, R. P. Doherty, N. G. Ndifor-Angwafor, D. J. Riley and Y. Sun, Thin Solid Films, 2007, 515, 8679–8683 CrossRef CAS.
- P. Chettiyankandy, A. Chand, R. Ghosh, S. K. Sarkar, P. Das and S. Chowdhuri, J. Mol. Liq., 2019, 296, 111820 CrossRef CAS.
- A. Sugunan, H. C. Warad, M. Boman and J. Dutta, J. Sol-Gel Sci. Technol., 2006, 39, 49–56 CrossRef CAS.
- R. Parize, J. Garnier, O. Chaix-Pluchery, C. Verrier, E. Appert and V. Consonni, J. Phys. Chem. C, 2016, 120, 5242–5250 CrossRef CAS.
- R. Devaraj, K. Venkatachalam, K. Saravanakumar, P. M. Razad and K. Mahalakshmi, J. Mater. Sci.: Mater. Electron., 2016, 27, 12201–12208 CrossRef CAS.
- V. Strano, R. G. Urso, M. Scuderi, K. O. Iwu, F. Simone, E. Ciliberto, C. Spinella and S. Mirabella, J. Phys. Chem. C, 2014, 118, 28189–28195 CrossRef CAS.
- K. M. McPeak, M. A. Becker, N. G. Britton, H. Majidi, B. A. Bunker and J. B. Baxter, Chem. Mater., 2010, 22, 6162–6170 CrossRef CAS.
- K. M. McPeak, T. P. Le, N. G. Britton, Z. S. Nickolov, Y. A. Elabd and J. B. Baxter, Langmuir, 2011, 27, 3672–3677 CrossRef CAS PubMed.
- C. Ou, P. E. Sanchez-Jimenez, A. Datta, F. L. Boughey, R. A. Whiter, S. L. Sahonta and S. Kar-Narayan, ACS Appl. Mater. Interfaces, 2016, 8, 13678–13683 CrossRef CAS PubMed.
- A. S. Kamble, B. B. Sinha, K. Chung, M. G. Gil, V. Burungale, C. J. Park, J. H. Kim and P. S. Patil, Electrochim. Acta, 2014, 149, 386–393 CrossRef CAS.
- N. Caicedo, J. S. Thomann, R. Leturcq and D. Lenoble, CrystEngComm, 2016, 18, 5502–5511 RSC.
- G. Mirabello, A. Ianiro, P. H. H. Bomans, T. Yoda, A. Arakaki, H. Friedrich, G. de With and N. Sommerdijk, Nat. Mater., 2020, 19, 391–396 CrossRef CAS PubMed.
- J. J. M. Lenders, C. L. Altan, P. H. H. Bomans, A. Arakaki, S. Bucak, G. de With and N. A. J. M. Sommerdijk, Cryst. Growth Des., 2014, 14, 5561–5568 CrossRef CAS.
- M. W. P. van de Put, PhD Thesis, Technische Universiteit Eindhoven, 2015 Search PubMed.
- M. M. J. van Rijt, B. M. Oosterlaken, R. R. M. Joosten, L. E. A. Wijkhuijs, P. H. H. Bomans, H. Friedrich and G. de With, CrystEngComm, 2020, 22, 5854–5861 RSC.
- J. R. Fisher and H. L. Barnes, J. Phys. Chem., 1972, 76, 90–99 CrossRef CAS.
- K. He, N. F. Chen, C. J. Wang, L. S. Wei and J. K. Chen, Cryst. Res. Technol., 2018, 53, 1700157 CrossRef.
- T. Park, K. E. Lee, N. Kim, Y. Oh, J. K. Yoo and M. K. Um, J. Mater. Chem. C, 2017, 5, 12256–12263 RSC.
- G. Amin, M. H. Asif, A. Zainelabdin, S. Zaman, O. Nur and M. Willander, J. Nanomater., 2011, 2011, 1–9 CrossRef.
- T. Morimoto, H. Yanai and M. Nagao, J. Phys. Chem., 1976, 80, 471–475 CrossRef CAS.
- B. Sun, X. P. Yang, D. Zhao and L. Q. Zhang, Comput. Mater. Sci., 2018, 141, 133–140 CrossRef CAS.
- S. Kanaparthi and S. Govind Singh, Mater. Sci. Energy Technol., 2020, 3, 91–96 CAS.
- F. F. Franco, L. Manjakkal, D. Shakthivel and R. Dahiya, IEEE Sensor, 2019, 1–4, DOI:10.1109/SENSORS43011.2019.8956763.
- B. Chatterjee and A. Bandyopadhyay, Environ. Qual. Manag., 2016, 26, 89–105 CrossRef.
- L. Xu, Y. L. Hu, C. Pelligra, C. H. Chen, L. Jin, H. Huang, S. Sithambaram, M. Aindow, R. Joesten and S. L. Suib, Chem. Mater., 2009, 21, 2875–2885 CrossRef CAS.
- K. Ozawa, T. Hasegawa, K. Edamoto, K. Takahashi and M. Kamada, J. Phys. Chem. B, 2002, 106, 9380–9386 CrossRef CAS.
- T. Hasegawa, Y. Shirotori, K. Ozawa, K. Edamoto and K. Takahashi, Appl. Surf. Sci., 2004, 237, 352–357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supporting discussion and figures. See DOI: 10.1039/d1ce00222h |
| This journal is © The Royal Society of Chemistry 2021 |