
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uncovering new transition metal Zintl phases by cation substitution: the crystal chemistry of Ca3CuGe3 and Ca2+nMnxAg2−x+zGe2+n−z (n = 3, 4)†‡
Siméon
Ponou
ab,
Gordon J.
Miller
b and
Anja-V.
Mudring
 *a
*a
aDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16C, 114 18 Stockholm, Sweden. E-mail: anja-verena.mudring@mmk.su.se
bDepartment of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, 50011 Iowa, USA
First published on 16th February 2021
Abstract
High-temperature solid-state reactions of the respective elements afforded the new transition metal Zintl phases Ca3CuGe3 (Sc3NiSi3 type, monoclinic C2/m – i7, Pearson code mC28), Ca6MnxAg2−x+zGe6−z (own type, monoclinic P21/m – e14, Pearson code mP28) and, Ca5MnxAg2−x+zGe5−z (Ca5MgAgGe5 type, orthorhombic Pnma – c12, Pearson code oP48) as evidenced by single-crystal X-ray diffraction. They are additional representatives of the recently discovered homologous series Ca2+nM2+zGe2+n−z, already reported with M = Ag, Mg. These new phases were rationally prepared, after speculation that Cu and Mn could replace the isovalent Ag and Mg, respectively, to yield isostructural phases. Their crystal chemistry is discussed using established ‘structure directing rules’. Their structures are best described according to the Zintl–Klemm formalism as (Ca2+)(2+n)[M2+zGe2+n−z)]2(2+n)− featuring (poly-)germanide oligomers, [Gen](2n+2)− with n = 1–5. These Zintl anions interact with the highly polarizing small M (Cu, Ag, Mn) cations through their terminal Ge atoms, while the central Ge atoms are in trigonal prismatic coordination with the active metal Ca. Electronic structure calculations using density functional theory (DFT) were conducted on the idealized fully ordered model of “Ca3MGe3” (Sc3NiSi3 type) with M = Cu, Ag for an analysis of the chemical bonding and structure stabilizing factors. Our findings suggest that new transition metal Zintl phases can be obtained through partial to complete replacement of the highly polarizing small s-block cations (Li, Mg) in the Ca–(Li,Mg)–(Ge,Si) system by their isovalent transition metals like Ag, Cu, and Mn. However, due to differences in coordination requirements and possible strong metal–metal bonding between the d-block elements, the resulting transition metal phases may not be isostructural with their Li and Mg counterparts, even when featuring the same type of Zintl anions.
Introduction
Polar intermetallic compounds (PICs) form a large class of extended solids, combining electropositive ‘active’ metals (s- or f-block metals mainly) with electronegative post-transition p-block elements and/or late transition (noble) metals.1,2 Interest in polar intermetallic compounds was originally driven by their structural diversity and exotic bonding features.2–5 In fact, they represent intermediates between semiconductors and typical metallic systems.5,6 Hence, their bonding pictures are often very complex and, it is virtually impossible to apply simple heuristic concepts to reliably deduce their compositions, structural features or the nature of the bonding. This is, because unlike molecular systems, PICs show a much higher degree of electronic flexibility, allowing them to violate the electronic stability factors (valence rules), despite their dominant effects on the structure stability. Hence, for fundamental reasons also, it is very important to study the different mechanisms of structure stability in PICs. The Zintl concept has been very efficient to rationalize the structure and bonding of many PICs involving only main group elements.7 Even for structures that could not be formally classified as Zintl phases, the implementation of this rather simplistic concept provided interesting insights into their structure directing forces. Transition metal Zintl phases (TMZP) represent a special class of PICs, referring to transition metal (TM) containing compounds that are either isostructural with classical main group Zintl compounds or new TM structures for which the Zintl–Klemm formalism provides insights into their electronic structure and bonding.8Because of increased structural complexity and tunability of their electronic structure in comparison with classical Zintl phases, semiconducting TMZPs provide the ideal prototype of phonon glass electron crystal (PGEC) materials for application in thermoelectric (TE) energy generation from heat waste.8,9 The Zintl anionic components provide the diverse electronic structures as the “electron crystal” and, the cations play the role of the “phonon scattering center” yielding an extremely low lattice thermal conductivity. New TMZPs with complex crystal structure and semiconducting properties are important for the realization of better performing bulk TE materials.8 In the course of our research efforts to uncover new structurally complex TMZPs for potential application as thermoelectric, we have tested an empirical approach based on cation replacement of highly polarizing small cations Li and Mg in classical Si- and Ge-containing Zintl phases by corresponding isovalent TMs like Ag, Cu, and Mn to generate their TM-based analogues. We first considered the large family of main group ternary and quaternary classical Zintl phases AE/M/Tt (AE = Ca, Sr, Ba; M = Li, Mg; Tt = Si, Ge) as one possible system for a case study. These AE/M/Tt systems were intensively investigated by Nesper and co-workers.10–12 Previously, the complete replacement of small polarizing Li by Ag in the Ca–M–Ge germanide systems resulted in a new series of structures, Ca2+nM2+zGe2+n−z (M = Ag, Mg), which are built up by linear intergrowth of slabs cut from the parent structures, CaM1+zGe1−z (AlB2 or TiNiSi type) and CaGe (CrB type).13–16 Our next attempt was toward similar isovalent replacement of Mg by divalent TM elements like Mn and Cd.
We report herein on the existence and crystal structure of some new TMZPs, Ca3CuGe3 (monoclinic, C2/m – i7, mC28) and Ca6MnxAg2−x+zGe6−z (monoclinic P21/m – e14, mP28), as well as Ca5MnxAg2−x+zGe5−z (orthorhombic Pnma – c12, oP48). Their crystal chemistry can be understood within the Zintl–Klemm concept, and empirical ‘structure directing rules’ established by Nesper.10–12 In addition, the bonding characteristics of Ca3CuGe3 and, for comparison, the analogue Ca3Ag1+zGe3−z (x ∼ 1/3) reported previously, are investigated with the help of electronic band structure calculations obtained by density functional theory (DFT) using the LMTO code on idealized fully ordered models “Ca3MGe3” (Sc3NiSi3 type with M = Cu, Ag).13
Experimental
Materials and synthesis
The starting materials for the synthesis were the elements, Ca (granule, 99.5%), Ge (50 μ powder, 99.999%), Ag (60 μ powder, 99.9%), Cu (granules, 99.995+%), Cd (pieces, 99.9999%) and Mn (pieces, 99.98%) all from ABCR (Karlsruhe, Germany), which were stored in an Argon-filled glove-box and used as received. The mixtures (to give a total of ca. 400 mg, see below for stoichiometric ratios) of the elements were arc-sealed in Nb or Ta ampules under Ar atmosphere. Both elements were found to be suitable as container materials. The sealed Nb or Ta ampoules containing the mixtures were enclosed in fused silica glass tubes under a vacuum of ca. 10−2 mbar. The reactions were carried out inside a tubular furnace by slowly heating (60 °C per hour) to 980 °C and holding for one hour, then cooling to 870 °C at 2 °C min−1, and annealing for at least five days; at this point, the furnace was switched-off to allow the products to cool to room temperature.The reaction of the mixture Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu:Cd:Ge = 5:1:2:5, was designed to target the Cd/Cu analogue of the recently reported Ca5MgAgGe5.14 An excess amount of Cd was intended to compensate loss during synthesis because of its volatility. However, we obtained a ternary phase with the composition Ca3CuGe3 in high yield, indicating that Cd was acting essentially as a flux. Powder X-ray diffraction shows accompanying phases, but we were not able to identify them. For the Mn/Ag compound, a mixture with an atomic ratio Ca:Mn:Ag:Ge = 6:2:1:5 yielded the targeted structure Ca5MnxAg2−x+zGe5−z. However, the resulting moisture sensitive product is multiphasic with large amounts of the target compound, having lower Mn content than the starting mixture, but also binary phase impurities, CaGe and Ca5Ge3. Increasing the Ag content in the mixture to Ca:Mn:Ag:Ge = 6:2:2:5 yielded another homologue, Ca6MnxAg2−x+zGe6−z, with more extensive Ge/Ag mixing, and lower quality of the single crystals. Longer reaction times resulted essentially in thermodynamically more stable binary and ternary phases like CaGe and CaAgGe (TiNiSi-type),17 often Mn substituted, as the reaction product. Another mixture with nominal composition Ca:Mn:Ag:Ge = 12:5:3:11 yielded a product containing single crystal of both homologues Ca5MnxAg2−x+zGe5−z (block shape crystals) and Ca6MnxAg2−x+zGe6−z (needle shape crystals). Hence, quantitative syntheses of the pure title compounds were unsuccessful and their phase widths could not be precisely determined.
:Cu:Cd:Ge = 5:1:2:5, was designed to target the Cd/Cu analogue of the recently reported Ca5MgAgGe5.14 An excess amount of Cd was intended to compensate loss during synthesis because of its volatility. However, we obtained a ternary phase with the composition Ca3CuGe3 in high yield, indicating that Cd was acting essentially as a flux. Powder X-ray diffraction shows accompanying phases, but we were not able to identify them. For the Mn/Ag compound, a mixture with an atomic ratio Ca:Mn:Ag:Ge = 6:2:1:5 yielded the targeted structure Ca5MnxAg2−x+zGe5−z. However, the resulting moisture sensitive product is multiphasic with large amounts of the target compound, having lower Mn content than the starting mixture, but also binary phase impurities, CaGe and Ca5Ge3. Increasing the Ag content in the mixture to Ca:Mn:Ag:Ge = 6:2:2:5 yielded another homologue, Ca6MnxAg2−x+zGe6−z, with more extensive Ge/Ag mixing, and lower quality of the single crystals. Longer reaction times resulted essentially in thermodynamically more stable binary and ternary phases like CaGe and CaAgGe (TiNiSi-type),17 often Mn substituted, as the reaction product. Another mixture with nominal composition Ca:Mn:Ag:Ge = 12:5:3:11 yielded a product containing single crystal of both homologues Ca5MnxAg2−x+zGe5−z (block shape crystals) and Ca6MnxAg2−x+zGe6−z (needle shape crystals). Hence, quantitative syntheses of the pure title compounds were unsuccessful and their phase widths could not be precisely determined.
Powder X-ray diffraction
For the phase identification, powder X-ray diffraction (PXRD) data were collected on a Stoe StadiP diffractometer with a Ge(111) monochromator using CuKα1 radiation (λ = 1.54056 Å) equipped with a linear position sensitive detector (PSD).Energy-dispersive spectroscopy (EDS)
The chemical compositions of the same single crystals of the title compounds used to collect single crystal X-ray data were verified by scanning electron microscopy (SEM) using a field emission scanning electron microscope (JSM-7000F, JEOL, Japan) operating at 15 kV and equipped with an energy dispersive X-ray spectrometer EDX system (INCAx-sight, Oxford Instruments, UK). The analysis confirmed the presence of all elements. No other contaminant elements were detected, but quantitative evaluations were difficult because of the rapid decomposition of the single crystals when exposed to air during transfer to the microscope chamber.Single-crystal X-ray diffraction
For single crystal X-ray diffraction experiments, several crystals were selected from crushed samples, mounted on glass fibers, and sealed in glass capillaries inside an Argon filled glove-box. Single-crystal X-ray diffraction data collection at room temperature was performed for detailed structural analyses at ambient temperature on an Oxford Diffraction Xcalibur3 diffractometer with CCD detector, using graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å), operated at 50 kV and 40 mA, and a detector-to-crystal distance of 50 mm. Absorption correction based on a semi-empirical “multi-scan” approach was applied to the integrated reflections using the program CrysAlis RED.18 Additionally, some single-crystal X-ray diffraction (SXRD) measurements were performed on a Bruker D8 Venture diffractometer operating at 50 kV and 1 mA equipped with a Photon 100 CMOS detector, a flat graphite monochromator and a Mo Kα IμS microfocus source (λ = 0.71073 Å). The raw frame data were collected using the Bruker APEX3 software package,19 while the frames were integrated with the Bruker SAINT program19 using a narrow-frame algorithm for integration of the data, and were corrected for absorption effects via the multiscan method (SADABS).20 The atomic thermal factors were refined anisotropically for all positions. Charge flipping,21 as implemented in Superflip,22 was applied for structure solution and full-matrix least-squares refinement on F2 was carried out using the program SHELXL.23 For Ca3CuGe3, Cd mixing with Cu was assessed but the refined composition, Ca3Cd0.03(1)Cu0.97(1)Ge3, indicated no significant Cd inclusion within standard deviation. In the case of Ag/Mn or Ag/Ge mixed sites, the correct assignment could not be based on the electron density alone. We considered also the interatomic distances and, more important, the local coordination environment of the sites, to determine the element mixing with Ag. Anionic Ge atoms are supposed to prefer the site with coordination geometry closer to trigonal prismatic (mono-capped), while cationic Mn atoms are assumed to prefer the site closer to tetragonal geometry (albeit very distorted). This assessment is also in agreement with the interatomic distances, since in the final models, Mn/Ag–Ge distances are longer than Ge–Ge distances as expected.The crystallographic information including fractional coordinates and selected bond lengths of the compounds are listed in Tables 1–8.
| Empirical formula | Ca3CuGe3 | Ca6Mn0.67(1)Ag1.79(1)Ge5.54(1) |
|---|---|---|
| a w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Max (Fo2, 0) + 2 × Fc2)/3. | ||
| Formula weight | 403.14 | 872.53 |
| Crystal color and habit | Dark silver needle | Dark metallic needle |
| Temperature | 293(2) K | |
| Crystal system, space group | Monoclinic, C2/m (no. 12) | Monoclinic, P21/m (no. 11) |
| Unit cell parameters | a = 10.6701(1) Å | a = 10.8208(3) Å |
| b = 4.4452(1) Å; β = 110.73(1)° | b = 4.4979(1) Å; β = 110.01(3)° | |
| c = 14.2961(2) Å | c = 14.3546(4) Å | |
| Unit cell volume/Z | 634.18(2) Å3/4 | 656.48(3) Å3/2 |
| Density calculated | 4.206 g cm−3 | 4.41 g cm−3 |
| Abs. coeff. (Mo Kα) | 19.673 mm−1 (λ = 0.71073 Å) | 18.026 mm−1 (λ = 0.71073 Å) |
| F(000) | 740 | 796 |
| Crystal size | 0.10 × 0.03 × 0.03 mm3 | 0.08 × 0.02 × 0.02 mm3 |
| Theta range | 1.523–36.53° | 3.769–32.14° |
| Index range | −17 ≤ h ≤ 17, −7 ≤ k ≤ 7,−23 ≤ l ≤ 23 | −12 ≤ h ≤ 16, −6 ≤ k ≤ 5,−21 ≤ l ≤ 19 |
| Reflections collected | 15281 (Rσ = 0.019) |
6027 (Rσ = 0.034) |
| Independent reflections | 1689 (Rint = 0.032) | 2349 (Rint = 0.029) |
| Data completeness to theta = 25.242° | 99.8% | 98.7% |
| Absorption correction | Empirical | Empirical |
| Parameters | 44 | 89 |
| Goodness-of-fit on F2 | 1.061 | 1.058 |
| Observed reflns [I > 2σ(I)] | 1549 | 1819 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0210/wR2 = 0.0587 | R 1 = 0.0508/wR2 = 0.1320 |
| Final R indices (all data) | R 1 = 0.0244/wR2 = 0.0599 | R 1 = 0.0642/wR2 = 0.1370 |
| Weighting parametersa | a = 0.0309/b = 3.6501 | a = 0.0691/b = 5.2963 |
| Extinction coefficient | 0.0056(3) | 0.0084(7) |
| Residual map (e− Å−3) | 1.475/−0.706 | 3.009/−1.963 |
| Atom | Site | x | y | z | U eq. (Å−2) |
|---|---|---|---|---|---|
| Cu1 | 4i | 0.74968(4) | 0 | 0.10417(3) | 0.0115(1) |
| Ge1 | 4i | 0.12928(3) | 0 | 0.04363(2) | 0.0086(1) |
| Ge2 | 4i | 0.95812(3) | 0 | 0.26702(2) | 0.0077(1) |
| Ge3 | 4i | 0.89123(3) | 0 | 0.42438(2) | 0.0072(1) |
| Ca1 | 4i | 0.45705(6) | 0 | 0.11593(4) | 0.0097(1) |
| Ca2 | 4i | 0.25301(6) | 0 | 0.27115(4) | 0.0086(1) |
| Ca3 | 4i | 0.60001(6) | 0 | 0.42058(4) | 0.0081(1) |
Electronic structure calculations
For the isostructural analogues Ca3CuGe3 and Ca3Ag1+zGe3−z, the electronic structures and the chemical bonding were investigated on the basis of the density-functional theory (DFT) using the tight-binding linear-muffin-tin-orbital (TB-LMTO-ASA) approach and the local-density approximation (LDA) within the program LMTO47c.24 Since the crystal structure of Ca3Ag1+zGe3−z exhibits one mixed occupied Ge/Ag site, a hypothetical ordered model “Ca3AgGe3” was used for calculations. In this model the Ge/Ag mixed site is fully occupied by Ge which is the major component [exp. 0.33(1) Ag]. The radii of the muffin-tin spheres were determined by an automatic procedure.25 The k-space integration was performed by the tetrahedron method on a set of 325 irreducible k points and a basis set with Ca 4s/(4p)/3d, Ge 4s/4p/(3d), Cu 4s/4p/3d and Ag 5s/5p/4d (down-folded orbitals in parentheses).26 Crystal orbital Hamilton populations (COHPs)27 were used for the analysis of relative bond strengths. The Fermi level in all figures is taken as the reference (0 eV) energy level. The COHP curves are drawn by reversing their values with respect to the energy scale (i.e., –COHP vs. E). Since the COHP is an energy partitioning function, negative/positive values indicate bonding/antibonding interactions.| Atom | Site | Occupancy | x | y | z | U eq. (Å−2) |
|---|---|---|---|---|---|---|
| Ge1 | 2e | 1 | 0.1296(1) | ¼ | 0.9562(1) | 0.011(1) |
| Ge2/Ag2 | 2e | 0.54/0.46(1) | 0.4903(1) | ¼ | 0.8814(1) | 0.021(1) |
| Ge3 | 2e | 1 | 0.2764(1) | ¼ | 0.7230(1) | 0.010(1) |
| Ge4 | 2e | 1 | 0.3552(1) | ¼ | 0.5726(1) | 0.009(1) |
| Ge5 | 2e | 1 | 0.1426(1) | ¼ | 0.4237(1) | 0.010(1) |
| Ge6 | 2e | 1 | 0.2162(1) | ¼ | 0.2704(1) | 0.011(1) |
| Ag1/Mn1 | 2e | 0.68/0.32(1) | 0.3908(1) | ¼ | 0.0404(1) | 0.024(1) |
| Ag3/Mn3 | 2e | 0.65/0.35(1) | 0.0063(1) | ¼ | 0.1017(1) | 0.020(1) |
| Ca1 | 2e | 1 | 0.7811(2) | ¼ | 0.8801(1) | 0.014(1) |
| Ca2 | 2e | 1 | 0.8542(2) | ¼ | 0.4250(1) | 0.010(1) |
| Ca3 | 2e | 1 | 0.5098(2) | ¼ | 0.2767(1) | 0.011(1) |
| Ca4 | 2e | 1 | 0.7154(2) | ¼ | 0.1209(1) | 0.012(1) |
| Ca5 | 2e | 1 | 0.9905(2) | ¼ | 0.7279(1) | 0.011(1) |
| Ca6 | 2e | 1 | 0.6482(2) | ¼ | 0.5793(1) | 0.010(1) |
| Empirical formula | Ca5Mn0.64(1)Ag1.41(1)Ge4.95(1) | Ca5Mn0.71(1)Ag1.33(1)Ge4.96(1) |
|---|---|---|
| a w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Max (Fo2, 0) + 2 × Fc2)/3. | ||
| Formula weight | 747.16 | 742.74 |
| Crystal color and habit | Dark metallic regular block | Dark metallic regular block |
| Temperature | 293(2) K | 293(2) K |
| Crystal system/space group | Orthorhombic, Pnma (no. 62) | Orthorhombic, Pnma (no. 62) |
| Unit cell parameters | a = 22.9432(9) Å, | a = 23.0029(7) Å, |
| b = 4.4763(2) Å, | b = 4.4744(1) Å, | |
| c = 10.8109(4) Å | c = 10.8489(3) Å | |
| Unit cell volume/Z | 1110.29(8) Å3/4 | 1116.61(5) Å3/4 |
| Density calculated | 4.47 g cm−3 | 4.418 g cm−3 |
| Abs. coeff. (Mo Kα) | 18.589 mm−1 (λ = 0.71073 Å) | 18.454 mm−1 |
| F(000) | 1363 | 1356 |
| Crystal size | 0.08 × 0.06 × 0.04 mm3 | 0.06 × 0.05 × 0.02 mm3 |
| Theta range | 3.77–32.30° | 2.075–42.856° |
| Index range | −33 ≤ h ≤ 33, −6 ≤ k ≤ 6,−11 ≤ l ≤ 15 | −34 ≤ h ≤ 43, −8 ≤ k ≤ 4,−20 ≤ l ≤ 20 |
| Reflections collected | 12853 (Rσ = 0.0324) |
27200 (Rσ = 0.0278) |
| Independent reflections | 2068 (Rint = 0.036) | 4313 (Rint = 0.0371) |
| Data completeness to theta = 25.242° | 99.6% | 98.9% |
| Absorption correction | Multi scan | Multi scan |
| Parameters | 77 | 77 |
| Goodness-of-fit on F2 | 1.110 | 1.016 |
| Observed reflns [I > 2σ(I)] | 1598 | 3610 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0289/wR2 = 0.0576 | R 1 = 0.0235/wR2 = 0.0408 |
| Final R indices (all data) | R 1 = 0.0453/wR2 = 0.0600 | R 1 = 0.0342/wR2 = 0.0442 |
| Weighting parametersa | a = 0.0282/b = 1.4157 | a = 0.0097/b = 1.2192 |
| Extinction coefficient | 0.0026(1) | 0.0017(1) |
| Residual map (e− Å−3) | 1.486/−1.031 | 1.374/−1.949 |
Results and discussion
Synthesis and characterization
All compounds were obtained by reactions of the corresponding elements at elevated temperatures under protective argon atmosphere. All attempts toward the phase-pure synthesis of the title compounds were unsuccessful, and the products typically consist of mixtures of the targeted intergrowth structures with diverse phases, including the simple parent structures, mainly CaGe, as well as for Ca2+nMnxAg2−x+zGe2+n−z (n = 3, 4) ternary Mn-substituted CaAgGe,17 and unreacted Mn. It is likely that the intergrowth phases, and the neat parent structures are located on an extremely flat energy surface and may be in or close to thermal equilibrium. Since these intergrowth structures are defined by the rate of TM (Ag, Cu, Mn) intercalation into the pristine CaGe structure, the equilibrium principle may apply to the starting mixtures for a given reaction temperature. This should explain why it is difficult to prepare a target compound from the stoichiometric mixture. The quantitative synthesis of corresponding Ca/(Li, Mg)/Ge phases was also challenging.10–12 As their structures are so closely related, it is difficult to distinguish the homologous structures between each other or with their parent-structures from X-ray powder diffraction patterns. Therefore, the characterization of our structures is limited essentially to single crystal diffraction and qualitative chemical composition by EDS. The samples are also exceptionally sensitive to air and moisture and decompose to a dark red powder with the typical smell of germanes.| Atom | Site | Occupancy. | x | y | z | U eq. (Å−2) |
|---|---|---|---|---|---|---|
| Ge1 | 4c | 1 | 0.97467(2) | ¼ | 0.34305(2) | 0.01199(5) |
| Ge2/Ag | 4c | 0.965/0.035(3) | 0.92363(2) | ¼ | 0.95475(2) | 0.01150(6) |
| Ge3 | 4c | 1 | 0.83476(2) | ¼ | 0.09713(2) | 0.00962(4) |
| Ge4 | 4c | 1 | 0.74756(2) | ¼ | 0.94970(2) | 0.00940(4) |
| Ge5 | 4c | 1 | 0.65693(2) | ¼ | 0.08948(2) | 0.01040(4) |
| Ag1/Mn1 | 4c | 0.81/0.19(1) | 0.02113(2) | ¼ | 0.11150(2) | 0.01560(5) |
| Mn2/Ag2 | 4c | 0.52/0.48(1) | 0.55418(2) | ¼ | 0.96359(2) | 0.01589(6) |
| Ca1 | 4c | 1 | 0.07236(2) | ¼ | 0.83745(4) | 0.01239(7) |
| Ca2 | 4c | 1 | 0.92913(2) | ¼ | 0.67087(4) | 0.01258(7) |
| Ca3 | 4c | 1 | 0.16343(2) | ¼ | 0.11771(4) | 0.01026(6) |
| Ca4 | 4c | 1 | 0.34210(2) | ¼ | 0.11452(4) | 0.01073(6) |
| Ca5 | 4c | 1 | 0.25391(2) | ¼ | 0.83987(4) | 0.01041(6) |
| Atom pairs | Distances | –iCOHP | Atom pairs | Distances | –iCOHP | ||
|---|---|---|---|---|---|---|---|
| Cu1 | –Ge1 (×2) | 2.5598(2) | 1.98 | Ge1 | –Ge1 | 2.5945(6) | 2.38 |
| –Ge2 | 2.5886(5) | 1.95 | –Cu1 (×2) | 2.5598(2) | |||
| –Ge1 | 2.8398(5) | 1.18 | –Cu1 | 2.8398(5) | |||
| –Ca1 (×2) | 3.0985(5) | 0.32 | –Ca2 | 3.0468(6) | 0.78 | ||
| –Ca1 | 3.1390(7) | 0.28 | –Ca1 (×2) | 3.0829(5) | 0.70 | ||
| –Ca1 | 3.1858(7) | 0.44 | –Ca1 (×2) | 3.2757(5) | 0.58 | ||
| –Ca2 (×2) | 3.2526(5) | 0.36 | –Ca1 | 3.2773(7) | 0.43 | ||
| Ge2 | –Ge3 | 2.5897(5) | 2.30 | Ge3 | –Ge3 | 2.5516(6) | 2.45 |
| –Cu1 | 2.5886(5) | –Ge2 | 2.5897(5) | ||||
| –Ca1 (×2) | 3.0965(5) | 0.91 | –Ca2 (×2) | 3.1027(4) | 0.84 | ||
| –Ca3 (×2) | 3.1143(5) | 0.88 | –Ca3 | 3.0885(7) | 1.07 | ||
| –Ca2 | 3.1261(7) | 0.99 | –Ca3 (×2) | 3.1169(5) | 0.86 | ||
| –Ca2 (×2) | 3.1344(5) | 0.66 | –Ca3 (×2) | 3.1609(5) | 0.62 | ||
Crystal chemistry
During extended investigations of multinary AE/M/Tt phases of Si and Ge (Tt) with heavier alkaline-earth elements (AE = Ca, Sr, Ba) and smaller, highly polarizing (M = Li or Mg), Nesper et al. empirically derived a set of “structure-directing rules” that determine very different roles of the cations in these often complex structures.10,11 According to these rules: (i) the Zintl anions are always planar (when comprising three or more atoms) and are ecliptically stacked; (ii) the central atoms of the Zintl anions are always coordinated by a trigonal prism mainly formed by the larger cations (AE); (iii) the terminal atoms of the Zintl anions are always coordinated by a square antiprism of cations AE and M, with the outer periphery formed by lighter cations M; and (iv) the highly polarizing, small cations M determine the number of highly charged terminal groups in the Zintl anions. In our previous reports on the homologous series Ca2+nM2+zGe2+n−z (M = Mg, Ag),13–16 we demonstrated that new TMZPs can be obtained by partial to complete replacement of the small polarizing s-block metal Li with isovalent transition metal Ag, while the ‘structure-directing rules’ remain applicable. However, whilst the chemical composition and crystal structure of the targeted TMZPs did not match that of the transition metal-free parent compounds, the same type of planar Zintl anions were observed.The three title compounds are new members of the homologous series Ca2+nM2+zGe2+n−z with M = Cu, Ag and Mn. Hence, they are alkaline-earth (AE) analogues of the homologous series with general formula R2+nT2X2+n, which was first rationalized by Zhao and Parthé (1989), and described with rare-earth (RE) and early TM (group 3) metals.28 Within the Zintl–Klemm concept, a formal charge transfer is assumed from the active metal Ca to the anionic network according to (Ca2+)(2+n)[M2+zGe2+n−z)]2(2+n)−.
| Atom pair (×n) | Distance/Å | Atom pair (×n) | Distance/Å | ||
|---|---|---|---|---|---|
| Ge1 | –M3 (×2) | 2.660(1) | Ge2/Ag2 | –Ge3 (×1) | 2.632(1) |
| –M1 (×1) | 2.667(1) | –M1 (×2) | 2.643(1) | ||
| –M3 (×1) | 2.838(1) | –M1 | 2.838(1) | ||
| –Ca5 | 3.103(2) | –Ca1 | 3.154(2) | ||
| –Ca1 (×2) | 3.156(1) | –Ca4 (×2) | 3.156(1) | ||
| –Ca4 (×2) | 3.219(1) | –Ca3 (×2) | 3.195(1) | ||
| –Ca1 | 3.549(2) | –Ca4 | 3.467(2) | ||
| Ge3 | –Ge4 | 2.579(1) | Ge4 | –Ge5 | 2.548(1) |
| –Ca2 (×2) | 3.085(1) | –Ca3 (×2) | 3.116(1) | ||
| –Ca5 | 3.118(2) | –Ca6 (×2) | 3.124(1) | ||
| –Ca4 (×2) | 3.154(1) | –Ca6 | 3.139(2) | ||
| –Ca3 (×2) | 3.226(1) | –Ca2 (×2) | 3.201(1) | ||
| Ge5 | –Ge6 | 2.583(1) | M1 | –Ca4 (×2) | 3.150(1) |
| –Ca2 (×2) | 3.118(1) | –Ca3 | 3.190(2) | ||
| –Ca5 (×2) | 3.119(1) | –Ca4 | 3.301(2) | ||
| –Ca2 | 3.128(2) | –Ca1 (×2) | 3.358(2) | ||
| –Ca6 (×2) | 3.202(1) | M3 | –Ca1 (×2) | 3.163(1) | |
| Ge6 | –M3 | 2.694(1) | –Ca4 | 3.252(2) | |
| –Ca6 (×2) | 3.114(1) | –Ca1 | 3.280(2) | ||
| –Ca1 (×2) | 3.125(1) | –Ca5 (×2) | 3.314(1) | ||
| –Ca3 | 3.147(2) | ||||
| –Ca5 (×2) | 3.178(1) | Ca(2) | Ca(3) | 3.604(2) | |
| Atom pair (×n) | Distance/Å | Atom pair (×n) | Distance/Å | ||
|---|---|---|---|---|---|
| Ge1 | –M2 (×2) | 2.6709(4) | Ge2/Ag | –Ge3 (×1) | 2.556(1) |
| –M1 (×1) | 2.7177(7) | –M1 (×2) | 2.668(1) | ||
| –M2 (×1) | 2.7876(7) | Ge2 | –M1 | 2.813(1) | |
| –Ca4 | 3.079(1) | –Ca2 | 3.081(1) | ||
| –Ca2 (×2) | 3.146(1) | –Ca3 (×2) | 3.104(1) | ||
| –Ca1 (×2) | 3.165(1) | –Ca1 (×2) | 3.168(1) | ||
| Ge3 | –Ge4 | 2.558(1) | Ge4 | –Ge5 | 2.570(1) |
| –Ca5 (×2) | 3.102(1) | –Ca3 (×2) | 3.115(1) | ||
| –Ca4 | 3.124(1) | –Ca4 (×2) | 3.117(1) | ||
| –Ca1 (×2) | 3.171(1) | –Ca5 | 3.130(1) | ||
| –Ca3 (×2) | 3.224(1) | –Ca5 (×2) | 3.194(1) | ||
| Ge5 | –M2 | 2.717(1) | M1 | –Ca1 (×2) | 3.145(1) |
| –Ca2 (×2) | 3.113(1) | –Ca1 | 3.189(1) | ||
| –Ca5 (×2) | 3.124(1) | –Ca2 (×2) | 3.437(1) | ||
| –Ca4 (×2) | 3.145(1) | M2 | –Ca2 (×2) | 3.186(1) | |
| –Ca3 | 3.166(1) | –Ca2 | 3.225(1) | ||
| M2 | –Ca4 (×2) | 3.362(1) | –Ca1 | 3.277(1) | |
The ternary phase Ca3CuGe3 (thereafter, 4_Cu) is isostructural with the previously reported Ag analogue Ca3Ag1+zGe3−z (4_Ag), crystallizing with the Sc3NiSi3 type structure, corresponding to the n = 4 member of the homologous series (for that reason compound identifiers like 4_Cu were chosen).13 Perhaps because of the small difference in atomic scattering factors between Cu and Ge atoms, Cu/Ge partial mixing similar to Ag/Ge in the latter could not be detected. Since CaCuGe and CaAgGe are isostructural (CaAuGe type), monovalent Cu+ is assumed and, consequently, these partial mixings are expected from the Zintl–Klemm electron counting approach with x = 1/3 ideally. A perspective view of the crystal structure is provided in Fig. 1a, showing a linear intergrowth of two chemically distinct domains corresponding to fragments cut from the parent structures, CaCuGe (ref. 29) and CaGe.30 The anionic substructure of 4_Cu consists of [Ge4]10− tetramers in CrB-related slabs and [Ge2]6− dumbbells in AlB2-related slabs with Cu atoms located at the interface. The Zintl–Klemm concept results in an ionic formulation 2 Ca3CuGe3 ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) (Ca2+)6(Cu+)2{[Ge4]10−[Ge2]6−}, indicating that the system is apparently one electron short per Ca3CuGe3 formula unit. However, with divalent Cu2+ it would be electron precise according to 2 Ca3CuGe3 (Ca2+)6(Cu2+)2{[Ge4]10−[Ge2]6−}, satisfying the Zintl–Klemm electron counting scheme. However, the true electronic bonding situation might be far from this extreme formulation, as frequently observed for Zintl phases.
(Ca2+)6(Cu+)2{[Ge4]10−[Ge2]6−}, indicating that the system is apparently one electron short per Ca3CuGe3 formula unit. However, with divalent Cu2+ it would be electron precise according to 2 Ca3CuGe3 (Ca2+)6(Cu2+)2{[Ge4]10−[Ge2]6−}, satisfying the Zintl–Klemm electron counting scheme. However, the true electronic bonding situation might be far from this extreme formulation, as frequently observed for Zintl phases.
 | ||
| Fig. 1 Perspective view of the homologous structures 4_Cu and 4_Mn along approximately the monoclinic b-axis: (a) Ca3CuGe3, with [Ge2]6− dimers and [Ge4]10− tetramers, in the two different blocks, and (b) its Mn-substituted derivative Ca6MnxAg2−x+zGe6−z with isolated Ge4− entities in TiNiSi related slabs, and coexistence of [Ge4]10−/[Ge5]12− oligomers in the CrB related slabs due to Ge/Ag mixing. The thermal ellipsoids are at 99% probability level. | ||
In the Ag analogue Ca3Ag1+zGe3−z (z = 1/3), partial Ag/Ge mixing in Ge2 dimers is ascribed to charge balancing,13 but similar Cu/Ge mixing will be hard to determine by X-ray diffraction due to little difference in atomic form factors. As listed in Table 6, the Ge–Ge bond lengths in 4_Cu (2.552 to 2.595 Å) are close to the value in the parent-structure CaGe (dGe–Ge = 2.592 Å), and similar Ge–Ge distances (2.54 to 2.59 Å) observed in the analogue 4_Ag.13 Also, the Cu–Ge distances in 4_Cu (2.56 to 2.84 Å) are in the same range as in CaCuGe (2.51 to 2.83 Å).29
Ca6MnxAg2−x+zGe6−z (thereafter, 4_Mn) represents a new structure type (P21/m), which is a hettotype of the Sc3NiSi3-type (C2/m). In fact, it is best viewed as a Mn-substituted derivative of Ca3Ag1+zGe3−z (thereafter, 4_Ag). Upon partial replacement of monovalent Ag by divalent Mn, an alteration of the structure occurs: the Ge–Ge bond of the expected Ge2 dumbbells in the TiNiSi-related slabs are broken up by the additional valence electrons (Fig. 1b). A klassengleiche symmetry reduction of index two (k2) from C2/m to P21/m originates from switching one Ge position with M (Mn, Ag) atoms. A direct group-subgroup tree from C2/m to P21/m is provided in the ESI.‡31,32 As a result, there are two distinct Ge positions in the TiNiSi-related slabs, of which the one at the slab's interface is mixed with Ag atoms (Fig. 2). In addition, two M positions result (Table 3), surprisingly with nearly the same Ag/Mn ratio of roughly 2/1. Albeit much lower values of Mn occupancy were expected at the central M1 position (corresponding to the Ge position in 4_Ag) because of unfavourable Ag–Mn interactions. In fact, a similar Mn for Ag substitution was attempted in the parent structure CaAgGe, yielding very modest 7% Mn inclusion (CaMn0.07Ag0.93Ge); however with strict site preference to avoid Mn–Ag direct contacts in the structure. In the case of the 4_Mn structure, only a modest trend in the expected direction is visible experimentally. As shown in the perspective view of the crystal structure of Ca6MnxAg2−x+zGe6−z (4_Mn) provided in Fig. 1b, the TiNiSi-related slabs consist of ‘isolated’ Ge4− only. Meanwhile, the interface between CrB- and TiNiSi-related slabs consists of both Ge/Ag and Ag/Mn mixed positions. In comparison, only Ag (or Cu) atoms are found at the interface in their Sc3NiSi3 structures, which feature Ge2 dumbbells (Fig. 1a). The occurrence of Ge/Ag mixing at the interface is an unprecedented feature in the crystal chemistry of the series Ca2+nM2+zGe2+n−z series, and may be important for the targeted synthesis of new members of the homology.
 | ||
| Fig. 2 Structural details of 4_Mn emphasizing the atomic local ordering model; Zintl anions include [Ge]4− monomers, and the co-existence of [Ge5]12−/[Ge4]10− pentamer/tetramers due to Ge/Ag (54/46) mixing at one terminal Ge2 position; rather similar Ag/Mn ratio (M1 = 68/32) and (M3 = 65/35). The atom numbering is according to Table 3. | ||
In fact, similar complete replacement of one Ge atom by Mg was described for the Zintl phase Eu8Mg16Ge12 in which the star-like 26-electron moiety [Ge4]10− is replaced by [MgGe3]10− units.33 Since the Ge/Ag ratio (0.54/0.46(1)) in 4_Mn is close to 50% within standard deviation, we can assume a local ordering according to the “coloring problem” approach as Ca6Mn0.67Ag1.79Ge5.54 0.54[Ca6Mn0.67Ag1.33Ge6] (with Ge at the interface) + 0.46[Ca6Mn0.67Ag2.33Ge5] (with Ag at the interface) as schematised in Fig. 2. Hence, the anionic substructure consists of a mixture of [Ge5]12− pentamers and [Ge4]10− tetramers in CrB-related slabs, while only isolated Ge4− are found in the TiNiSi-related slabs. As such, 4_Mn may be viewed as an unanticipated intermediate between n = 4 and n = 5 members of the structure series Ca2+nM2+zGe2+n−z. Additionally, it demonstrates the intricate process of aliovalent replacement of monovalent Ag by divalent Mn, while maintaining the structural architecture of the system. Thus, the insertion of Mn atoms not only results in displacement of Ag atoms, it also triggers simultaneous displacement of Ge by Mn and Ag atoms (breaking-up the Ge2 dumbbells), and unexpected displacement of Ag by Ge atoms at the interface.
Hence, the response of Mn insertion may provide some useful insights into the poorly understood interplay between intercalation and conversion processes in intermetallic battery materials.34 Interestingly, all these structural and chemical alterations are strictly localized in the TiNiSi-related slabs, while the CrB-related slabs remain undisturbed. The Zintl–Klemm electron-counting scheme offers some clues to understand the driving forces behind the structural alterations. The corresponding ionic formulation is as follows: (Ca2+)6(Mn2+)0.67[Ge4−]{Ag+1.33[Ge5]12−}0.54{Ag+2.33[Ge4]10−}0.46, indicating that the phase is roughly charge balanced within standard deviation for the refined occupancies. However, the fact that two Ag/Mn mixed positions have similar occupancies, despite very different coordination environments (see Fig. 2), can hardly be ascribed to similar atomic sizes between Ag (1.44 Å) and Mn (1.37 Å) atoms as compared to the significantly larger Mg (1.60 Å).35 The elongated thermal ellipsoid on the Ag3/Mn3 position with direct interactions to the Ge2/Ag2 position is a clear sign that Ag–Mn direct interactions are not thermodynamically driven, and are rather unfavourable. Since X-ray diffraction offers an average picture of the atomic ordering, we could use the “coloring problem” approach to construct local ordering models that avoid or, at least, minimize Ag–Mn direct interactions.36 The Mn maximum occupancy is 35%, whereas Ge is 54% at the mixed position, so that favourable Ge–Mn and Ag–Ag interactions can be realized almost exclusively, while thermodynamically unfavourable Ag–Mn direct interactions can be considered merely incidental, meaning kinetically driven. Selected bond distances in 4_Mn structure are listed in Table 7, the Ge–Ge bond lengths (2.548 to 2.583 Å) are very close to the value in the Mn-free structure Ca3Ag1+xGe3−x (2.546 to 2.588 Å).13 The Ge–M distances are longer as expected, while Ca–Ge and Ca–M interactions are longest.
Orthorhombic Ca5MnxAg2−x+zGe5−z (thereafter, 3_Mn) is isostructural to the previously reported Ca5MgAgGe5.14 From the refinement of two single crystals (Table 4), obtained from different batches, we observed significantly higher Ag content overall, and slight Ge/Ag mixing (roughly 4 to 5% Ag occupancy) at the interface, not observed in the Mg analogue. The Ge/Ag mixing is quite marginal in 3_Mn when compare to the 4_Mn case (vide supra). Like the Mg analogue, the 3_Mn structure corresponds to the n = 3 member of the homology and it features isolated Ge4− and tetrameric [Ge4]10− Zintl anions, instead of the expected [Ge3]8− trimeric chain (Fig. 3). A more detailed crystal structure and chemical bonding description of the 3_Mn structure may be found in the report on the prototype Ca5MgAgGe5.14 The two Ag/Mn positions in 3_Mn have significantly different atomic ratios indicating differential fractional site occupancy, with Mn preferring the Mg site in the prototype as expected. The Ag/Mn mixing at the Ag position is comparatively smaller (17 to 20% of Mn) than the Ag/Mg mixing (22% Mg at Ag position) in the Mg analogue. Considering the refined compositions (Ca5Mn0.71(1)Ag1.33(1)Ge4.96(1) and Ca5Mn0.64Ag1.41Ge4.95(1)), if one neglects Ge/Ag mixing (4–5% Ag) at the interface at first approximation, the closed-shell isolated Ge4− and tetrameric [Ge4]10− units result in the ionic formulation (Ca2+)5(Mn2+)x(Ag+)2−x[Ge4−][Ge4]10− (x = 0.64 to 0.71). This suggests that the system is 1.29 to 1.36 valence electron (ve) deficient per formula unit. In comparison, the Mg analogue Ca5MgAgGe5 is one valence electron short, but, surprisingly, DFT band structure calculations indicated that the bonding is fully optimized within its anionic substructure.14
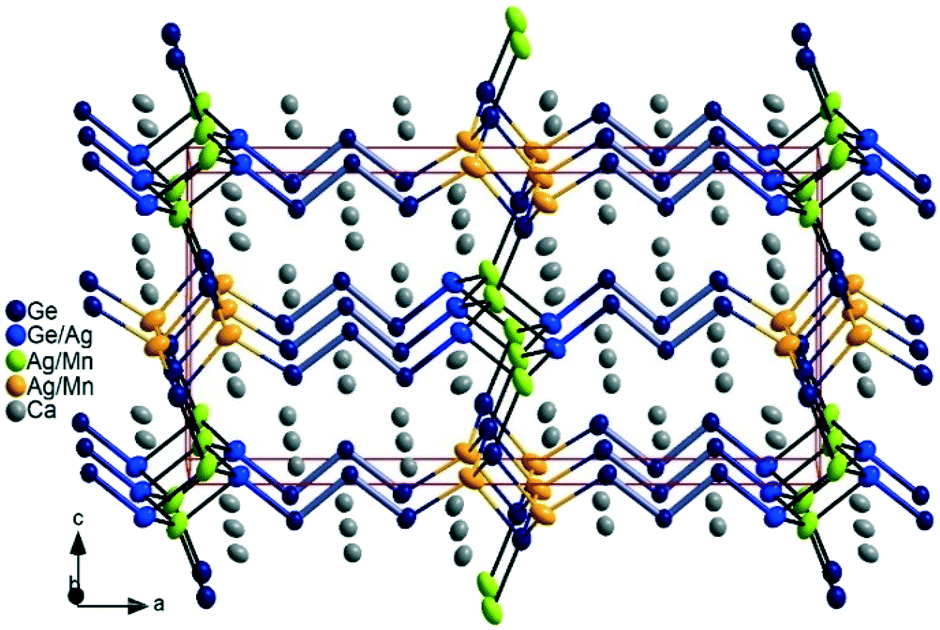 | ||
| Fig. 3 Perspective view of the Ca5MnxAg2−x+zGe5−z structure in approximately b-direction, emphasizing the Zintl anions [Ge]4− monomers, and [Ge4]10− tetramers showing slight Ge/Ag mixing at one terminal Ge position. The thermal ellipsoids are at 99% probability level. | ||
Hence, weak Ge/Ag mixing at the interface is possibly an attempt to reduce the electron shortage to the ‘ideal’ value of one electron. Indeed, in this marginal scenario, when Ag replaces Ge atoms at the interface, the ionic formulation becomes (Ca2+)5(Mn2+)0.64(Ag+)2.36(Ge4−)[Ge3]8−, resulting in rather 1.64 ve excess per formula unit. Unlike the 4_Mn system, unfavourable Mn–Ag direct interactions are minimized through differential Ag/Mn site occupancy in the 3_Mn structure. Selected bond distances in 3_Mn structure are listed in Table 8, the Ge–Ge bond (2.556 to 2.558 Å) and Ge–M distances are very close to the values in the 4_Mn (see Table 7). In the isostructural phase Ca5MgAgGe5, Ge–Ge distances (2.557 to 2.581 Å) are very comparable.
As depicted in Fig. 4, various oligomeric germanides [Gen](2n+2)− with n = 1–5 are observed in the title compounds, often simultaneously. These Zintl-anions are always ecliptically stacked and bridged by small cationic M (Cu, Ag, Mn) atoms located at the interface. Hence, these M metals act as highly polarizing cations coordinating only terminal Ge atoms of the Zintl-anions. This results in 3D open anionic frameworks [M2+zGe2+n−z)]2(2+n)− with large channels that are filled with (2n + 2)Ca atoms, forming the typical trigonal prismatic coordination of central Ge atoms and square antiprismatic coordination at terminal Ge atoms. This means that all title structures follow Nesper's ‘structure directing rules’.10,11 Close structural relationships between the homologous phases are also nicely revealed, but with [Ge2]6− dimers observed only in 4_Cu in the AlB2-related slabs. Thus, on the one hand, the 4_Mn homologue can be derived from 4_Cu by simply breaking down those dimers and switching the position of one Ge atom with one M atom. On the other hand, one can derive 4_Mn from 3_Mn by just increasing the length of the [Gen](2n+2)− oligomer in the CrB-type slabs from n = 3 to n = 4. In fact, only [Ge2]6− dimers are observed in the AlB2-type related slabs in RE analogues of the homology RE2+nT2X2+n.
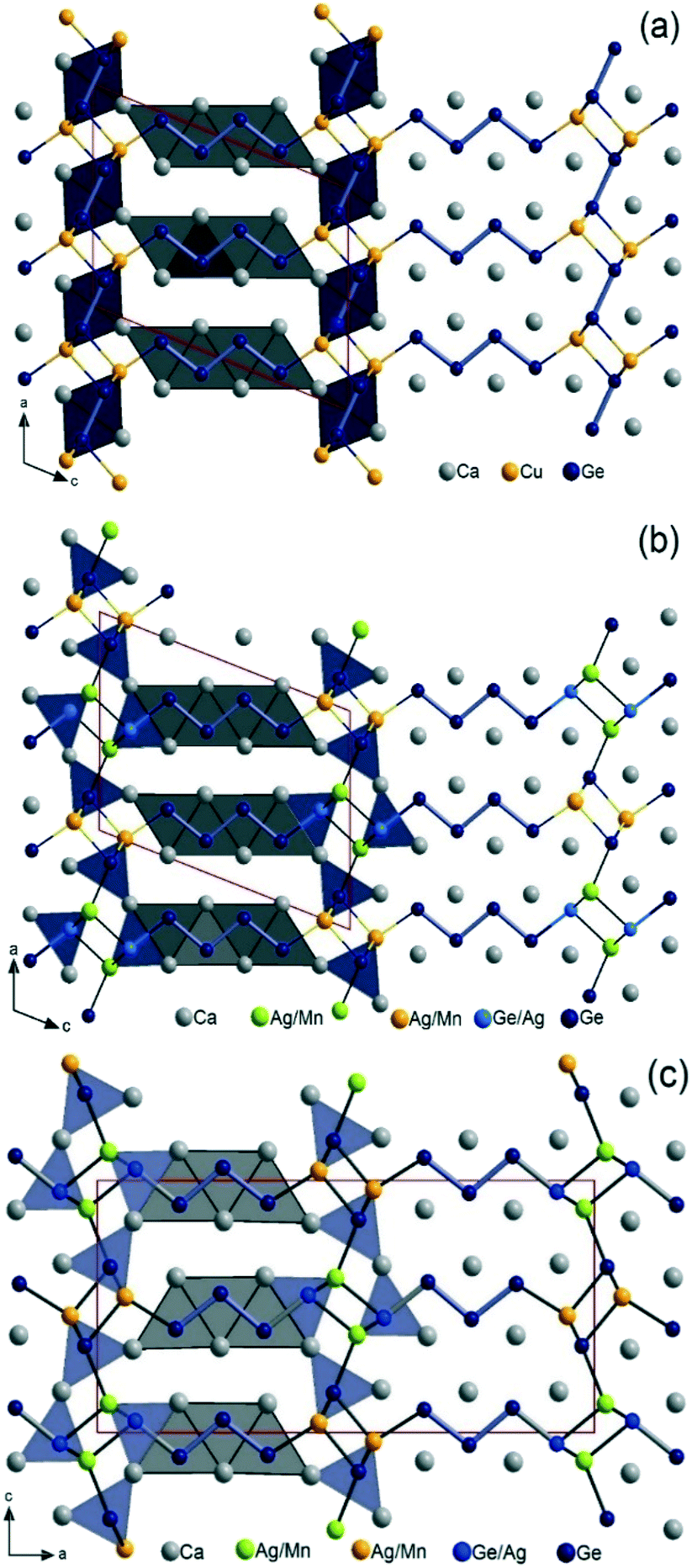 | ||
| Fig. 4 Combined ball-and-stick and polyhedral views of the projected structures along the b-direction, showing the relationship between the different homologues (a) Ca3CuGe3 (C2/m, n = 4), (b) Ca6MnxAg2−x+zGe6−z (P21/m, n = 4.5), and (c) Ca5MnxAg2−x+zGe5−z (Pnma, n = 3). | ||
Hence, the series Ca2+nM2+zGe2+n−z (M = Cu, Ag, Mg, Mn) demonstrates a high level of structural responsiveness to chemical substitution, that may have huge influences on magnetic exchange interactions when paramagnetic elements are involved. With the discovery of ferromagnetism in p-type Mn-doped semiconductors, often referred as diluted magnetic semiconductors (DMSs) and dilute magnetic oxides (DMOs), much attention is being paid to the possible applications of diluted magnetic systems for spintronics.37 Still, a more systematic understanding of the spin–spin interactions is crucial for any future development of spintronic devices. In this context, selected tetrelide Zintl phases involving paramagnetic transition metals like Mn may be interesting systems, because they contain local magnetic moments, which can interact via the extended π states of the polyanions, giving rise to a plethora of interesting phenomena. Therefore, we are now planning to extend our investigations to divalent rare-earth like Eu and Yb. An interesting prospect will be to achieve coupling between structural and magnetic degrees of freedom in those systems, a coupling which is crucial for realization of interesting physical phenomena associated with magneto-structural transformations.38
Electronic structures and bonding
The electronic structures of the two isostructural analogues Ca3MGe3 (M = Cu, Ag) were calculated using the LMTO code on hypothetical fully ordered models. The DOS (density of states) and COHP (crystal orbital Hamilton population) plots for both phases are depicted in Fig. 5. The DOS plots are virtually identical; the most dissimilar features are mainly ascribed to the metal M valence d-orbitals. While Ag-4d orbitals are highly localized below the valence band with enhanced soft-core character, the Cu-3d orbitals fall within the valence band and remain reasonably localized (narrow) as well. The absences of energy gaps at the Fermi level indicate metallic properties just as the parent structures CaAgGe and CaGe are all metallic.13,39 The Fermi levels are very close to and just below local minima of the DOS (pseudo-gaps), which may indicate narrow electron-deficient systems as predicted by the Zintl–Klemm concept. Interestingly, the Ca contribution to the occupied region of the DOS is very significant, and becomes largely dominant above the pseudo-gap in agreement with incomplete formal valence electron transfer. The Zintl–Klemm concept predicts the compounds to be charge balanced for 30 ve per f.u. The ordered models “Ca3MGe3” correspond to 29 ve per f.u, and, within the rigid band approximation, the pseudo-gap should be close to 0.56 eV according to IDOS. The pseudo-gap is close to 0.56 eV indeed, and the COHP curves indicate that it corresponds to optimized bonding within the anionic framework (MGe3), but resulting from a combination of Ge–Ge antibonding and Ag–Ge bonding interactions. According to COHP curves of the Ge–Ge bonds, antibonding states start well below EF, which agrees with the reported electronic structure of [Ge4]10− with an antibonding HOMO (πp*) level. As discussed previously for the homologue Ca7Ag2+xGe7−x (n = 5), this bonding feature can effectively address the destabilizing effects of the incomplete charge transfer on Ge–Ge covalent bonds. Hence, the particular electronic structures of [Gen](2n+2)− oligomers allow electronic back donation from Ge to Ca and, additionally, strengthening of Ge–Ge covalent bonds by depopulation of those πp* states.15 The fact that these π* states overlap with Ca 3d orbitals could explain the absence of a real band gap above EF, predicting the compounds to be metallic.4 | ||
| Fig. 5 Calculated DOS and COHP curves for “Ca3MGe3” with (a) M = Cu and (b) M = Ag. The DOS, partial DOS, and COHP curves are plotted on the same energy scale, and the Fermi level is set at 0 eV as the energy reference. | ||
Relative bond strengths are estimated by means of the integrated crystal orbital Hamilton populations (–ICOHPs), and they are very consistent with the bond lengths observed from single crystal X-ray diffraction analyses (see Table 6). Thus, Ge–Ge bonds are strongest with the maximum for the Ge–Ge bond between central Ge atoms in Ge4 tetramers (2.45 and 2.53 eV for 4_Cu and 4_Ag, respectively), followed by Ge2 dumbbells (2.38 eV for 4_Cu and, 2.51 eV for 4_Ag), in agreement with essentially covalent-type interactions. The second strongest bonds are the shortest Ag–Ge (1.99 eV) and Cu–Ge (1.98 eV) bonds, also consistent with predominant polar-covalent character.5,6 All Ca–Ge bonds are comparatively weaker, reaching maximum values of 0.91 and 0.70 eV in 4_Cu, and 0.88 to 0.61 eV in 4_Ag.
Nevertheless, the respectable values of ICOHP for Ca–Ge interactions indicate significant contribution to overall covalent bonding in this system, meaning incomplete charge transfer.4–6
The virtually identical electronic structures of the two analogues cannot explain why Ge/Cu mixing is not observed. Besides breaking up of the Ge2 dumbbells, another noteworthy impact of Ge/Ag mixing is to generate Ag–Ag direct interactions, not observed in a fully ordered model. The most accepted paradigm is that this type of metallophilic interactions between coinage metals is the result of electron correlation and, is strengthened by relativity.40 This means that similar Cu–Cu interactions will be comparatively weaker and, perhaps, less likely to occur. This scenario could be evaluated by building superstructure models within the coloring problem approach,36 and investigating their electronic and total energies; but this exceeds the scope of this report.
Conclusions
The crystal structures of three compounds that are new representatives of the structure series Ca2+nM2+zGe2+n−z (M = Cu, Ag, Mg, Mn) could be determined from single crystal X-ray diffraction data. Ca3CuGe3 is a new representative of the n = 4 homologue, and is isostructural with the Ag analogue adopting the Sc3NiSi3-type (monoclinic C2/m). Partial substitution of monovalent Ag by divalent Mn atoms results in either Ca6MnxAg2−x+zGe6−z (own type, monoclinic P21/m) representing an unanticipated intermediate between the n = 4 and n = 5 members, or Ca5MnxAg2−x+zGe5−z, as the second representative of the n = 3 member, isostructural with the Mg analogue Ca5MgAgGe5 (orthorhombic Pnma). The two Mn compounds demonstrate that it is possible to replace diamagnetic Mg and Ag atoms by paramagnetic Mn atoms through combined isovalent and aliovalent substitution reactions, respectively. The Zintl–Klemm concept provides critical insight into the bonding features of these homologous structures, qualifying them as transition metal Zintl phases. However, LMTO band structure calculations predict that Ca3CuGe3 is metallic, just like the parent structures CaCuGe and CaGe. Oligomeric germanides [Gen](2n+2)− with n = 1–5 are observed in the three title compounds, often simultaneously. Hence, new TMZPs with magnetic properties can be obtained by replacing the highly polarizing small s-block cations (Li, Mg) in the family AE–(Li, Mg)–(Si, Ge) by selected TMs like Cu, Ag, and even paramagnetic Mn. Importantly, mixing two TMs as small polarizing cations, in one single phase, may open new perspectives for the discovery novel functional materials. This is because of the potential to yield combined chemical and magnetic frustration, resulting from the co-existence of inherently incompatible metal–metal bonding motifs within the same crystal structure.Author contributions
All authors contributed to the scientific content of the manuscript and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at Stockholm University was financially supported through the Carl-Tryggers Stiftelse and Energimyndigheten (contract no. 46595-1). AVM would like to acknowledge support by the Royal Swedish Academy of Science through the Göran Gustafsson prize in chemistry. Work at Iowa State University was supported by the National Science Foundation (NSF DMR 10-05765 and 12-09135). S. P. also thanks Prof. Sven Lidin for inspiring and splendid mentorship. A.-V. M. and G. J. M. would like to honor a great and inspiring colleague.Notes and references
- G. J. Miller, Metal-Rich Compounds of the d-Metals, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 311–357, DOI:10.1016/B978-0-08-097774-4.00214-X.
- V. Smetana, M. Rhodehouse, G. Meyer and A.-V. Mudring, Gold Polar Intermetallics: Structural Versatility through Exclusive Bonding Motifs, Acc. Chem. Res., 2017, 50, 2633–2641 CrossRef CAS PubMed.
- Yu. Grin, The Intergrowth Concept as a Useful Tool to Interpret and Understand Complicated Intermetallic Structures, in Modern Perspectives in Inorganic Crystal Chemistry, ed. E. Parthé, Kluwer Academic Publishers, Norwell, MA, 1992 Search PubMed.
- A.-V. Mudring and J. D. Corbett, Unusual Electronic and Bonding Properties of the Zintl Phase Ca5Ge3 and Related Compounds. A Theoretical Analysis, J. Am. Chem. Soc., 2004, 126(16), 5277–5281 CrossRef CAS PubMed.
- F. R. Wagner, D. Bende and Yu. Grin, Heteropolar bonding and a position-space representation of the 8-N rule, Dalton Trans., 2016, 45(8), 3236–3243 RSC.
- R. Freccero, P. Solokha, S. De Negri, A. Saccone, Y. Grin and F. R. Wagner, Polar-Covalent Bonding Beyond the Zintl Picture in Intermetallic Rare-Earth Germanides, Chem. – Eur. J., 2019, 26, 6600–6612 CrossRef PubMed.
- H. Schäfer, B. Eisenmann and W. Müller, Zintl Phases: Transitions between Metallic and Ionic Bonding, Angew. Chem., Int. Ed. Engl., 1973, 12, 694–712 CrossRef.
- S. M. Kauzlarich, S. R. Brown and G. J. Snyder, Zintl phases for Thermoelectric Devices, Dalton Trans., 2007, 2099–2107 RSC.
- J. Wang, X. C. Liu, S. Q. Xia and X. T. Tao, Ca1-xRExAg1-ySb (RE = La, Ce, Pr, Nd, Sm; 0 ≤ x ≤ 1; 0 ≤ y ≤ 1): Interesting Structural Transformation and Enhanced High-Temperature Thermoelectric Performance, J. Am. Chem. Soc., 2013, 135, 11840–11848 CrossRef CAS PubMed.
- F. Zürcher and R. Nesper, Cationic Channels with Partial Anion Occupation in the Zintl Phases Ba2Mg12Ge7.33 and Ba6Mg17.4Li2.6Ge12O0.64, Z. Anorg. Allg. Chem., 2002, 628, 1581–1589 CrossRef.
- A. Currao, J. Curda and R. Nesper, Can One Design Zintl Anions? Contributions from the System Sr/Mg/Si to the Topic Si2−, Z. Anorg. Allg. Chem., 1996, 622, 85–94 CrossRef CAS.
- I. Schellenberg, M. Eul, C. Schwickert, C. M. Kubata, E. C. Reyes, R. Nesper, U. Ch. Rodewald and R. Pöttgen, The Zintl Phases Eu3Mg5Si5 and Eu3Mg5Ge5, Z. Anorg. Allg. Chem., 2012, 638, 1976–1985 CrossRef CAS.
- S. Ponou and S. Lidin, Ca3Ag1+xGe3-x (x = 1/3): New Transition Metal Zintl Phase with Intergrowth Structure and Alloying with Aluminum Metal, Z. Anorg. Allg. Chem., 2013, 639, 35–40 CrossRef CAS.
- S. Ponou, S. Lidin, Y. Zhang and G. J. Miller, Valence State Driven Site Preference in the Quaternary Compound Ca5MgAgGe5: An Electron-Deficient Phase with Optimized Bonding, Inorg. Chem., 2014, 53, 4724–4732 CrossRef CAS PubMed.
- S. Ponou, I. Doverbratt, S. Lidin and G. J. Miller, Structure and Bonding of an Intergrowth Phase Ca7Ag2+xGe7–x (x ≈ 2/3) Featuring a Zintl-Type Polyanionic Chain, Eur. J. Inorg. Chem., 2016, 1, 169–176 CrossRef.
- S. Ponou, S. Lidin, D. Grüner and G. J. Miller, Conflict between the Electronic Factors and Structure-Directing Rules in the Intergrowth Structure of Ca4Ag2+xGe4–x with x = 1/2, Cryst. Growth Des., 2016, 16(10), 5946–5953 CrossRef CAS.
- S. Ponou, On a TiNiSi-Type Superstructure: Synthesis, Crystal and Electronic Structures of CaAgGe and its Mn-Substituted Derivative, Eur. J. Inorg. Chem., 2010, 26, 4139–4147 CrossRef.
- CrysAlis. CCD and CrysAlis. RED, Oxford Diffraction, 2006, p. 171.31.2 Search PubMed.
- APEX3, Bruker AXS Inc., Madison, WI, 2015 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, SUPERFLIP–a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. Oszlányi and A. Sütő, The Charge Flipping Algorithm, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 123–134 CrossRef PubMed.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(1), 3–8 Search PubMed.
- O. Jepsen, A. Burkhardt and O. K. Andersen, The Program TBLMTO-ASA, 4.7, Max-Planck-Institut für Festkörperforschung, Stuttgart, Germany, 1999 Search PubMed.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved Tetrahedron Method for Brillouin-Zone Integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(23), 16223–16233 CrossRef PubMed.
- W. R. L. Lambrecht and O. K. Andersen, Minimal basis sets in the linear muffin-tin orbital method: Application to the diamond-structure crystals C, Si, and Ge, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 2439–2449 CrossRef CAS PubMed.
- R. Dronskowski and P. Blöchl, Crystal Orbital Hamilton Populations (COHP): Energy-Resolved Visualization Of Chemical Bonding In Solids Based on Density-Functional Calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- J. T. Zhao and E. Parthé, Sc3Ni2Si3 with Hf3Ni2Si3-type structure, an intergrowth of CrB-, ThCr2Si2-and W-type slabs, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1853–1856 CrossRef.
- D. Kussmann, R. D. Hoffmann and R. Pöttgen, Syntheses and Crystal Structures of CaCuGe, CaAuIn, and CaAuSn – Three Different Superstructures of the KHg2 Type, Z. Anorg. Allg. Chem., 1998, 624, 1727–1735 CrossRef CAS.
- P. Eckerlin, H. J. Meyer and E. Wölfel, Die Kristallstruktur von CaSn und CaGe, Z. Anorg. Allg. Chem., 1955, 281, 322–328 CrossRef CAS.
- H. Bärnighausen, Group-subgroup Relations between Space Groups: a useful Tool in Crystal Chemistry, MATCH, 1980, 9, 139 Search PubMed.
- U. Müller, Crystallographic Group-Subgroup Relations and Their Use in Crystal Chemistry, Z. Anorg. Allg. Chem., 2004, 630, 1519–1537 CrossRef.
- A. Slabon, E. Cuervo-Reyes, C. Kubata, C. Mensing and R. Nesper, Exploring the Borders of the Zintl-Klemm Concept: On the Isopunctual Phases Eu5+xMg18–xGe13 (x = 0.1) and Eu8Mg16Ge12, Z. Anorg. Allg. Chem., 2012, 638(12–13), 2020–2028 CrossRef CAS.
- R. Benedek, J. T. Vaughey and M. M. Thackeray, Modeling and Design of Intermetallic Electrodes for Lithium Batteries, in Materials for Electrochemical Energy Conversion and Storage, ed. A. Manthiram, P. N. Kumta, S. K. Sundaram and G. Ceder, The American Ceramic Society, Ohio, 2006, DOI:10.1002/9781118370858.ch24.
- J. Emsley, The Elements, Clarendon Press, Oxford University Press, Oxford, New York, 1998 Search PubMed.
- G. J. Miller, The “Coloring Problem” in Solids: How It Affects Structure, Composition and Properties, Eur. J. Inorg. Chem., 1998, 5, 523–536 CrossRef.
- T. Story, R. R. Galazka, R. B. Frankel and P. A. Wolff, Carrier-concentration–induced ferromagnetism in PbSnMnTe, Phys. Rev. Lett., 1986, 56, 777–779 CrossRef CAS PubMed.
- G. J. Miller, Complex rare-earth tetrelides, RE5(SixGe1−x)4: New materials for magnetic refrigeration and a superb playground for solid state chemistry, Chem. Soc. Rev., 2006, 35, 799–813 RSC.
- Z. Yang, D. Shi, B. Wen, R. Melnik, S. Yao and T. Li, First-principle studies of Ca–X (X=Si,Ge,Sn,Pb) intermetallic compounds, J. Solid State Chem., 2010, 183, 136–143 CrossRef CAS.
- P. Pyykkö, Strong Closed-Shell Interactions in Inorganic Chemistry, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed.
Footnotes |
| † Dedicated to Professor Sven Lidin on the Occasion of his 60th Birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic CIF files for Ca3CuGe3 (4_Cu): CCDC-2031725, for Ca6Mn0.67(1)Ag1.79(1)Ge5.54(1) (4_Mn): CCDC-2031727 and for (Ca5MnxAg2+x+zGe5−z, 3_Mn) Ca5Mn0.64(1)Ag1.41(1)Ge4.95(1): CCDC-2031726, and Ca5Mn0.71(1)Ag1.33(1)Ge4.96(1): CCDC-2055289. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00094b |
| This journal is © The Royal Society of Chemistry 2021 |