
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the role of solution additive anions in Mg2+ dehydration: implications for mineral carbonation†
Dimitrios Toroza,
Fu Songa,
Gregory A. Chass *abc and
Devis Di Tommaso*a
*abc and
Devis Di Tommaso*a
aSchool of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: g.chass@qmul.ac.uk; d.ditommaso@qmul.ac.uk
bDepartment of Chemistry and Chemical Biology, McMaster University, Hamilton, Ontario L8S 4M1, Canada
cDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China
First published on 10th March 2021
Abstract
Simulations of hydrated Mg2+, in the absence and presence of several solution additive anions, show that in pure liquid water Mg(H2O)62+ is the only stable coordination state; yet anions may stabilise undercoordinated five-hydration configurations. Solution composition can lower the barrier to Mg2+ dehydration and subsequent incorporation into the lattice of Mg-carbonates, promoting low-temperature crystallisation.
Formed via aqueous mineral carbonation of Mg2+ ions, the crystallisation of magnesite (MgCO3) represents an industrially effective route to CO2 storage and utilisation, generating stable, inert, non-hazardous, and ready-to-use carbonate-based materials,1 revenues of which are expected to reach $1 trillion per year by 2030.2 Mg2+ sources are widespread and plenty and include Mg-silicate deposits (>100
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Gt)3 and alkaline industrial residues.4 However, CO2 mineralisation is limited by the slow rates of MgCO3 precipitation.5
000 Gt)3 and alkaline industrial residues.4 However, CO2 mineralisation is limited by the slow rates of MgCO3 precipitation.5
Magnesite production is an energy-intensive process due to the high temperatures (120 ≤ T ≤ 600 °C) required to prevent the formation of metastable hydrated phases nesquehonite (MgCO3·3H2O) and hydromagnesite, (Mg5(CO3)4(OH)2·4H2O).6 The high T conditions necessary to promote the direct precipitation of anhydrous MgCO3, increased solid mass and volume of the hydrated Mg-carbonate phases per mole of CO2 sequestered and their inferior mechanical properties, all have negative impacts on the industrial viability and profitability of CO2 mineralisation.7
Due to the highly hydrated character of Mg2+ in aqueous solutions (ΔhydG° = −455 kcal mol−1),8 the difficulty in precipitating magnesite has long been ascribed to the slow kinetics of Mg2+ dehydration.9 However, geological records show that magnesite, MgCO3, and dolomite, CaMg(CO3)2, form during weathering of ultramafic rocks (Mg-rich and low silica) and sedimentary processes (precipitation and/or replacement) taking place at low-T,10 which are different from the conditions necessary to stimulate the synthesis of the anhydrous forms of Mg-carbonates. This conundrum, known as the “dolomite problem”, represents one of the most long-standing questions in low-temperature geochemistry.11
Natural solutions are far from pure water, being rich in ions, making solution environments highly influential on the rate determining Mg2+ dehydration step. Resolving the catalytic role of composition is imperative in determining what catalyses MgCO3 formation in sedimentary environments. The goal of this communication is to provide a computational characterisation of the mechanism of Mg2+⋯H2O dissociation reaction in the presence of several solution additive anions. We reason that the root of these effects may reside in the ability of organic ligands and inorganic anions that are typically present in aqueous environments to activate Mg2+ dehydration, and subsequent nucleation and growth steps, by influencing the hydration structure of Mg2+.
Here, we report a computational characterisation of the rate determining Mg2+ (de)hydration process. We conducted simulations of Mg2+ in pure liquid water and in solutions containing ions (X) commonly found in groundwater (F−, Cl−, NO3−, HCO3−, CO32−, SO42−) including bisulfide (HS−)12 and carboxylate (CH3COO−) ions thought to catalyse carbonation.13 The computational details of the molecular dynamics (MD) and enhanced sampling metadynamics (MetaD) simulations and of the electronic structure analyses adopted are reported in ESI,† together with the details of the electrolyte solutions considered (number of ions and H2O molecules, equilibrated cell length).
Fig. 1A shows the free energy landscape, computed using MetaD, of a hydrated Mg2+ (isolated metal ion, without any counterions) as a function of the ion–water distance, d(Mg–H2O), and ion–water coordination number, CN(Mg–H2O). Letters A, B, C and D correspond to representative configurations on the free energy landscape. The octahedral six-fold coordination with water (configurations A and C) represents the only thermodynamically stable state for hydrated Mg2+ in pure liquid water. Configuration B corresponds to the metastable five-fold trigonal bipyramidal coordination state, which lies on the lowest energy pathway between the six-fold configurations A and C. Configuration D is another five-coordinated intermediate Mg(H2O)52+ on the free energy surface of hydrated Mg2+. Our metadynamics results are consistent with gas-phase density functional theory (DFT) calculations of Mg2+ hydrates containing up to 18 water molecules, which showed that the process of going from the six- to the five-coordinated states (the two lowest energy hydrated structures) is highly unfavorable.14 The arrangement of H2O molecules around the five-coordinated intermediate B also shows that the Mg2+ dehydration proceeds with the assistance of out-of-shell solvent water molecules, in agreement with static electronic structure calculations of small hydrated Mg2+ cluster models.15
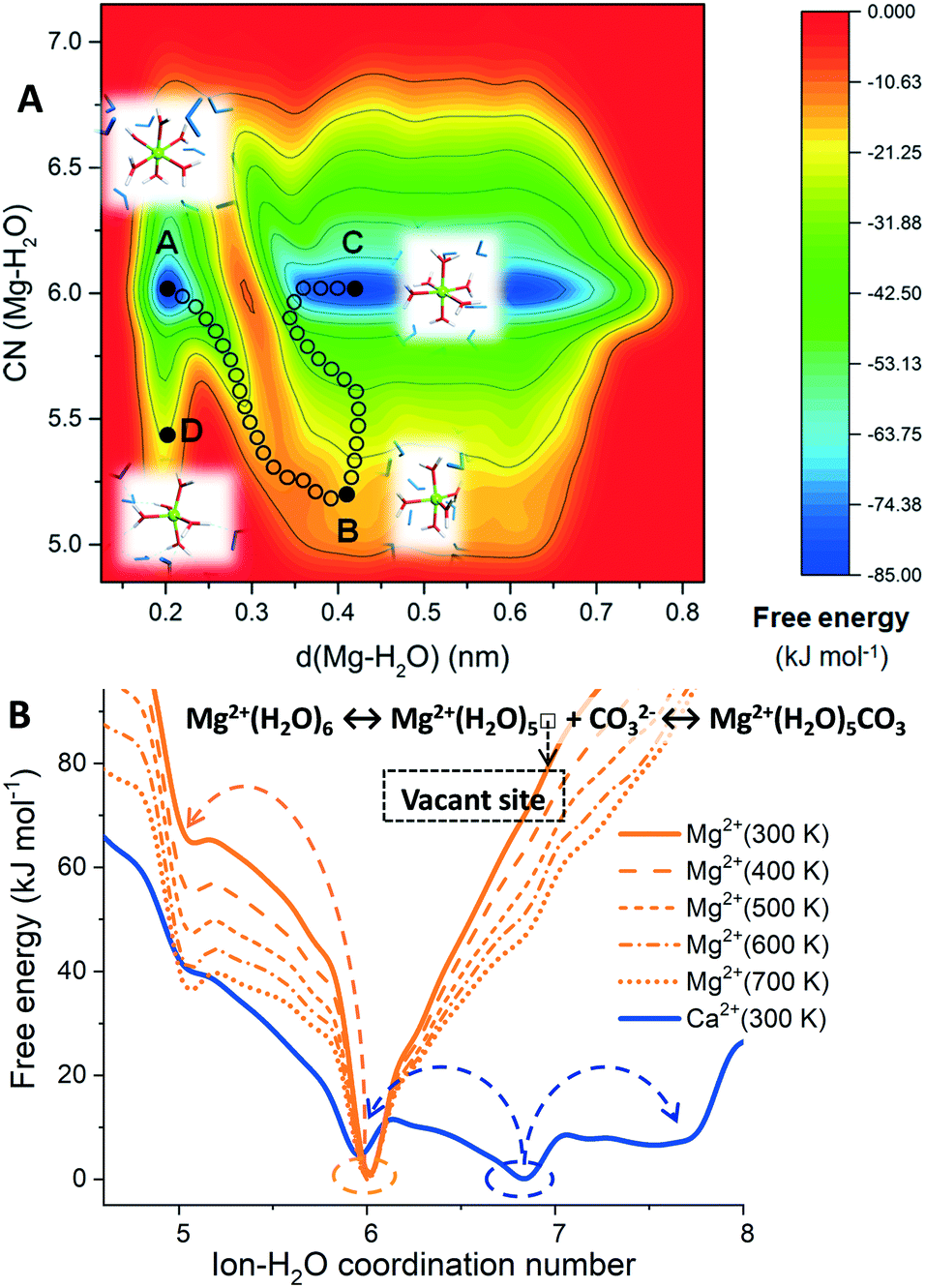 | ||
| Fig. 1 (A) Free energy contour plot of hydrated Mg2+ as a function of the Mg–H2O coordination number, CN(Mg–H2O), and of the Mg–H2O distance, d(Mg–H2O), of one of the six water molecules (H2O*) that at the start of the MetaD simulation is part of the first hydration shell of Mg2+. A and C correspond to the six-coordinated state. B and D are the five-coordinated intermediate Mg(H2O)52+ when H2O* is inside or outside of the first hydration shell of Mg2+, respectively. (B) Free energy profiles of hydrated Ca2+ and Mg2+ ions as a function of the ion–water coordination number. For the hydrated Mg2+, MetaD simulations were conducted at temperatures ranging from 300 K to 700 K. | ||
In Fig. 1B, the free energy profiles of hydrated Mg2+ as a function of the ion–water coordination number are compared to that of Ca2+. From these profiles, we can extract thermodynamic information on the accessible coordination states for these cations. For the calcium ion, the seven-coordinate Ca(H2O)72+ is the most likely hydration state and not the six-coordinate as for Mg2+, but the six- and eight-fold coordination states are also accessible at room temperature (Fig. 1B, in blue). The calcium incorporation in calcite or dolomite may occur through dissociative (7 → 6) or associative (7 → 8) pathways. The hydrated Mg2+ has a very stable minimum corresponding to six-fold coordination with water, Mg(H2O)62+. On the other hand, the five-coordinated intermediate, Mg(H2O)52+, is inaccessible at 300 K due to the 65 kJ mol−1 activation barrier between six- and five-coordinated configurations of Mg2+, which is significantly larger than the thermal energy at 300 K (kT = 2.5 kJ mol−1). Consequently, water exchange is drastically retarded in the first hydration shell of Mg2+. MD simulations showed no water exchange around Mg2+ (no dehydration) after 100 ns compared to aqueous Ca2+ which underwent ∼50 water exchanges in only 180 ps (facile dehydration) (Fig. S2†). Even a single H2O dissociated from Mg2+ would still translate to a ∼36000-fold retardation compared to Ca2+. The subsequent steps of nucleation and growth are likewise susceptible to Mg-(re)hydration. Consequently, the precipitate rates of MgCO3 and CaMg(CO3)2, are six and four orders of magnitude slower, respectively than CaCO3 at 300 K.16
The generation of a vacant site at the central magnesium ion, to which carbonate can bind to initiate the MgCO3 nucleation process, requires the displacement of a water molecule from the first coordination shell of Mg2+. However, the Mg2+ dehydration is a highly activated process, restricted by a free energy barrier of 65 kJ mol−1 for the transformation from the six to the five-coordinated states (Fig. 1B). By increasing the temperature from 300 K to 700 K, a gradual stabilisation of Mg(H2O)52+ occurs and the activation barrier between six- and five-coordinated Mg2+ decreases (Fig. 1B, in orange), which is consistent with the experimental evidence that higher temperatures promote the direct precipitation of magnesite.6,7
Towards resolving the bases of the faster kinetics of MgCO3 precipitation observed within the nanoconfined space of adsorbed H2O films, compared to the bulk solution, Miller et al. proposed a reduction in coordinating water molecules (fewer than six) for Mg2+.17 Similarly, Mergelsberg conjectured that the greater salinity in natural systems may stabilise the five-coordinated intermediate.18 A substantial proportion of Mg(H2O)52+ in solution will lower the barrier to cationic dissolution, and its incorporation in the crystal lattice of Mg-carbonates.
In electrolyte solutions, ions are not purely hydrated and can form contact ion pairs (CIPs), when the cation and anion are in direct contact, or solvent-shared ion pairs (SSHIPs), when separated by one water molecule. Ion pairs interspersed by two or more water molecules are labelled solvent-separated ion pairs (SSIP). The tendency of Mg2+ and a counterion (X) of forming CIP, SSHIP or SSIP depends on the competition between Mg2+⋯H2O and Mg2+⋯X interactions. The strength of ion-pairing was assessed by computing, using MetaD, the free energy profiles as a function of the Mg2+⋯X distance and compare them to the free energy profile of removing a water molecule from hydrated Mg2+ (Fig. 2A). Key features of the free energy profiles for the formation of Mg2+⋯X CIPs, the standard Gibbs energy of activation (Δ‡G) and Gibbs free energy of reaction (ΔG), are summarised in Table S2 (ESI†). Fig. 2B reports the distribution of ion pairs in the solutions, where we have sorted additive anions based on their ability to form a CIP with Mg2+.
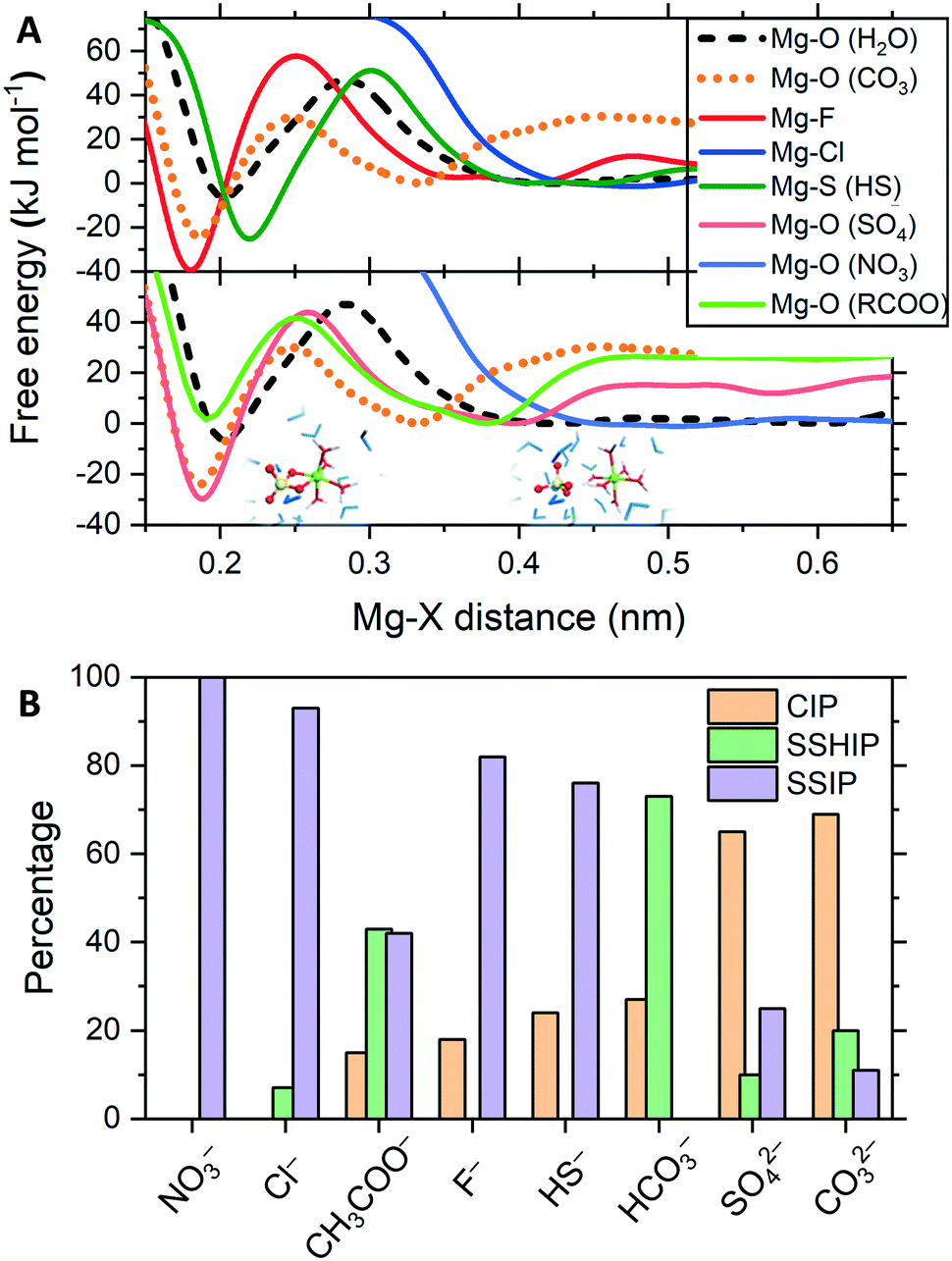 | ||
| Fig. 2 (A) The free energy profiles as a function of the distance between Mg2+ and the centre of mass of the additive X (CO32−, F−, Cl−, HS−, SO42−, NO3−, CH3COO−) are compared with the free energy profile for the removal of H2O from Mg(H2O)62+. Structures of Mg(η2-SO4)(H2O)4, with SO42− coordinated in a bidentate mode and the hydration number four, and of Mg2+ and SO42− interspersed by one water molecule. (B) Distribution of Mg2+ and X contact ion pairs (CIP), and solvent shared (SSHIP) and solvent separated (SSIP) ion pairs obtained from the analysis of the MetaD simulations. | ||
The free energies for the Mg2+⋯CO32− (ΔG = −26 kJ mol−1) and Mg2+⋯SO42− (−30 kJ mol−1) pairing are significantly lower than Mg(H2O)62+ (−7 kJ mol−1), whilst the Gibbs energy of activation of these CIPs are lower than Mg2+⋯H2O dissociation (Δ‡G = +48 kJ mol−1). Consequently, both Mg2+⋯CO32− and Mg2+⋯SO42− CIPs are thermodynamically and kinetically favourable compared to Mg(H2O)62+. That is why in Fig. 2B, carbonate and sulfate display a high propensity towards CIP. Such a result corroborates the significant body of knowledge in low-temperature geochemistry showing the prerequisite for dolomite precipitation in sedimentary environments is the low concentrations of sulfate ions, and that such a low concentration is due to the activity of sulfate reducing bacteria (forming bisulfide ions).19 Using AFM, King et al. showed that even small amounts of sulfate in solution is detrimental to the growth of both anhydrous and hydrated Mg-carbonate phases.20 The authors interpreted the detrimental effect of sulphate ions in solution to Mg-carbonation to the formation of Mg2+⋯SO42− ion pairs, which lower the activity of the free Mg2+ ion, reducing the solution supersaturation with respect to magnesite. Our results also agree with Raman spectra of aqueous solutions of MgSO4 reported by Rudolph et al., where the changes to the ν1-SO42− mode at ∼980 cm−1 was associated with the formation of an Mg2+⋯SO42− contact ion pairs.21 Based on our simulations, sulfate ions may inhibit the nucleation and growth of magnesite because ion pairing of Mg2+⋯SO42− is competitive to Mg2+⋯CO32−.
The formation of the Mg2+⋯F− CIP is thermodynamically very favourable (ΔG = −41 kJ mol−1 at r(Mg–F) = 0.18 nm in Fig. 2A). However, the lower propensity towards ion-pairing observed in our simulations is likely to be caused by the very high Gibbs energy of activation for the formation of the Mg2+⋯F− CIP (Δ‡G = 60 kJ mol−1 at r(Mg–F) = 0.25 nm in Fig. 2A). Such a value is significantly higher than removing an H2O molecule from the Mg(H2O)62+ complex (Δ‡G = 47 kJ mol−1 at r(Mg–O) = 0.29 nm in Fig. 2A). Similar behaviour is observed for bisulfide. The Mg2+⋯HS− is thermodynamically favourable (ΔG = −26 kJ mol−1) but the high activation barrier (Δ‡G = 51 kJ mol−1) limits the formation of CIPs.
Due to the absence of a minimum on the free energy profile, Cl− and NO3− form almost exclusively SSIPs. For these ions, no disturbance in the inner hydration shell of Mg2+ occurs priory to the energetically costly replacement of a water molecule with one Cl or O (nitrate) atom. For MgCl2(aq), such a result confirms recent broadband dielectric relaxation spectroscopic measurements on the aqueous solution of MgCl2 where no evidence for the significant formation of contact ion pairs was observed.22 Also, a combined Raman spectroscopic study and MD simulation using an ad hoc force field for aqueous MgCl2 solutions also supported the absence of Mg2+⋯Cl pairing.23Fig. 2B shows that CH3COO− and HCO3− have a propensity for SSHIP because the free energy difference between contact, Mg2+⋯X, and solvent shared, Mg2+⋯H2O⋯X, pairs are close to zero (Table S2†). The values of ΔG = 1 kJ mol−1 and Δ‡G = 41 kJ mol−1 for the carboxylate group, deemed responsible for Mg2+ dehydration, imply that CH3COO− does not inhibit MgCO3 pairing. The low propensity of the acetate anion towards pairing with Mg2+ observed in our simulations qualitatively agrees with the positive value of the free energy for water ↔ formate (HCOO−) substitution exchange reaction [Mg(H2O)6]2+ + HCOO− → [Mg(H2O)5(HCOO)]+ + H2O computed using DFT and continuum dielectric methods.24
To determine the influence of additive anions on the kinetics of Mg2+ dehydration, we computed the free energy as a function of the Mg2+–water coordination number, CN(Mg–H2O), for solutions containing counterions. Exploring the dynamical aspects of the CN(Mg–H2O) reaction coordinate allows us to examine the ability of solution additive anions to influence the transition between coordination states of Mg2+ during the dynamics of cation solvation. Fig. 3A compares the free energy profiles of Mg2+ in pure liquid water with that of Mg2+ in solutions containing anions with a propensity for SSHIP or SSIP as they are not competitive to CIP MgCO3 formation.
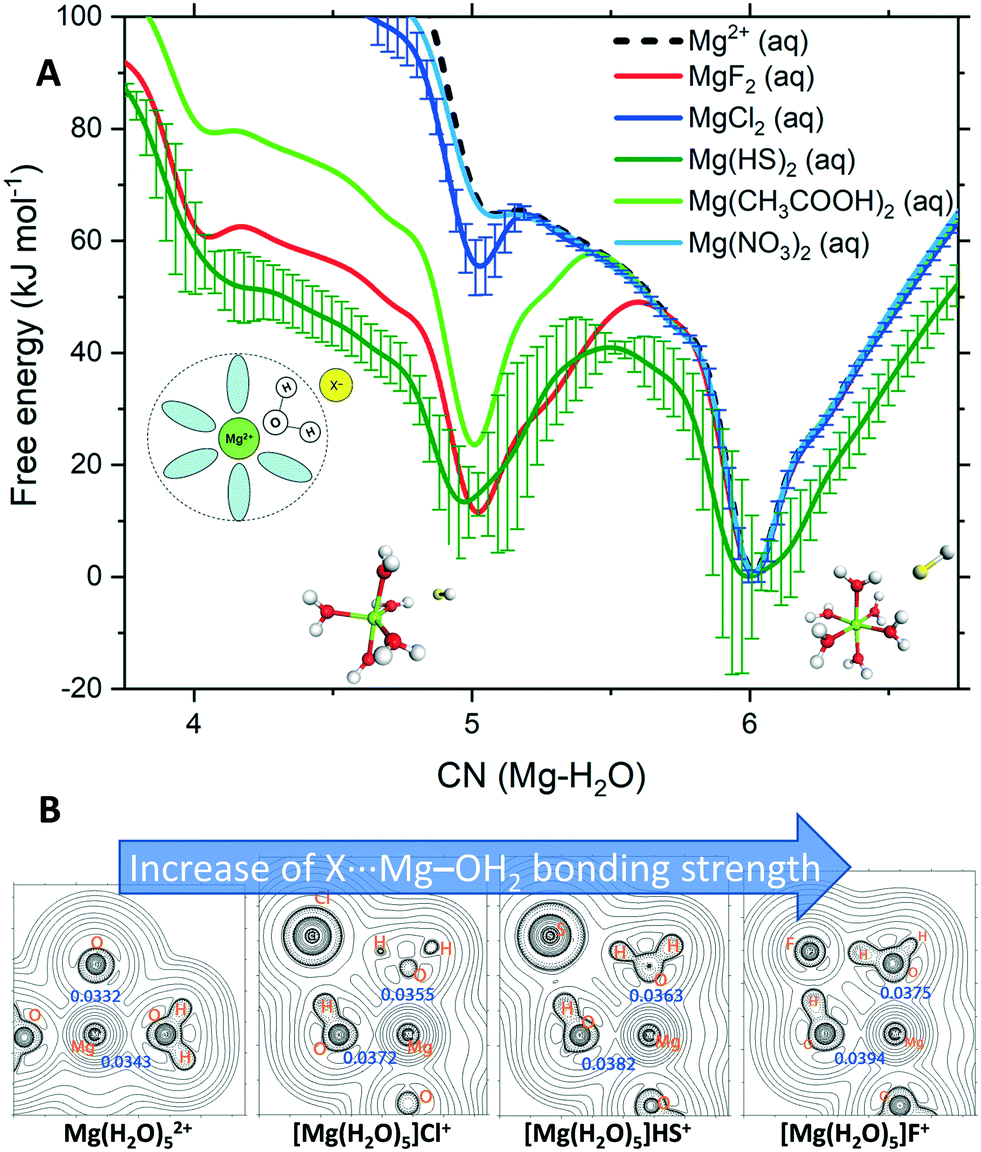 | ||
| Fig. 3 (A) Free energy as a function of the Mg2+–H2O coordination number, CN(Mg–H2O), for hydrated Mg2+ (single Mg2+, no counterions) and solvated Mg2+ with a counterion in the second hydration shell. The structures are the five- and six-coordinated states in Mg(HS)2(aq). Standard deviation computed from the average of the profiles of four independent MetaD simulations (300 K). Error bars are presented for the cases with the largest uncertainty, others have been removed for clarity (see Fig. S3†). (B) 2-D Laplacians of electronic density (∇2ρ) in the plane of the Mg, O, H and X atoms in Mg(H2O)52+ and [Mg(H2O)5]·X+ (X = F−, Cl−, HS−). Values of ρb (in e bohr−3) at selected bond critical points are provided in blue. | ||
In these MetaD simulations, we have kept the separation between Mg2+ and the counterion at ∼4.5 Å, which corresponds to a solvent shared Mg2+⋯H2O⋯X ion pair. According to Fig. 3A, the nitrate ion does not affect the kinetics of Mg2+–H2O dissociation because the free energy profile of Mg(NO3)2(aq) overlaps that of hydrated Mg2+ (isolated metal ion, without any counterions). In both solutions, the Mg2+ six-coordinated state is the only one accessible at room T. Chloride shows a small stabilisation of the five-coordinated Mg2+. In comparison, the bisulfide ion has a dramatic effect on the thermodynamics and kinetics of Mg2+ dehydration. In Mg(HS)2(aq), the five-coordinated Mg(H2O)52+ state is as stable as its six-coordinate compliment Mg(H2O)62+ and the Gibbs free energy barrier to jump between these two states is 49 kJ mol−1 compared to 66 kJ mol−1 in pure liquid water. Our simulations thus show, for the first time, that additive anions such as HS− forming SSHIP with Mg2+ can promote the dehydration of the Mg2+ cation. Such a process may generate a large proportion in the aqueous solution of undercoordinated hydrated Mg2+ states with a vacant coordination site to which CO32− can bind, initiating the process of MgCO3 nucleation or promoting the Mg2+ incorporation into the crystal lattice. Our result supports McKenzie's hypothesis that bisulfides, delivered by sulfate-reducing bacteria, promote natural dolomite formation by catalysing Mg2+–H2O dissociation.12 Similarly, the dehydration of Mg2+via surface-bound carboxyl groups has been proposed by Kenward et al. to explain the low-T precipitation of dolomite on carboxylated polystyrene spheres.25 The result for the carboxylate ion in Fig. 3A confirms Kenward's hypothesis since CH3COO− in the second hydration shell of Mg2+ stabilises the five-hydration state in a more energetically favourable process than in pure liquid water, thus promoting Mg-carbonate nucleation. The fluoride ion shows similar stabilisation behaviour (Fig. 3A). Further, the role of fluoride in facilitating Mg2+ dehydration may help to explain natural deposits of thin alternating layers of fluorite and dolomite, known as zebra rocks.26 For the solutions containing Mg2+⋯X CIPs, the free energy profiles show stabilisation of configurations with three and four water molecules in the first shell (Fig. S4†). Therefore, Mg2+ is dehydrated even when directly coordinated to the solution additive anion.
The electronic structure of Mg(H2O)n2+ and [Mg(H2O)n]·X+ (n = 5, 6) were determined using Bader's atoms-in-molecules analyses of the wavefunctions generated from their DFT geometry-optimised structures (Fig. S5†). The rho-b (ρb/e bohr−3) values (Fig. 3B, in blue) indicate a progressive increase in the electron density at the bond critical point of the Mg–O bond with the axial-H2O group interacting with the incoming X ion. Such a bond strengthening is concomitant with the weakening of other Mg–O bonds, the opposing axial in particular (Fig. S4†); an anti-type effect in classical inorganic coordination. In Fig. 3B, the 2-D Laplacians of the electronic density in the plane of the Mg, O, H and X atoms show iso-density contour lines progressively widening ‘saddle points’ between Mg2+ and O. These trends point to a continuing stabilisation of the five-coordinate species, with the allosteric weakening of the other Mg–O bonds, effectively readying the complex for subsequent dehydration. The widening of the free-energy potentials for the five-coordinate minimums (Fig. 3A) support such a conclusion, implying more structurally flexible geometries.
Conclusions
We have computationally characterised the mechanism of Mg2+–H2O dissociation in the absence and presence of several solution additive anions. The slow kinetics of Mg2+ dehydration in pure liquid water is due a high-energy metastable five-coordinated intermediate. Anions such as bisulfide, carboxylate and fluoride ions can stabilise undercoordinated Mg2+ hydration configurations, even when they are in the second hydration shell of Mg2+. Wavefunction analyses revealed the changes in bonding responsible for the equilibration of five and six hydrated states, with insight into the mechanisms by which these can inter-change and open-up coordination sites on the central Mg2+ ion. The characterisation of the rate-determining Mg2+ dehydration process in a series of solutions, as covered in our work, contributes to resolving the catalytic role of solution composition in promoting Mg2+ dehydration, and subsequent MgCO3 formation in natural and industrial environments.This project is funded through the ACT programme (Accelerating CCS Technologies, Horizon2020 Project No 294766). Financial contributions made from Department for Business, Energy & Industrial Strategy together with extra funding from NERC and EPSRC research councils (UK), ADEME (FR), MINECO-AEI (ES). We thank the UK MMM Hub (EP/P020194/1), HEC MCC (EP/L000202, EP/R029431), and Queen Mary's Research-IT for computing resources. GAC thanks McMaster University and Hong Kong Univ. for his adjunct and honorary positions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 Search PubMed
.
- Mission Innovation, Accelerating Breakthrough Innovation in Carbon Capture, Utilization, and Storage, Houston, Texas, 2017.
- K. S. Lackner, Annu. Rev. Energy Environ., 2002, 27, 193–232 Search PubMed
- E. R. Bobicki, Q. Liu, Z. Xu and H. Zeng, Prog. Energy Combust. Sci., 2012, 38, 302–320 Search PubMed
- D. E. Giammar, R. G. Bruant Jr. and C. A. Peters, Chem. Geol., 2005, 217, 257–276 Search PubMed
- E. J. Swanson, K. J. Fricker, M. Sun and A.-H. A. Park, Phys. Chem. Chem. Phys., 2014, 16, 23440–23450 Search PubMed
- V. Prigiobbe, M. Hänchen, M. Werner, R. Baciocchi and M. Mazzotti, Energy Procedia, 2009, 1, 4885–4890 Search PubMed
- J. Burgess, Metal ions in solution, Ellis Horwood Ltd., Chichester, 1978 Search PubMed
- F. L. Sayles and W. S. Fyfe, Geochim. Cosmochim. Acta, 1973, 37, 87–99 Search PubMed
- M. Kralik, P. Aharon, E. Schroll and D. Zachmann, in Monograph Series on Mineral Deposits, 1989, vol. 28, pp. 197–223 Search PubMed
- H. Zimmermann, Int. Geol. Rev., 2000, 42, 481–490 Search PubMed
- J. McKenzie and C. Vasconcelos, Sedimentology, 2009, 56, 205–219 Search PubMed
- I. M. Power, P. A. Kenward, G. M. Dipple and M. Raudsepp, Cryst. Growth Des., 2017, 17, 5652–5659 Search PubMed
- C. W. Bock, G. D. Markham, A. K. Katz and J. P. Glusker, Inorg. Chem., 2003, 42, 1538–1548 Search PubMed
- Z. Qian, H. Feng, C. Wang and J. Chen, Inorg. Chim. Acta, 2010, 363, 3627–3631 Search PubMed
- G. D. Saldi, G. Jordan, J. Schott and E. H. Oelkers, Geochim. Cosmochim. Acta, 2009, 73, 5646–5657 Search PubMed
- Q. R. S. Miller, J. P. Kaszuba, H. T. Schaef, M. E. Bowden, B. P. McGrail and K. M. Rosso, Chem. Commun., 2019, 55, 6835–6837 Search PubMed
- S. T. Mergelsberg, S. N. Kerisit, E. S. Ilton, O. Qafoku, C. J. Thompson and J. S. Loring, Chem. Commun., 2020, 56, 12154–12157 Search PubMed
- P. A. Baker and M. Kastner, Science, 1981, 213, 214–216 Search PubMed
- H. E. King and C. V. Putnis, Geochim. Cosmochim. Acta, 2013, 109, 113–126 Search PubMed
- W. W. Rudolph, G. Irmer and G. T. Hefter, Phys. Chem. Chem. Phys., 2003, 5, 5253–5261 Search PubMed
- S. Friesen, G. Hefter and R. Buchner, J. Phys. Chem. B, 2019, 123, 891–900 Search PubMed
- K. M. Callahan, N. N. Casillas-Ituarte, M. Roeselová, H. C. Allen and D. J. Tobias, J. Phys. Chem. A, 2010, 114, 5141–5148 Search PubMed
- T. Dudev, J. A. Cowan and C. Lim, J. Am. Chem. Soc., 1999, 121, 7665–7673 Search PubMed
- J. A. Roberts, P. A. Kenward, D. A. Fowle, R. H. Goldstein, L. A. González and D. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14540–14545 Search PubMed
- U. Kelka, M. Veveakis, D. Koehn and N. Beaudoin, Sci. Rep., 2017, 7, 14260 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and data. See DOI: 10.1039/d1ce00052g |
| This journal is © The Royal Society of Chemistry 2021 |