
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid state structure and properties of phenyl diketopyrrolopyrrole derivatives†
Joshua
Humphreys
ab,
Flavia
Pop‡
 ab,
Paul A.
Hume
c,
Alanna S.
Murphy
ab,
William
Lewis§
b,
E. Stephen
Davies
b,
Stephen P.
Argent
b and
David B.
Amabilino
*ab
ab,
Paul A.
Hume
c,
Alanna S.
Murphy
ab,
William
Lewis§
b,
E. Stephen
Davies
b,
Stephen P.
Argent
b and
David B.
Amabilino
*ab
aThe GSK Carbon Neutral Laboratories for Sustainable Chemistry, The University of Nottingham Jubilee Campus, Triumph Road, Nottingham NG7 2TU, UK
bSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: David.Amabilino@nottingham.ac.uk
cMacDiarmid Institute for Advanced Materials and Nanotechnology and School of Chemical and Physical Sciences, Victoria University of Wellington, Wellington 6010, New Zealand
First published on 4th February 2021
Abstract
The solid state supramolecular interactions of diketopyrrolopyrrole derivatives (DPPs) and their correlation with thin film optical properties are of particular interest because of the applications of these materials in organic electronics. In this study, we report the single crystal X-ray structures of several phenyl DPP derivatives, containing 4-methoxyphenyl, 4-hydroxyphenyl and 4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl aryl units, and show how subtle changes in the substituent chains at side or end positions of the chromophore can lead to very different packing. They are compared to their phenyl counterpart to explore how the nature of both the alkyl chain and the aryl unit influence the optical properties that have been measured in solid and solution states. Importantly, for the three families of N-substituted compounds studied, the structures are changed by the conformation of the molecules and are apparently dominated by crystal packing effects where edge-to-face interactions are favoured rather than π stacking, with only one of the compounds showing a flat form, promoted by intermolecular contacts between the aromatic regions. It is therefore possible that the twist between DPP and phenyl units in crystals of DPPs results from edge-to-face interactions (rather than steric interactions between the N-substituent and the protons attached to the aromatic ring) that might be overcome in more extended structures. Hydrogen bonding dominates the packing to generate chains of DPP units for phenol derivatives. Remote bulky groups do affect the core conformation. The emission of the materials as thin films is dominated by local effects in the packing of the materials that are unique for each case as the structures are distinct from one another. Charge mobility (as calculated from the crystal structures) is not favoured because of twisted conformations and large displacement, but the sometimes high emission and large Stokes shift could make the materials interesting for other purposes, such as light emitters.
Introduction
Diketopyrrolopyrrole derivatives (DPPs) are an increasingly important family of organic semiconductors,1–6 in part because of the synthetic possibilities through which their molecular properties can be modified to produce materials with distinct functionality. Ciba-Geigy first developed their synthesis and commercialised the materials as pigments for the automotive industry,3,7–9 owing to the high thermal stability and low solubility of the unsubstituted lactam materials (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H in Fig. 1). These properties arise from intermolecular interactions in the solid state, principally hydrogen bonding between the lactam units, and π⋯π stacking between the largely planar chromophores.3,10–15 More recent work has attempted to modify these intermolecular interactions in order to alter solubility, crystal packing and optoelectronic properties.16–21 Therefore it is surprising that a correlation between their solid state conformations and supramolecular arrangements and optical properties remains largely unexplored.
H in Fig. 1). These properties arise from intermolecular interactions in the solid state, principally hydrogen bonding between the lactam units, and π⋯π stacking between the largely planar chromophores.3,10–15 More recent work has attempted to modify these intermolecular interactions in order to alter solubility, crystal packing and optoelectronic properties.16–21 Therefore it is surprising that a correlation between their solid state conformations and supramolecular arrangements and optical properties remains largely unexplored.
 | ||
| Fig. 1 Chemical structures of the DPP compounds that are the subject of this paper. | ||
DPP-derived materials have found use in a plethora of applications,1–6 most notably organic electronics, sensing and bio-imaging, owing to their high absorption and emission across the visible spectrum.7,22–24 As components in organic electronics, DPPs are most often incorporated as donor materials in organic photovoltaic devices,25–31 although examples that are electron deficient are under development to rival “Y6” and related non-fullerene acceptors.5,25,32–36
The most widely investigated derivatives of DPP contain thiophene (ThDPP),30,31,37–39 because of their efficient synthesis and ready polymerisation. In contrast, phenyl based materials (PhDPP, Fig. 1) have received less attention,25 partly because the N-alkylation of the lactam core causes a twist of the aryl rings out of DPP plane, thus interrupting extended conjugation. This twisting arises from steric interactions between the hydrogen atoms at the 2-position of the phenyl ring with the N-substituent on the DPP, leading to less favourable intermolecular interactions when compared to thiophene and furan based materials, whose planarity is retained after alkylation.21 Whilst twisting in the PhDPPs is unfavourable for applications that require highly conjugated molecules it does lead to molecules with relatively wide band gaps (>1.9 eV),40 and as such these materials have other potential applications, namely morphological and optoelectronic additives in materials for devices.41–44
Generally, solutions of PhDPPs show improved quantum yields when compared with thiophene and furan variants, attributed to the heavy atom effect quenching fluorescence in the latter pair.21 Despite this feature, DPPs find little use in lighting applications, a result of the strong intermolecular interactions quenching emission in the solid-state.45 However N-alkylation of PhDPP derivatives breaks the hydrogen bonding and modulates the π⋯π stacking interactions, leading to emission in the solid state. This behaviour opens up new potential applications for these materials as exciplex emitters in organic light emitting diodies,46 in addition to their existing use as biological imaging agents in two photon fluorescence microscopy and optical analyte sensing applications.1,47–55
In this study, we investigate the solid state packing and optical properties of three families of PhDPPs (Fig. 1) that are distinguished by the nature of the N-substituent on the heterocycle, where it is either the n-hexyl chain (Hex),56 branched (S)-2-methyl-butyl (2-MB)23 or tert-butyloxycarbonyl (Boc) group.57 These lateral groups have varying degrees of conformational space available to them, as well as differing orientational flexibility with respect to the DPP core because of their varying volume and branched nature. The hexyl chain can adopt orientations defined by the torsion around bond that unites it to the lactam nitrogen atom. On the other hand, the all-trans conformation expected for the hexyl unit affords the possibility of van der Waals interactions between the chains, an interaction that would be expected to be less dominant for the other substituents.
In addition, the bulk at the 4-position of the phenyl ring was changed, from a proton in 4-hydroxyphenyl variants (PhOHDPP) where hydrogen bonding can also play a role, to the 4-methoxyphenyl unit (PhOMeDPP), the 4-(tert-butyloxycarbonyl)phenyl (PhOBocDPP) and the 4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl unit (PhOTHPDPP). We anticipated that this group could also affect the stacking of the DPP units.
Of the compounds in these series that were prepared, seven crystallised and had their structures solved, and they are reported here and compared with the known PhDPP N-Hex21,58 and PhDPP N-Boc59–61 in order to understand the influence of the nature of the alkyl chain and the aryl unit on the solid-state packing and optical properties.
Results and discussion
Synthesis
The parent PhDPP compounds were synthesised using the established succinate method from their aryl carbonitrile precursor (see ESI† for full details).7,21,56,62 Reaction of the lactam to give the N-alkylated derivatives for the phenyl, 4-methoxyphenyl and 4-((tetrahydro-2H-pyran-2-yl)oxy)phenyl materials employed basic substitution at the lactam unit to increase solubility of the material using a metal carbonate and an appropriate alkyl halide in a polar solvent at high temperature.7,21,56,63 Access to the 4-hydroxyl materials was through deprotection of their methoxy counterparts with BBr3.56 The Boc decorated materials were synthesised from PhOMeDPP N-H and PhOHDPP N-H utilising standard Boc protection conditions.64The PhDPP, PhOMeDPP and PhOTHPDPP materials exhibit high solubility in a range of solvents including toluene, tetrahydrofuran, dichloromethane and acetone. The demethylation of the alkylated PhOMeDPPs to give the alkylated PhOHDPPs led to a decrease in solubility, presumably as a result of the hydrogen bonding between the molecules. However, these materials maintained high solubility in THF as well as more polar solvents such as DMF and DMSO and were also more soluble than PhOHDPP N-H. The branched chain derivative, PhOHDPP N-2MB, displays excellent solubility in acetone, unlike its hexyl counterpart. However, when compared with PhOMeDPP N-Boc, the introduction of two additional Boc units on the aryl ring of PhOBocDPP N-Boc leads to a loss of solubility in many common organic solvents, with significant solubility only observed in dichloromethane and chloroform.
Single crystal structure analysis
Single crystals of the new compounds were obtained through slow evaporation of concentrated solutions. The structural data and crystallisation conditions for the new compounds are shown in Table 1. In the discussion that follows, the structures and packing parameters are compared to the known PhDPP N-Hex and PhDPP N-Boc crystals.21,58–61,65| Compound | Crystallisation conditions | Space group | Z′ |
|---|---|---|---|
| PhOHDPP N-Hex | THF |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
0.5 |
| Slow evaporation | |||
| PhOMeDPP N-Hex | Xylene | P21/c | 0.5 |
| Slow evaporation | |||
| PhOTHPDPP N-Hex | DCM | P21/c | 0.5 |
| Slow evaporation | |||
| PhOHDPP N-2MB | Acetone | P21 | 1 |
| Low temperature | |||
| PhOMeDPP N-2MB | THF | P21 | 1 |
| Slow evaporation | |||
| PhOMeDPP N-Boc | Toluene | P21/n | 0.5 |
| Slow evaporation | |||
| PhOBocDPP N-Boc | DCM | I2/a | 0.5 |
| Slow evaporation |
The shapes of the PhDPP molecules can be defined by three angles around the core (Fig. 2, see also ESI† Fig. S1 and S2) that characterise the conformations of the compounds. These correspond to the dihedral angle formed by the twist of the plane of phenyl ring out of the plane of the DPP core, the torsion angle between the N-substituent and the lactam moiety (C(O)–N–C–C/O) and the angle of the α-carbon atom in the N-substituting chain with respect to the plane of the DPP core (Fig. 2).
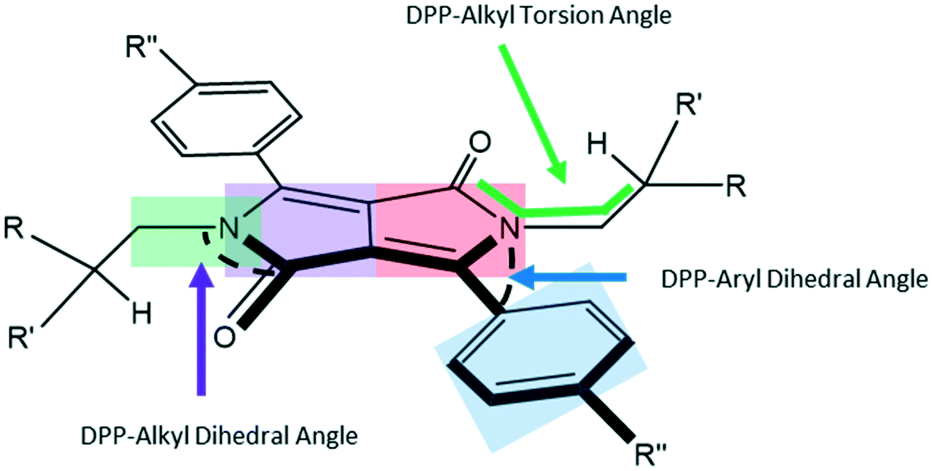 | ||
| Fig. 2 Two key dihedral angles that define the conformation of the PhDPP molecules. Highlighted areas indicate the atoms selected to define the planes between which the dihedral angles are measured. Green and purple planes DPP-alkyl, red and blue planes DPP-aryl. | ||
Generally, the molecules have a twist between the aromatic planes, often assigned to the modest steric clash between the α-methylene groups of the alkyl chain and the C–H at the 2-position of the aryl ring with respect to the DPP core. An exception is PhOMeDPP N-Boc that shows an essentially planar DPP-phenyl core. The generally large dihedral angles between the rings range from 31 to 52° (Table 2). There is no apparent correlation between the choice of aryl substituent or alkyl chain and the dihedral angle in this data set (see ESI† for graphs), indicating that the general crystal packing forces are the major driving force behind this conformational feature, and most of the compounds have edge-to-face type interactions that are apparently preferred and could favour this packing of the molecules, although there are generally multiple apparently weak interactions (ESI† Tables S1–S10).
| Compound | C–H⋯carbonyl distance (Å) | DPP-aryl dihedral anglea (°) | DPP-R torsionb (°) | DPP-R dihedral anglec (°) | Vertical displacement (Å) | Longitudinal displacement (Å) | Lateral displacement (Å) | Overall displacement (Å) | π interaction type | Centroid⋯centroid distance (Å) | C–H⋯centroid distance (Å) | C–H⋯centroid angle (°) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Defined as the dihedral angle of the planes of the DPP and aryl ring. b The torsion angle formed by the DPP amide to substituent: C(O)–N–C–C/O. c The dihedral angle formed between the DPP plane the C–N–C (see Fig. 2 and S2†). | ||||||||||||
| PhDPP N-Hex 21 | 2.600 | 34.25 | 78.45 | 7.88 | 4.13 | 3.27 | 1.71 | 3.69 | CH–π (Ph–Ph) | 2.900 | 131.38 | |
| CH–π (Ph–Lac) | 3.038 | 122.83 | ||||||||||
| PhOHDPP N-Hex | 2.503 | 31.33 | 81.95 | 5.91 | 4.07 | 3.01 | 1.51 | 3.37 | CH–π (Ph–Lac) | 3.049 | 119.15 | |
| PhOMeDPP N-Hex | 2.842 | 40.81 | 96.93 | 16.42 | 3.55 | 10.81 | 1.18 | 10.87 | CH–π (Ph–Lac) | 4.221 | 2.826 | 172.23 |
| Parallel displaced (Ph–Ph) | ||||||||||||
| PhOTHPDPP N-Hex | 3.000 | 48.70 | 95.32 | 6.54 | 3.68 | 8.80 | 0.97 | 8.85 | Parallel displaced (Ph–Ph) | 3.813 | ||
| Sandwich (OTHP-Lac) | 4.249 | |||||||||||
| PhOHDPP N-2MB | 2.882, 3.099 | 44.54, 51.75 | −104.27106.70 | 12.21, 1.39 | 4.57 | 10.35 | 0.51 | 10.35 | CH–π (Ph–Ph) | 4.791 | 3.209 | 134.66 |
| Parallel displaced (Ph–Ph) | ||||||||||||
| PhOMeDPP N-2MB | 2.625, 2.706 | 35.95, 37.38 | −105.93, 116.42 | 1.64, 8.13 | 3.72 | 10.38 | 0.58 | 10.40 | CH–π (Ph–Lac) | 4.065 | 3.195 | 167.07 |
| Parallel displaced (Ph–Ph) | ||||||||||||
| β PhDPP N-Boc 61 | 2.445 | 29.25 | 47.50 | 14.19 | 4.22 | 0.54 | 5.59 | 5.62 | Parallel displaced (Ph–Ph) | 4.415 | ||
| 2.654 | 34.48 | 53.58 | 14.65 | CH–π (Ph–Ph) | 4.349 | 155.76 | ||||||
| α PhDPP N-Boc A 60 | ||||||||||||
| 2.413 | 25.60 | 65.96 | 20.99 | 4.15 | 3.32 | 4.48 | 5.58 | Parallel displaced (Lac–Lac) A + B | 4.141 | |||
| α PhDPP N-Boc B 60 | 2.406 | 26.09 | −55.48 | 30.46 | 4.12 | 1.17 | 2.16 | 2.46 | ||||
| 2.759 | 42.26 | 49.07 | 16.04 | CH–π (Ph–Ph) A + B | 2.859 | 146.67 | ||||||
| CH–π (Ph–Ph) B only | 2.727 | 114.22 | ||||||||||
| PhOMeDPP N-Boc | 2.190 | 1.46 | 82.60 | 0.76 | 3.31 | 5.35 | 0.10 | 5.35 | Parallel displaced (Ph–Lac) | 3.432 | ||
| 14.99 | ||||||||||||
| PhOBocDPP N-Boc | 2.617 | 31.52 | 63.41 | 14.99 | 3.59 | 9.67 | 0.59 | 9.69 | Parallel displaced (Ph–Ph) | 3.721 | ||
| 6.54 | ||||||||||||
The aryl substituent does influence the overall shape in the crystals. The conformation of PhOHDPP N-2MB in its crystals displays the largest dihedral angles between DPP and phenyl moieties in the series, of 44.5°and 51.8° for the two symmetry independent units (the latter being the highest reported to date for PhDPP derivatives). PhOHDPP N-Hex has a considerably smaller dihedral angle (31.3°). The methoxy analogues show the opposite trend, the dihedral angles for both derivatives are closer in magnitude (40.8° for the N-Hex derivative and 36.0° and 37.4° for N-2MB). PhOTHPDPP N-Hex displays a dihedral of 48.7°, large in comparison to the other hexyl substituted species, which could be attributed to the volume of the OTHP unit placing constraints on packing.
The N-Hex series show an increase in the magnitude of the aryl dihedral as the volume of the R′ group increases, with the exception of PhOHDPP N-Hex, presumably owing to the hydrogen bonding present in that compound. The increased magnitude of the aryl dihedral for the PhOMe derivatives correlates with increase in volume of the alkylating group at the lactam nitrogen. The magnitude of the intramolecular C–H (6-position of the phenyl ring) carbonyl distance correlates well with the dihedral angle in all cases, giving confidence in the measurement (Table 2). In terms of the magnitude of the N–R angle for the alkyl species, most display a moderate twisting of the chain out of the plane. Interestingly PhOMeDPP N-Hex displays a gauche conformation in the hexyl chain and a much larger deviation out of the plane (16.4°) pointing almost vertically.
We showed that ThDPP N-alkylated derivatives had correlations between molecular displacement and angles of aryl rings and N–R bonds out of the plane.20 The relative magnitude of displacement between neighbouring molecules for these phenyl derivatives can be appreciated by comparing the structures shown in Fig. 3–5 and the trends in molecular displacement rationalised by consideration of the π⋯π stacking interactions in these materials (Fig. 6–8), where the nature of the substituents have a large influence.
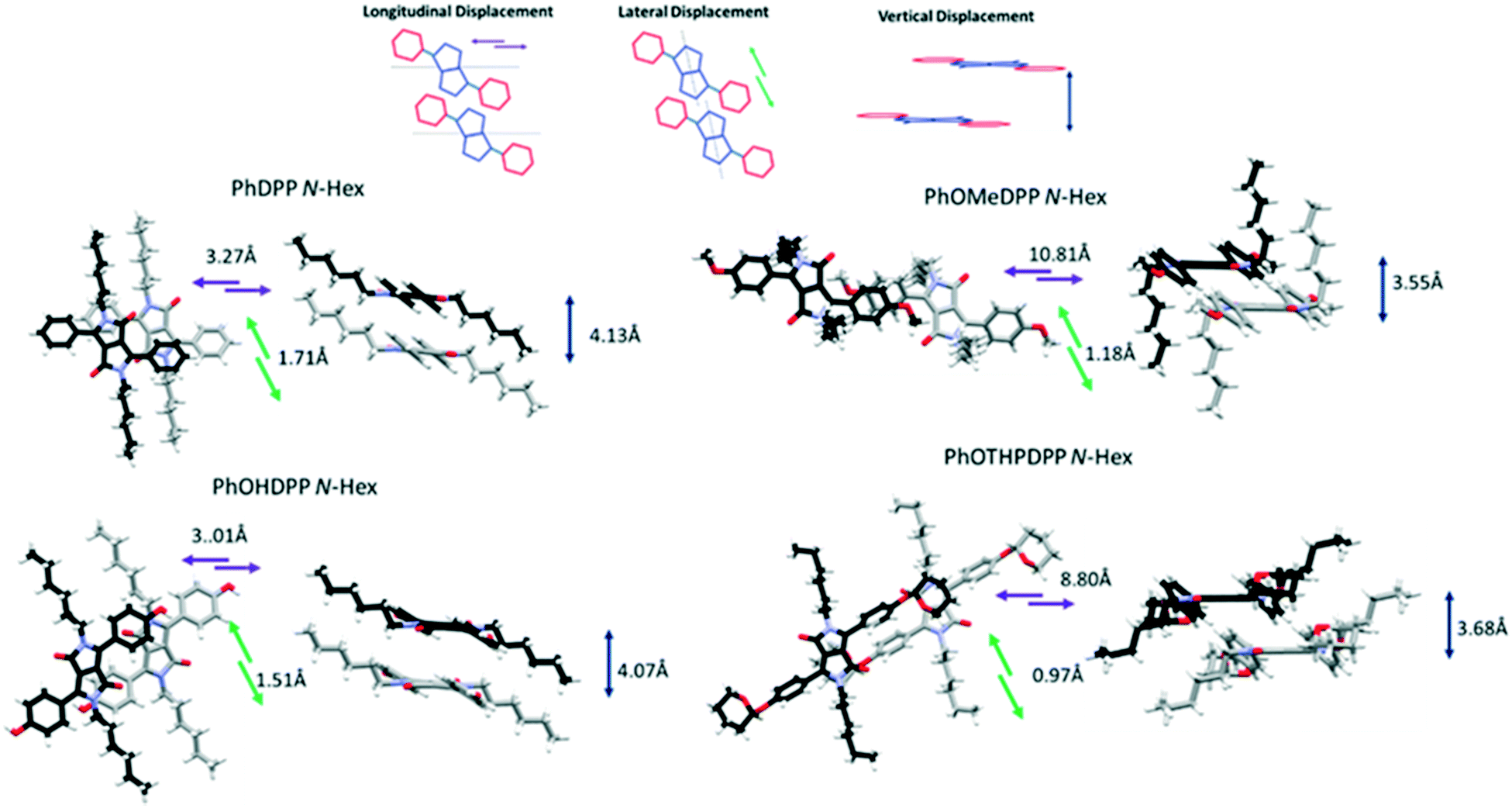 | ||
| Fig. 3 The longitudinal, lateral and vertical displacement of PhDPP N-Hex, PhOMeDPP N-Hex, PhOHDPP N-Hex, and PhOTHPDPP N-Hex. | ||
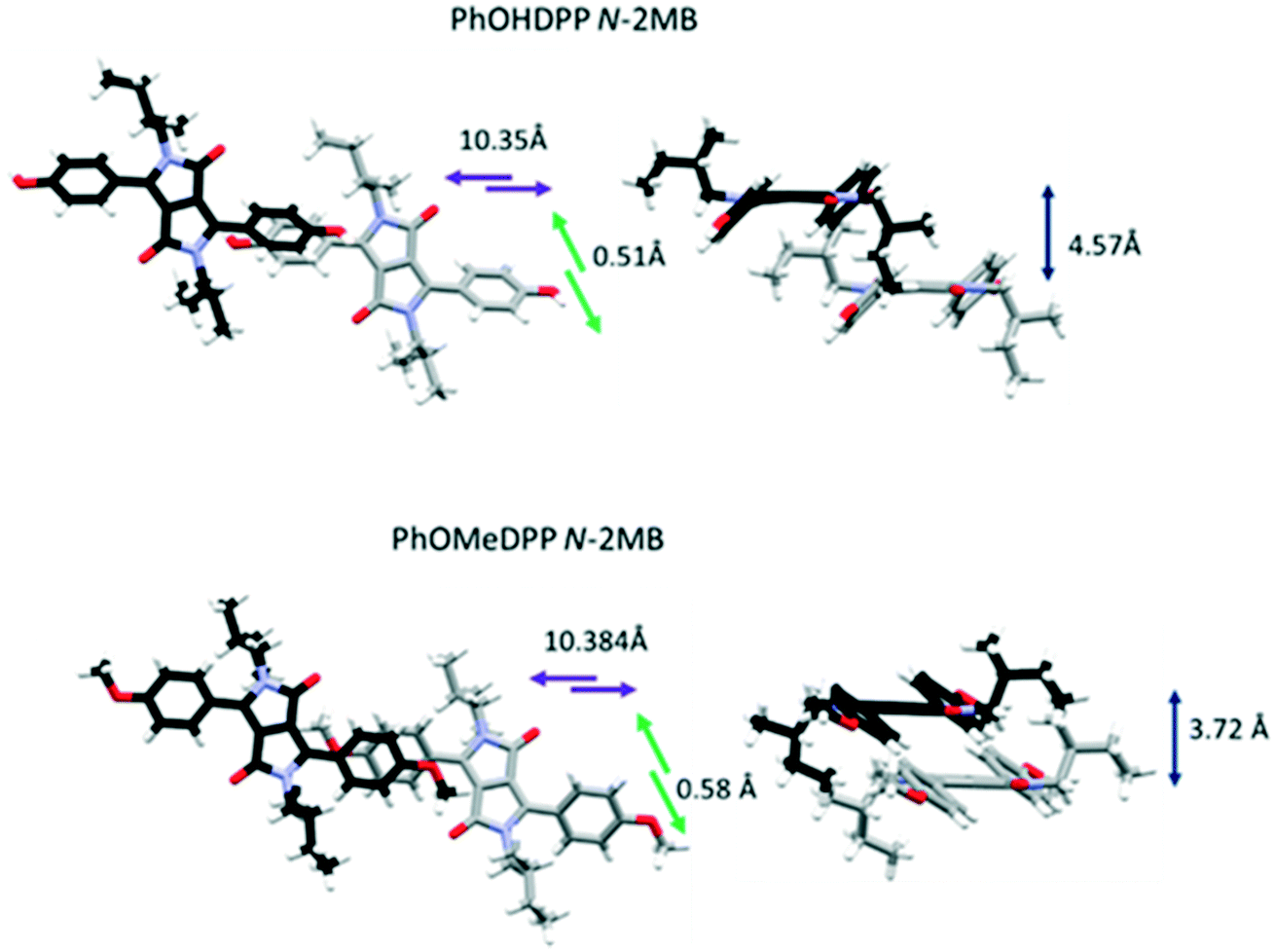 | ||
| Fig. 4 The longitudinal, lateral and vertical displacement of PhOMeDPP N-2MB, and PhOHDPP N-2MB. | ||
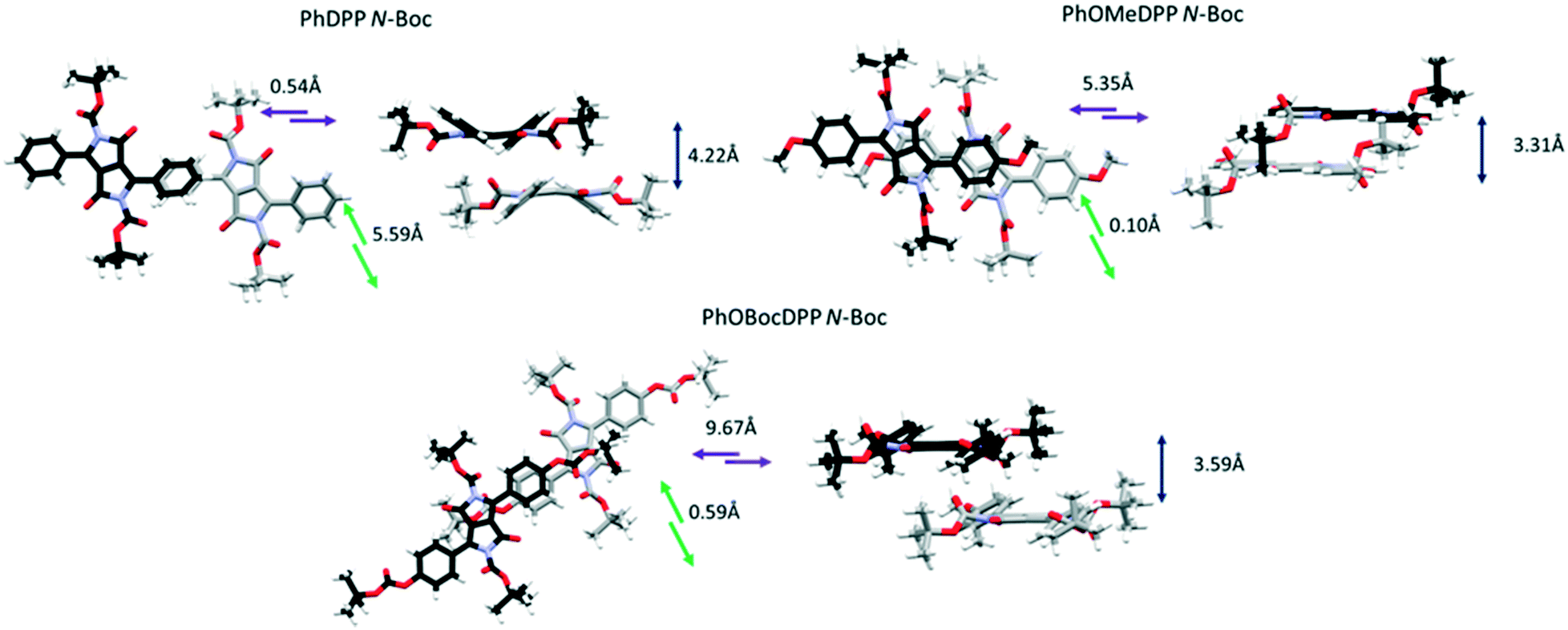 | ||
| Fig. 5 The longitudinal, lateral and vertical displacement of PhDPP N-Boc, PhOMeDPP N-Boc and PhOBocDPP N-Boc. | ||
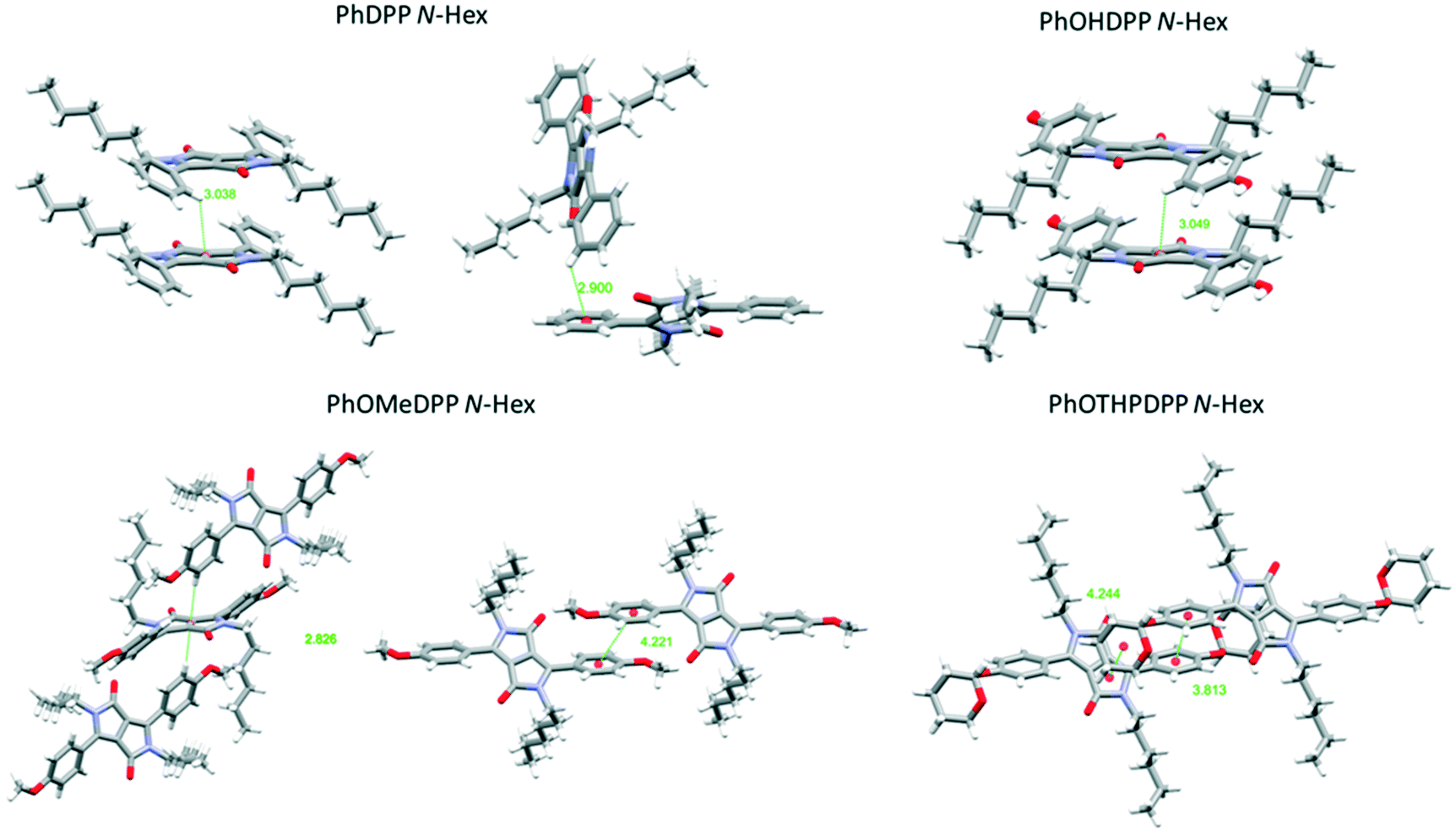 | ||
| Fig. 6 Views of the principle π⋯π stacking interactions of PhDPP N-Hex, PhOMeDPP N-Hex, PhOHDPP N-Hex, and PhOTHPDPP N-Hex. | ||
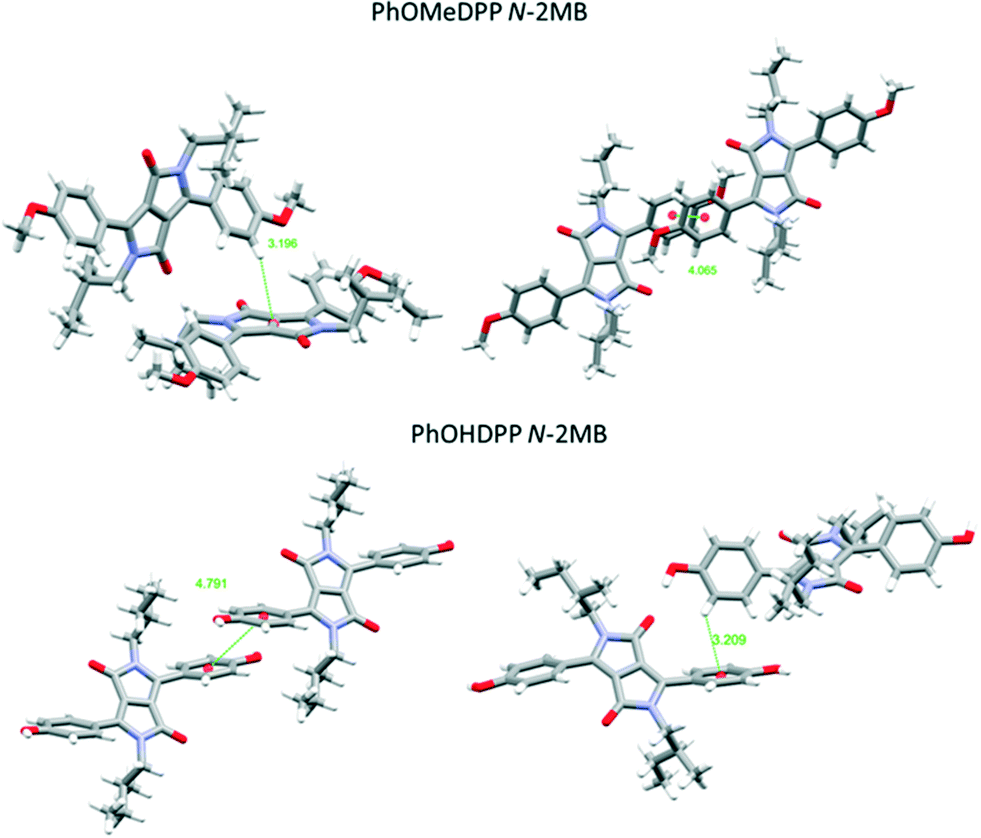 | ||
| Fig. 7 Views of the principle π⋯π stacking interactions of PhOMeDPP N-2MB and PhOHDPP N-2MB. | ||
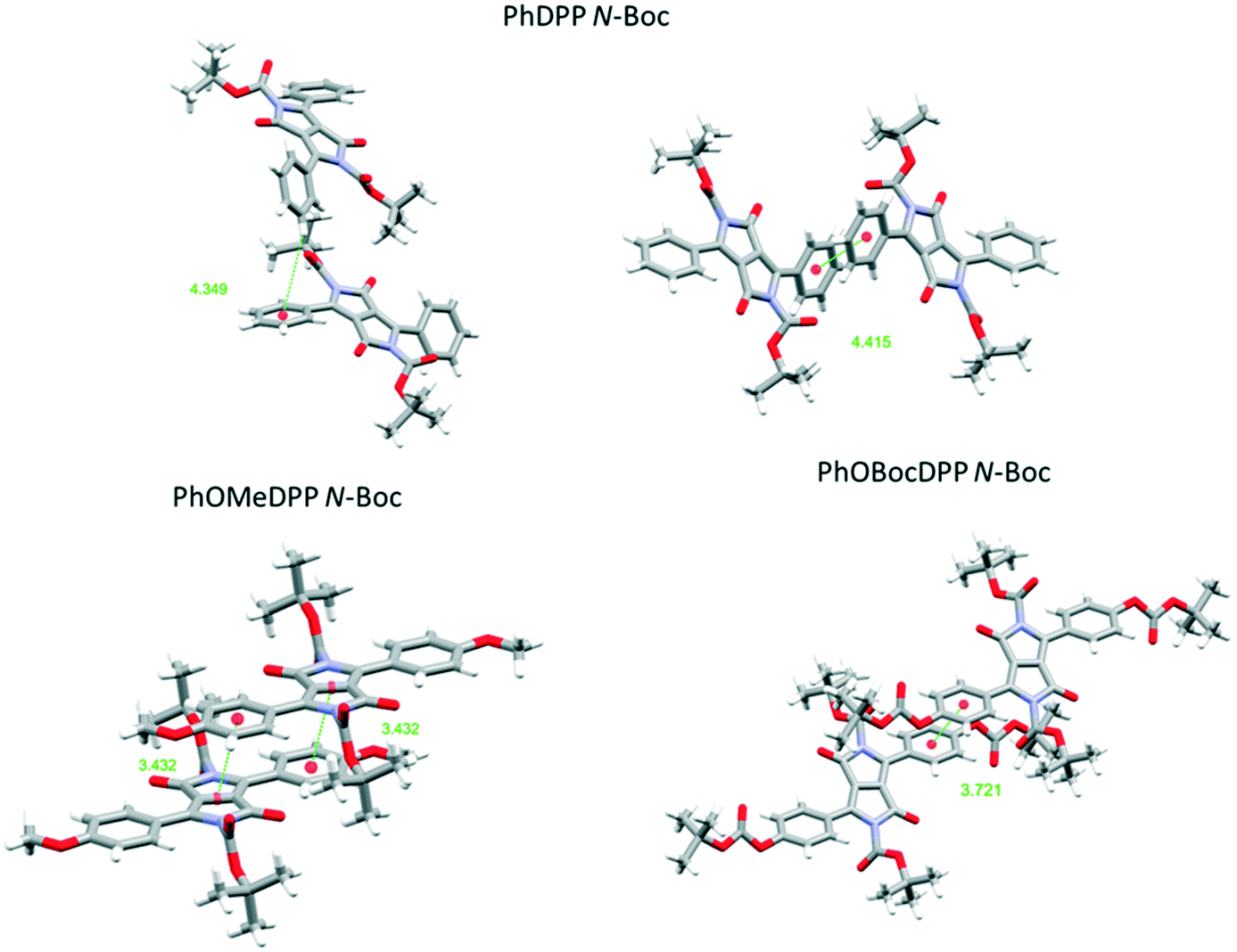 | ||
| Fig. 8 Views of the principle π⋯π stacking interactions of PhDPP N-Boc PhOMeDPP N-Boc and PhOBocDPP N-Boc. | ||
PhDPP N-Hex and PhOHDPP N-Hex both display moderate displacement vertically, longitudinally and laterally. In terms of π⋯π stacking interactions, PhDPP N-Hex displays T-shaped interactions between neighbouring columns and these occur between adjacent phenyl rings (2.90 Å) and also between phenyl (donor) and neighbouring lactam (acceptor) units (3.04 Å).58 These interactions are not perpendicular (131.4° and 122.8°), because of the twisting of the phenyl rings out of the plane. The π⋯π phenyl–phenyl distance is 5.54 Å with no overlap (ESI† Fig. S3).21PhOHDPP N-Hex also displays a long C–H⋯π interaction between the phenyl system and lactam core (3.05 Å) and the rings are also not perpendicular (119.1°). In addition, there are no significant π⋯π contacts, the distance is 5.28 Å between the two phenyl ring centroids (ESI† Fig. S4).
The larger the volume of R′ apparently leads to increased displacement but the presence of the phenol motifs hydrogen bond network results in reduced dihedral angles, smaller molecular displacement and a single marginally elongated π stacking interaction compared with PhDPP N-Hex (which possesses two shorter C–H⋯π interactions), suggesting hydrogen bonding is the dominant intermolecular interaction for this species.
The additional C–H⋯π (Ph–Ph) interaction of PhDPP N-Hex is the likely cause of slightly larger dihedral angle and molecular displacement. PhOMeDPP N-Hex displays the largest longitudinal displacement and subsequently overall displacement. This larger displacement allows for a short (2.83 Å), almost perpendicular (171.8°), CH–π interaction between the lactam core and two adjacent PhOMe rings. In addition, the larger displacement affords an additional parallel displaced (Ph–Ph) interaction of 4.22 Å, a much closer π–π contact than observed for PhDPP N-Hex or PhOHDPP N-Hex. Face-centred π⋯π stacking is disfavoured between the phenyl rings, and hence a displaced sandwich interaction65 is observed. PhOTHPDPP N-Hex, with its larger dihedral of 48.7°, also displays a relative large longitudinal displacement (8.80 Å). In contrast to the other hexyl species there are no CH–π interactions and instead a parallel displaced (Ph–Ph) π–π interaction (3.81 Å) is observed.
Turning to the packing of the chiral 2MB derivatives, these are especially relevant because they have very similar topology to the 2-ethyl hexyl unit that is widely used for N-alkylation of DPPs for optoelectronic applications.66–68 For PhOMeDPP N-2MB an edge to face interaction (3.20 Å) between the phenyl (donor) and lactam core (acceptor) is observed that is close to perpendicular (167.7°) (similar to PhOMeDPP N-Hex). A long parallel displaced (Ph–Ph) π–π interaction is observed (4.07 Å), which could result from the greater bulk of the 2MB chain compared to the hexyl counterpart (Fig. 4). The displacement is dominated by a longitudinal shift giving a value like the hexyl species, with the lateral displacement reduced. The hydrogen bond-containing PhOHDPP N-2MB gives similar longitudinal and lateral displacement to its methoxy counterpart, and larger than PhOHDPP N-Hex. PhOHDPP N-2MB has a CH–π interaction between two aryl rings and a parallel displaced (Ph–Ph) contact. As with PhOHDPP N-Hex, this interaction is angled (134.1°) and elongated compared with the methoxy analogue (3.21 Å). Moreover, the parallel displaced interaction is also at greater distance than the methoxy analogue with a large centroid–centroid distance (4.79 Å) and less overlap of aryl units. This less significant π interaction as seen for PhDPP N-Hex (5.54 Å) and PhOHDPP N-Hex (5.28 Å) correlates with a larger vertical displacement (>4 Å) (see Fig. 9 for a correlation, vide infra).
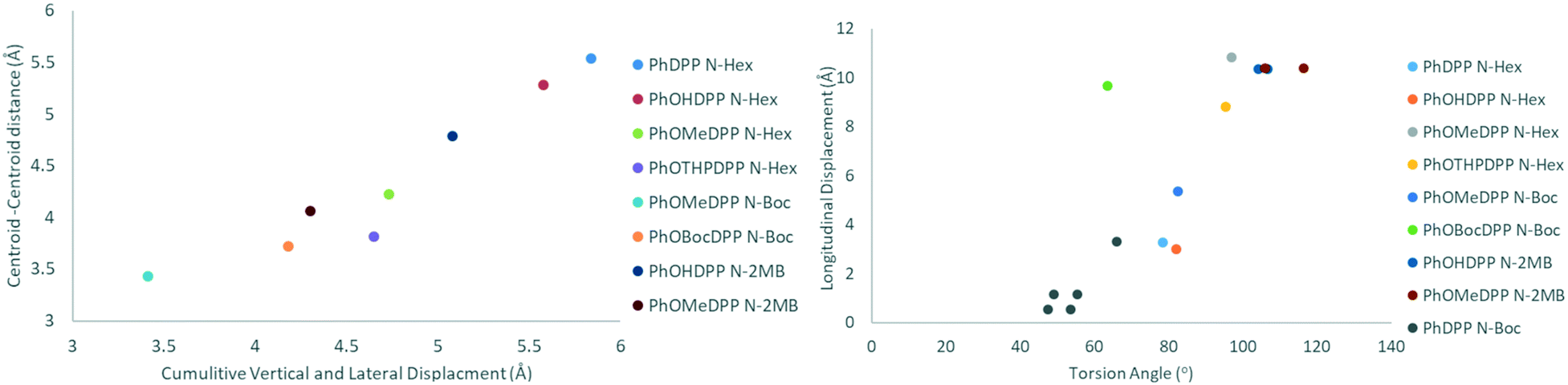 | ||
| Fig. 9 Plots of cumulative vertical and lateral displacement vs. the parallel displaced π π centroid–centroid distance and torsional angle vs. longitudinal displacement for the subjects of this paper. | ||
For the N-Boc family of compounds PhOMeDPP N-Boc is essentially planar (the DPP-aryl twist angle is 1.46°) in the resolved structure. In other DPP systems containing this unit polymorphism has been observed,69 and we also saw evidence of this phenomenon. Crystals of PhOMeDPP N-Boc grown from a dichloromethane solution had a different space group, that was similar to systems with dihedral angles of over 30°, although crystal quality did not allow precise resolution of the structure. This observation confirms the polymorphic behaviour of derivatives of DPP containing this unit, but no interconversion was observed that might have provided a clue as to the most stable form. PhOBocDPP N-Boc displays larger dihedral angles (31.5°), akin to the alkyl systems, possibly as a result of the large total volume of substituent groups around the core. The two polymorphs of PhDPP N-Boc also have dihedral angles of this order between the aromatic rings.
For PhDPP N-Boc, the β form possesses a small longitudinal displacement (0.54 Å) and more substantial lateral displacement (5.59 Å), that has not been observed in the previously discussed structures. This results in a parallel displaced interaction between phenyl rings (4.42 Å) along with a C–H⋯π interaction between phenyl rings of 4.35 Å, significantly larger than is seen for PhDPP N-Hex. For the α form a similar vertical displacement to the β form is present with an increase in longitudinal displacement and reduction of lateral displacement for both molecules in the unit cell. As with the β form both molecules display a parallel displaced interaction between phenyl rings (4.14 Å) and C–H⋯π interaction between phenyl rings of 2.86 Å, both shorter than the β form. For the unit with smaller displacement an additional shorter C–H⋯π interaction (2.73 Å) is also present. In contrast, PhOMeDPP N-Boc possesses the shortest vertical (3.31 Å) and lateral (0.10 Å) displacement of this series and displays a parallel displaced phenyl–lactam interaction at much shorter distances (3.43 Å) and greater overlap compared to its phenyl analogue and the alkyl variants, with an absence of C–H⋯π interactions. This closer packing can be attributed to the planarity of the system allowing proximity of neighbouring chromophores and hence smaller displacement. PhOBocDPP N-Boc also displays a parallel displaced interaction (3.72 Å) but in contrast to PhOMeDPP N-Boc it is between phenyl rings, akin to the previously described N-alkyl variants, suggesting the increased distortion prevents closer packing.
Despite having similar magnitude of aryl and alkyl dihedral angles, there are large differences in the π contacts of PhDPP N-Boc and PhOBocDPP N-Boc with the large lateral slip in the former seemingly contributing to the less substantial interactions, indicated by the α form possessing shorter interactions in comparison to the β. Our group previously showed that the thiophene equivalent ThDPP N-Boc possessed similar attributes to its phenyl counterpart with the N–R bond being twisted out of the plane by 25.0° and lateral displacement dominating (4.77 Å), with a parallel displaced slipped stacking of 4.14 Å.20
All N-alkyl species in this study, except PhOTHPDPP N-Hex, display a C–H⋯π interaction of some kind. PhOMeDPP N-2MB and PhOMeDPP N-Hex possess short vertical displacement and large longitudinal displacement resulting in short CH–π interactions close to a perpendicular orientation of the interaction between the two π systems. Their phenol counterparts display increased vertical displacement attributed to the introduction of hydrogen bonding and hence elongated CH–π interactions twisted from ideal orientation, suggesting hydrogen bonding dominates over π interactions in these systems. This is seen most evidently for PhOMeDPP N-2MB and PhOHDPP N-2MB where the latter has a large dihedral angle and the C–H⋯π interaction not only distorts but is no longer between donor and acceptor moiety's (Ph–Lac) but between adjacent aryl rings.
Overall, our analyses show that the introduction of an auxochrome reduces the vertical and lateral displacement in this series of compounds, the exception being the PhOHDPP derivatives which display relatively large vertical displacement, ascribed to the presence of the hydrogen bond network aiding in-plane proximity where the non-covalent network exists with less proximal packing between these sheets. It has been seen previous that for ThDPP longitudinal displacement is affected by donor–acceptor intermolecular interactions and aryl twisting, whereas lateral displacement correlates with distortion of the N–R bond out of the plane.20 The subjects of this paper however display no such trends, but there is a clear influence of the terminal groups on the packing in each family of laterally substituted DPPs.
There is a strong correlation between cumulative vertical and lateral displacement and the parallel displaced π–π centroid–centroid distance (Fig. 9). In addition, a moderate positive correlation is observed between longitudinal displacement and torsion angle. However, no significant correlation was found for displacement against DPP-aryl dihedral, DPP-alkyl dihedral and alkyl torsion angles for any of the sub-families (see ESI† Fig. S5–19 for all plots). Outliers arise where specific intermolecular interactions modify packing. For instance, hydrogen bonding means that PhOHDPP N-2MB has a smaller displacement than expected for the size of the dihedral angle.
Considering the known PhDPP N-Hex further, the parent system PhDPP NH70 (DPP-aryl dihedral 6.73°) displays a displaced sandwich interaction between lactam units and also between phenyl units. Upon mono alkylation, the aryl dihedral increases to 21.31° resulting in an interaction between the phenyl ring and lactam unit being somewhere between a phenyl lactam sandwich and a CH π interaction (2.92 Å, 96.9°). The disubstituted compound, has increased distortion of the DPP-aryl angle with a further twisting to a CH–π interaction (122.0°). PhDPP N-Me70 has a slightly less distorted DPP-aryl dihedral (30.5°), resulting in greater overlap of the phenyl unit with lactam core. The correlation between dihedral angle and volume of the alkylating group is witnessed for the PhOMeDPP family. N-Alkyl systems in the most part seem to be dominated by edge to face interactions accompanying aryl twist.
Other PhDPP derivative structures show that the substituent at the 4-position of the phenyl ring dictates greatly the crystal packing. For example, 4-chlorophenyl DPP shows a displaced sandwich interaction71 as do the 2,4 fluorobromo and the 3,4 fluorobromo derivatives.72 Incorporation of benzyl units (N-Bz) with 4-substituted halides73–75 results in an interaction similar to that seen for PhDPP N-Hex and PhOHDPP N-Hex, with greater overlap between phenyl and lactam units, less twisting present, and an interaction closer to a classic co-facial slipped structure rather than the observed distorted interaction observed for the latter pair. This is also observed for N-Bz with phenyl,76 4-butoxybiphenyl,77 pentafluorobenzyl78 and 4-(((dimethylamino)phenyl)ethynyl)phenyl79 aryl units amongst others and, coupled with the additional interactions of the benzyl group, gives rise to high mobility materials. Regarding PhOTHPDPP N-Hex, para substitution of the phenyl with pinacol boronate79 and morpholin-4-yl,80 although electron withdrawing, results in a similar interaction with the lactam unit as seen for OTHP, suggesting the conformational flexibility is the reason.
In all cases, there are interactions in addition to π⋯π stacking and hydrogen bonding that contribute to the crystal packing. PhDPP N-Hex possesses no apparent short intermolecular interactions arising from the hexyl chain, only carbonyl-aryl C–H interactions. In PhOMeDPP N-Hex, interactions occur between the oxygen of the methoxy unit and phenyl C–Hs and between the carbonyl and the C–Hs of the hexyl chain and between hexyl chains on adjacent molecules. Interdigitation of the hexyl chains results in the aforementioned increase in longitudinal displacement but serves as to reduce the vertical displacement, manifesting in shorter and more perpendicular CH–π interactions owing to the large twist of the chain out of the plane. PhOHDPP N-Hex possesses similar interactions; short contacts between the hexyl unit and hydroxyl oxygen replace those between the carbonyl and hexyl chain found in the methoxy analogue since the carbonyl is engaged in hydrogen bonding to the phenol but an absence of interactions between hexyl chains, as was the case for PhDPP N-Hex also, results in increased inter-planar distance preventing close packing and an absence of π contacts for both. For PhOTHPDPP N-Hex, there are interactions between the carbonyl unit and C–H's of the phenyl ring, in addition to interactions between the phenyl ring and hexyl chain, carbonyl oxygen and hexyl chain and the pyran oxygen and hexyl chain. PhDPP N-Hex and PhOHDPP N-Hex display similar layered structures (Fig. 10) plausible from the similarity in displacement and π interactions, with the chromophores situated quite far apart preventing close packed π stacks. The interactions of the hexyl chain of PhOMeDPP N-Hex result in a layered structure and substantial CH–π interaction of the phenyl ring and lactam core. The interaction of the hexyl chain with the oxygen of the OTHP ring in PhOTHPDPP N-Hex results in a bending of the hexyl chain resulting in the layered structure (Fig. 10). This suggests that the R′ group has a substantial influence on the packing and additional intermolecular interactions of N-hexyl variants. It is noted that interactions of the hexyl chain vary considerably between the four structures described here.
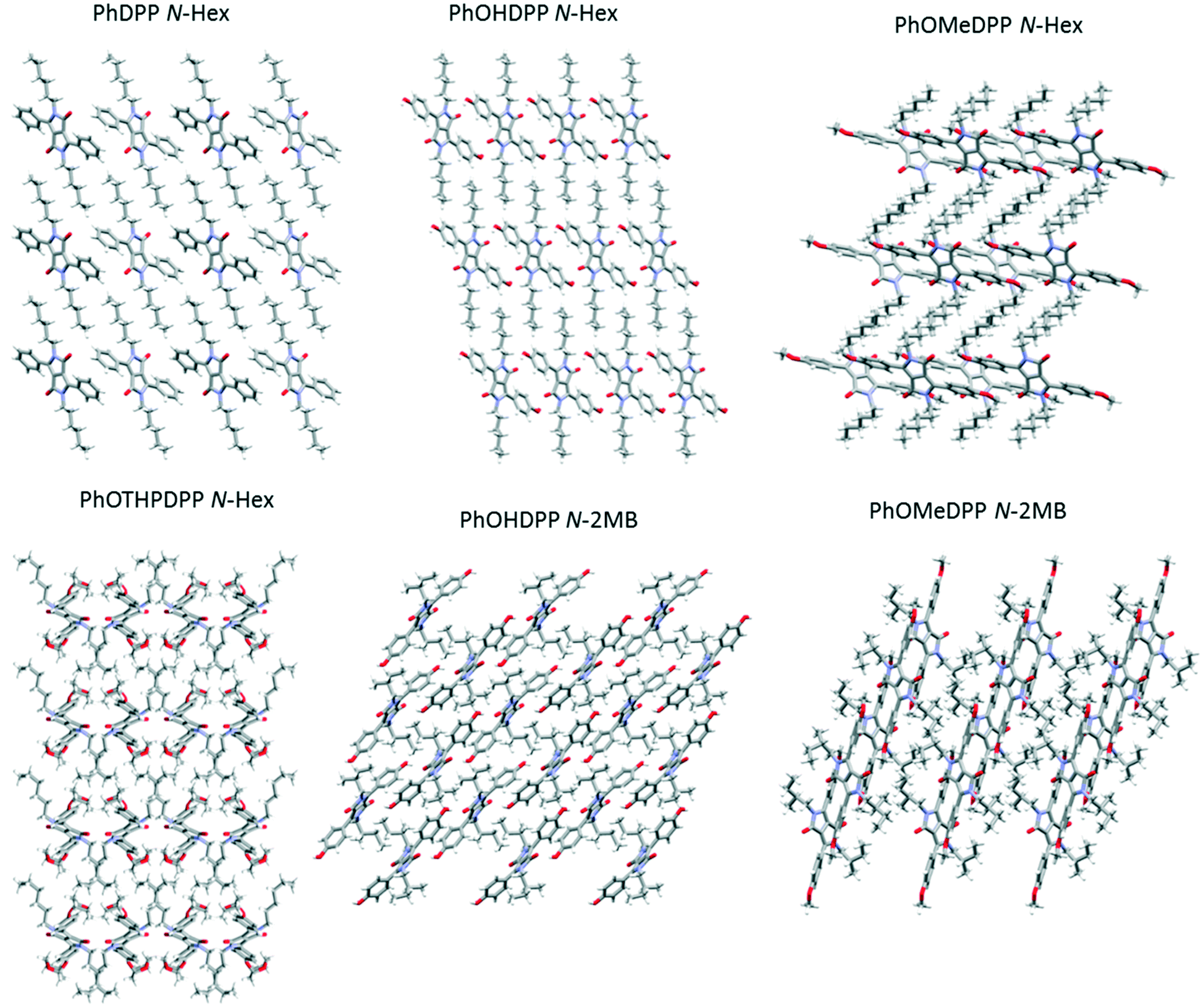 | ||
| Fig. 10 Views down the crystallographic axes of PhDPP N-Hex (b axis), PhOHDPP N-Hex (a axis), PhOMeDPP N-Hex (b axis), PhOTHPDPP N-Hex (c axis), PhOHDPP N-2MB (b axis), PhOMeDPP N-2MB (b axis) showing the alternate layers comprising the DPP core and the aliphatic regions. | ||
In PhOMeDPP N-2MB, interactions occur between the oxygen of the methoxy unit and phenyl C–H's, between the carbonyl and the C–H's of the 2MB chain, between the carbonyl and the C–H's of the phenyl and between the CH of the methoxy group and carbonyl. Unlike its hexyl variant, there is no interaction between 2MB chains on adjacent molecules. This results in the layered structure as seen in Fig. 10. For PhOHDPP N-2MB in addition to the hydrogen bonding and π interactions, the structure also possesses interaction of the carbonyl to aryl CH and interaction of the 2MB with the oxygen of the phenol and 2MB with lactam unit. As with PhOHDPP N-Hex, hydrogen bonding is influential, resulting in different interlayer packing (Fig. 10).
For the Boc series the α form of PhDPP N-Boc has interactions between the carbonyl and the aryl CH, the Boc CH and aryl and Boc carbonyl and CH of Boc. Both the α form and ThDPP N-Boc show interactions between the N-Boc units on adjacent molecules giving them the zig zag packing observed (Fig. 11). The β form has interactions between the carbonyl and the aryl CH, the Boc CH and aryl, and Boc carbonyl and aryl CH and there is an absence of interactions between Boc units giving rise to the lamellar packing. For PhOMeDPP N-Boc interactions between the carbonyl oxygen and C–H of the t-butyl unit, as well as interactions between the C–H's of the aryl unit with the methoxy unit are noted. For PhOBocDPP N-Boc interactions between the carbonyl oxygen and C–H of the N-t-butyl unit as well as additional interactions between the carbonyl of the N-Boc unit with C–H of the aryl unit, the carbonyl of the OBoc unit with C–H of the aryl unit and interaction of the OBoc and N-Boc units gives a cross shaped packing structure, previously seen for PhOMeDPP N-PMB81 and attributed to the twisting of the flanking phenyls.
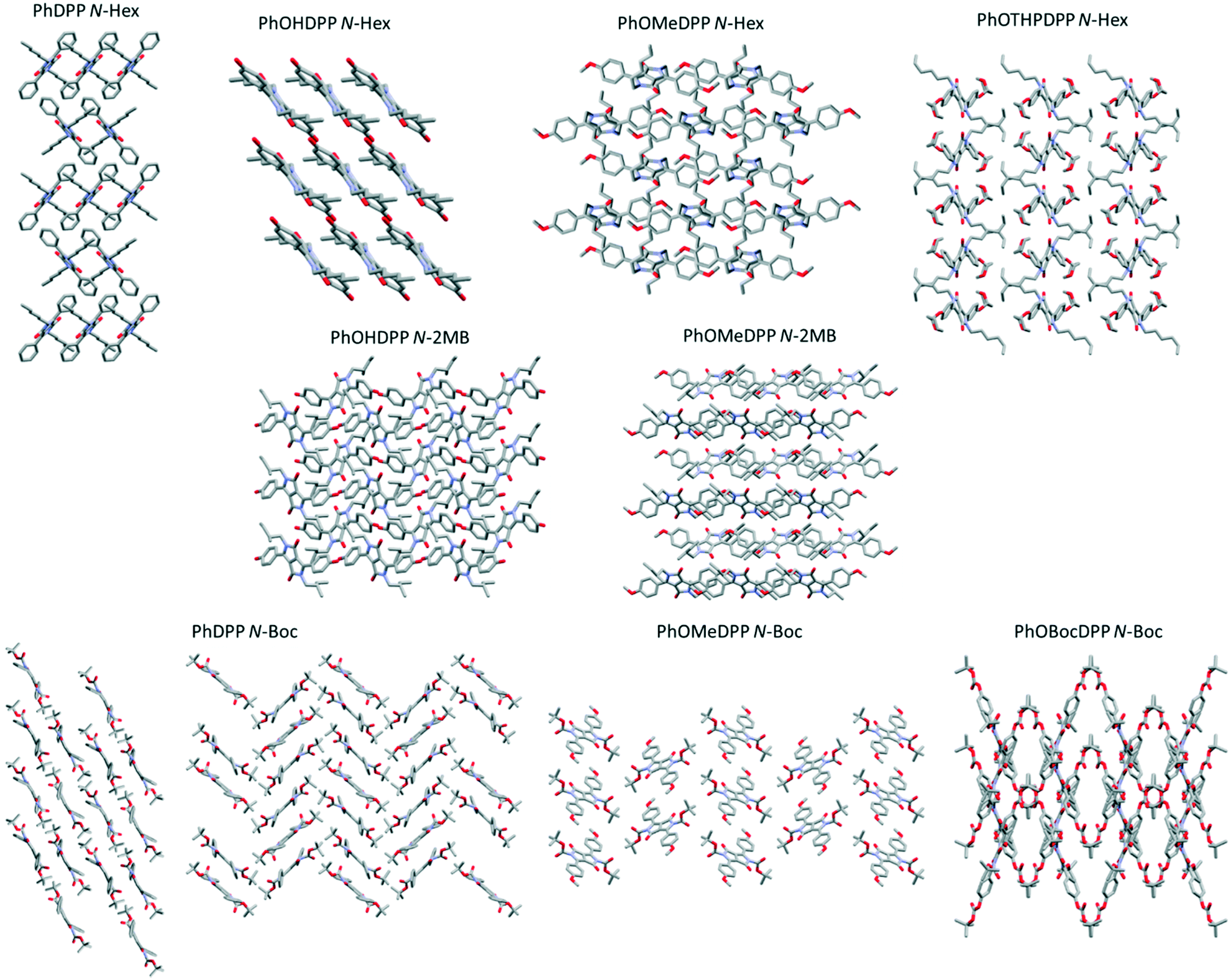 | ||
| Fig. 11 The infinite stacks for PhDPP N-Hex (herringbone), PhOMeDPP N-Hex (herringbone), PhOHDPP N-Hex (lamellar), PhOTHPDPP N-Hex (herringbone)., PhOHDPP N-2MB (herringbone), PhOMeDPP N-2MB (herringbone), PhDPP N-Boc (lamellar), PhDPP N-Boc (herringbone), PhOMeDPP N-Boc (herringbone) and PhOBocDPP N-Boc (herringbone) hydrogen atoms omitted for clarity. | ||
All these crystals comprise infinite stacks, most with a herringbone type arrangement (Fig. 11),21 except PhOHDPP N-Hex that has a lamellar 1D stacking arrangement. It has been previously shown that for DPP systems with this classic herringbone arrangement the experimental hole mobilities produced are moderate,82 whereas functionalisation to produce other packing motifs, such as co-facial, results in a marked increase in mobility. Therefore, the herringbone arrangement in this study are expected to have lower mobility than their lamellar packing counterparts, because of edge to face rather than face to face packing. The face to face π stacking of PhOMeDPP N-Boc makes it more interesting in this regard.
The chiral 2MB chain is structurally similar to 2-ethyl hexyl, one of the most widely investigated sidechains for N-alkylation of DPPs for optoelectronic applications.66–68 For small molecule derivatives, it has been shown that the mesomorph form has higher mobility versus the enantiopure forms and this is attributed to co-planarity and closer π stacks in the former.67,68 In oligothiophene variants, it has been shown that the enantiopure form has a small influence on photovoltaic performance and the largest influence on thin film morphology and solid state absorption.66 These findings all suggest that it is the branched nature of the chain that is responsible for the enhancement relative to linear N-alkyl chains.
Calculated electronic properties
Distortion of peripheral substituents from planarity is observed in the majority of systems in this study and is also seen in top performing organic semiconductors, most notably the acenes.83–85 Tetracene is planar in structure and displays moderate charge mobility, packing in a classic herringbone fashion.86,87 Introduction of peripheral phenyl groups results in twisting of the rings to minimise steric clashing and a slipped-co-facial packing of the π-conjugated tetracene backbone. This introduction of side peripheral groups, as observed for rubrene, results in closer π⋯π interactions and columnar stacks in two directions.68 The additional pathway results in a large improvement in mobility to give one of the highest performing p-type materials, with hole mobilities of up to 40 cm2 V−1 s−1.83,84Co-facial π⋯π stacking is thought to have the largest contribution to charge mobility in DPP derivatives, with lesser contributions from hydrogen bonding. PhDPP N-Hex, which exhibits a classic herringbone edge to face packing, has an experimentally determined charge mobility in the region of 2 × 10−4 cm2 V−1 s−1 for holes (3 × 10−3 cm2 V−1 s−1 best reported).19PhDPP N-MonoHex and the parent PhDPP N-H (ESI† Fig. S20), both of which exhibit co-facial π stacking, have mobilities of the order 1.0–1.5 × 10−2 cm2 V−1 s−1.3,19 For PhDPP N-Hex one charge pathway (π stack) is present, whereas PhDPP N-MonoHex has three charge pathways, with hydrogen bonding having little contribution. The exception is PhDPP NH, in which hydrogen bonding has been postulated as the largest single contribution to charge mobility.3
Careful choice of N-aryl-containing substituents gives PhDPP single crystals that can exhibit comparable or even greater charge transport values to rubrene.88,89 In particular, introducing benzyl derivatives at the lactam position results in co-facial π⋯π stacking and good potential for hole transport.85Table 3 gives the experimentally determined frontier orbital energy levels obtained through cyclic voltammetry measurements (ESI†) as well as the optical band gap from the solid-state absorption edge. Good agreement between electrochemical and solid-state data is observed, with the increase for PhOHDPP N-2MB attributed to stronger excitonic coupling in the solid state arising from the hydrogen bonded network. Table 3 also displays theoretical hole mobilities based on the crystal structures90–92 (separation distances and electronic couplings for unique neighbouring molecules are in the ESI†). While data for PhDPP N-Boc was from the literature,93 the mobility value of PhDPP N-Hex is close to that obtained experimentally,19 providing validation for the calculated values.
| Compound | HOMO [eV] | LUMO [eV] | Electronic Eg [eV] | Optical Eg [eV] | LUMO (optical) [eV] | Reorganisation energy [eV] | Calc. hole mobility [cm2 V−1 s−1] |
|---|---|---|---|---|---|---|---|
| a Determined from cyclic voltammetry measurements in solution of compound (1 mmol), of supporting electrolyte (tetrabutylammonium hexafluorophosphate, 0.1 M). Values referenced against the ferrocenium–ferrocene redox couple. b Acetonitrile solvent. c DMF solvent. d Dichloromethane solvent. e Computationally calculated values. f Thin films were formed on glass slides by drop casting of a concentrated THF solution (0.5 mg ml−1). g Thin films were formed on glass slides by drop casting of a concentrated DCM solution (0.5 mg ml−1). h Thin films of sufficient quality were not formed. | |||||||
| PhDPP N-Hex 21 | −5.40 (ref. 21) | −3.10 (ref. 21) | 2.30 (ref. 21) | 2.27f | −3.13f | 0.40 | 2.16 × 10−3 |
| PhOHDPP N-Hex | −4.98a,c (−5.24)e | −3.04a,c | 1.94a,c | n/a | n/ah | 0.47 | 2.60 × 10−3 |
| PhOMeDPP N-Hex | −5.25a,b (−5.21)e | −3.11a,b | 2.14a,b | 2.20f | −3.05f | 0.47 | 2.10 × 10−4 |
| PhOTHPDPP N-Hex | −5.36a,b (−5.17)e | −3.18a,b | 2.18a,b | 2.20f | −3.16f | 0.35 | 8.60 × 10−4 |
| PhOHDPP N-2MB | −4.93 (−5.24)e | −3.07 | 1.86a,c | 2.10f | −2.83f | 0.47 | 8.50 × 10−4 |
| PhOMeDPP N-2MB | −5.27a,b (−5.21e) | −3.10a,b | 2.17a,b | 2.23f | −3.04f | 0.47 | 5.80 × 10−4 |
| PhDPP N-Boc 95 | −5.49 (ref. 95) | −3.20 (ref. 95) | 2.29 (ref. 95) | n/a | n/a | n/a | n/a |
| PhOMeDPP N-Boc | −5.54a,d (−5.44)e | −3.21a,d | 2.33a,d | 2.39f | −3.15f | 0.54 | 1.91 × 10−2 |
| PhOBocDPP N-Boc | −5.82a,d (−5.72)e | −3.50a,d | 2.32a,d | 2.34g | −3.48g | 0.60 | 2.20 × 10−3 |
The derivatives with smaller dihedral angles and relatively moderate molecular displacement displayed the highest theoretical mobilities. PhOMeDPP N-Boc is virtually planar and gives the highest value for hole mobility, 1.9 × 10−2 cm2 V−1 s−1. PhOHDPP N-Hex has a small dihedral angle and the second highest mobility, 2.6 × 10−3 cm2 V−1 s−1. This value is significantly higher than its methoxy counterpart PhOMeDPP N-Hex, which exhibits a larger dihedral and considerably larger molecular displacement, therefore PhOHDPP N-Hex is expected to be the better performing material regardless of contributions from hydrogen bonding. PhOHDPP N-2MB illustrates the influence of hydrogen bonding on charge mobility with a moderate improvement versusPhOMeDPP N-2MB, despite the larger dihedral angles and increased displacement. However, the introduction of the hydrogen bonding groups does not give the substantial improvement in charge mobilities expected for these compounds. As discussed above, the influence of hydrogen bonding appears to have a larger contribution in NH unfunctionalized planar systems, whereby a synergistic relationship with π⋯π stacking maximises charge transfer integrals between neighbours. For N-alkylated derivatives the dominant charge transport pathway is likely to occur through π stacks and by increasing the dihedral angle the influence of hydrogen bonding appears reduced. This suggests that a combination of relatively planar chromophores and hydrogen bonding is therefore desired in future materials to strengthen nearest neighbour interactions. The other derivatives fit into a trend that larger dihedrals and displacement between neighbouring chromophores result in weaker intermolecular interactions and lower mobility.
Mobility of these systems follows a general trend with molecular overlap. The highest is the planar PhOMeDPP N-Boc with a slipped cofacial stacking, PhOHDPP N-Hex and PhDPP N-Hex display an interaction somewhere between an edge to face and face to face but have appreciable overlap of the phenyl rings with the lactam unit. PhOBocDPP N-Boc also possesses a parallel displaced interaction and hence moderate mobility. PhOMeDPP N-Hex and PhOMeDPP N-2MB have much lower mobility owing to the fact the interaction is truer to a CH–π interaction so overlap between π systems is lessened and hence mobility is low.
Hydrogen bonding interactions affect charge mobility. For PhOHDPP N-Hex, the phenol hydrogen atoms form hydrogen bonds with the carbonyl oxygen atom of the lactam core (H⋯O distance 1.863 Å, ESI† Fig. S21). This leads to one molecule of PhOHDPP N-Hex hydrogen bonding to two adjacent molecules. This double hydrogen bonding interaction results in stronger coupling between neighbouring chromophores and contributes to a relatively higher calculated charge mobility. For PhOHDPP N-2MB, each molecule hydrogen bonds to four adjacent molecules, with slightly longer carbonyl-phenol distances (1.918 and 1.910 Å, respectively) compared with the achiral compound, suggesting a weaker interaction with neighbouring chromophores and this results in lower charge mobility.
From the experimentally determined frontier orbital energy levels it can be observed that there is little difference in terms of energy levels of the incorporation of a 2MB chain or hexyl, whereas incorporation of the electron deficient Boc group results in more substantial lowering of the frontier orbital energies. The deeper HOMO–LUMO levels scale with decreasing electron richness (PhDPP < PhOTHP < PhOMe < PhOH) of the phenyl ring.
Spectroscopic properties: absorption and emission in solution and solid state
PhDPP derivatives typically give two band absorption spectra, a common feature for D–A systems,94 with an absorption range extending across the visible spectrum, an advantage for light harvesting applications. The low energy band is attributed to a charge transfer transition with the ground state density localised on the DPP core (HOMO–LUMO) and the high energy band is attributed to delocalised π–π* transitions along the backbone.21,95,96 The N-alkyl compounds possess a featureless low energy band in solution that is believed to result from the loss of vibronic structure arising from distortion of the molecule from planarity because of the substituent.22,97 In the solid state broadening of the absorption profile is observed owing to excitonic coupling between neighbouring chromophores, a feature that is substantially less than their thiophene counterparts.20The thin film absorption spectrum of PhDPP N-Hex (Fig. 12), shows broadening of the low energy absorption band3,12,14,98 with a blue shift in the absorption maxima compared with solution, indicating a contribution of H-type aggregation. In addition the band also shows bathochromic broadening, suggesting oblique stacking and indicating contributions from both transition dipole orientations, as shown by Davydov splitting of the absorption band.23,30,99–101 When there is larger molecular overlap, a greater hypsochromic shift of the solid state absorption band is observed, indicating H-type aggregation, with less molecular overlap indicating J-type aggregation and a bathochromic shift of the band.12,20PhOHDPP N-2MB (Fig. 12) also displays Davydov splitting23,30,99–101 and a substantial bathochromic shift of the absorption maxima resulting from hydrogen bonding interactions.3,14 Hence, the nature of the shift is a balance between the hydrogen bonding and stacking contributions. The significant bathochromic shift suggests J-aggregation and that head to tail stacking dominates. This hypothesis is supported by the large molecular displacement found in the crystal structure for this compound.
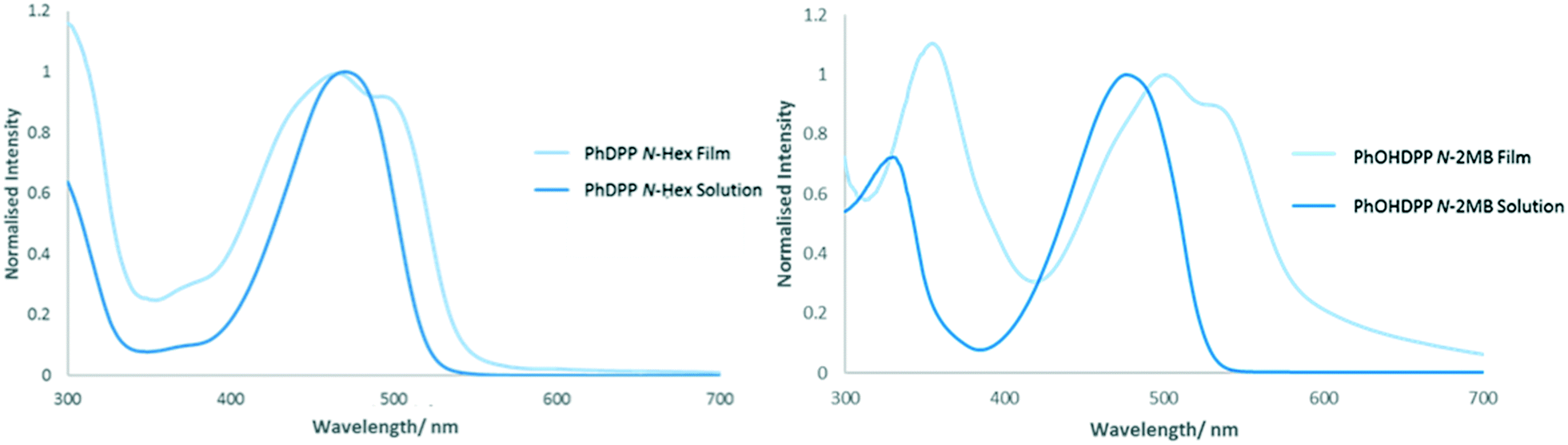 | ||
| Fig. 12 Solution (in THF) and solid-state absorption (normalised to maximum intensity) of PhDPP N-Hex and PhOHDPP N-2MB. The solid spectra are from thin films formed through drop casting of 0.5 mg ml−1 solution onto glass slide. | ||
Films of PhOMeDPP N-Hex (ESI† Fig. S22), display a bathochromic shift of the absorption band suggesting that J-type aggregation dominates and this correlates with the solid state structure which shows large displacement and a lack of molecular overlap, coupled with head to tail stacking. This stacking motif has previously shown favourable performance in photovoltaics, owing to faster charge transfer.30 For PhOHDPP N-Hex, films of sufficient quality for analysis were not obtained.
PhOMeDPP N-2MB (ESI† Fig. S23) displays similar behaviour to its hexyl analogue in THF and the bathochromic shift of the absorption band is attributed to J-type aggregation. In CH2Cl2, a shoulder appears on the absorption band and this is attributed to Davydov splitting,23,99 arising from excitonic coupling between neighbouring chromophores. In this scenario, there are contributions to the transition dipole moment from the stacking motif and aggregation,30,100,101 leading to two transitions of different intensity. The spectra are apparently dominated by J-aggregation, consistent with a bathochromic shift in the position of the absorption maximum. This large contribution from J-aggregation to the absorption band maximum in PhOMeDPP derivatives correlates well with literature.81
For PhOMeDPP N-Boc (ESI† Fig. S24), the absorption band is split and is either blue shifted (THF) or red shifted (CH2Cl2) depending on the solvent. This band splitting is characteristic of oblique stacking and suggests that both transition dipole orientations contribute to the absorption band. PhOTHPDPP N-Hex and PhOBocDPP N-Boc (ESI† Fig. S25 and S26) each display a bathochromic shift in absorption maxima, indicative of J-type aggregation, together with hypsochromic broadening of the band, suggesting a contribution from H-type aggregates in each.
The presence of auxochromes results in a bathochromic shift of the absorption band and the dominance of J-aggregation; a consequence of lone pair involvement in the conjugation. The polymorphic behaviour of PhOMeDPP N-Boc makes this the exception; in THF a larger contribution from a planar stacked polymorph is expected whilst CH2Cl2 should favour the twisted form.
Table 4 displays the optical properties of the materials. The high molar absorptivity in solution can be modulated by choice of solvent. The subjects of this paper display negative solvatochromism with increasing solvent polarity, correlating with the observed frontier orbitals electronic distribution which indicates a more polarised ground state. The optical band gap, determined from the position of the solid-state absorption edge, confirms that alkylation leads to breaking of effective conjugation. These band gaps are wider than ideal for incorporation into organic solar cells as donor species40,102 however this may be rectified in future by extending the conjugated backbone. Plots comparing the absorption maxima in the solid state with the DPP-aryl angle, vertical displacement and overall displacement display no prominent trends (ESI† Fig. S27–S34).
| Compound | Absorption λmax solution [nm] | ε [dm3 mol−1 cm−1] | Absorption λmax film [nm] | Absorption edge onset film [nm] | Optical Eg film [eV] |
|---|---|---|---|---|---|
| a Thin films were formed on glass slides via drop casting of a concentrated THF solution (0.5 mg ml−1). b Thin films were formed on glass slides via drop casting of a concentrated DCM solution (0.5 mg ml−1). | |||||
| PhDPP N-Hex | 470 | 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
465a | 547a | 2.27a |
| PhOHDPP N-Hex | 481 | 22000 |
n/a | n/a | n/a |
| PhOMeDPP N-Hex | 481 | 20600 |
494a | 553a | 2.25a |
| PhOTHPDPP N-Hex | 481 | 20300 |
498b | 558b | 2.23b |
| PhOHDPP N-2MB | 476 | 20700 |
501a | 592a | 2.10a |
| PhOMeDPP N-2MB | 479 | 30400 |
493a | 553a | 2.25a |
| PhDPP N-Boc 95 | 490 (ref. 95) | n/a | n/a | n/a | n/a |
| PhOMeDPP N-Boc | 460 | 24800 |
446a | 519a | 2.39a |
| PhOBocDPP N-Boc | 436 | 19900 |
457a | 531a | 2.34a |
As well as potential for light harvesting, phenyl DPPs display excellent emissive behaviour in solution (Table 5), indicated by the high fluorescence quantum yields, suggesting good viability for bioimaging and sensing applications. For PhDPP derivatives, solid state emission results from N-alkylation disrupting the intermolecular aromatic interactions that can lead to fluorescence quenching. This feature makes them promising candidates for solid state lighting applications. By contrast PhOHDPP materials are non-emissive in the solid state, a difference attributed to the hydrogen bond formed through the phenol motif to the DPP core.
| Compound | Absorption λmax solution [nm] | Emission λmax solution [nm] | Absorption λmax solid [nm] | Emission λmax solid [nm] | Stokes shift solution [nm] | Stokes shift solid [nm] | Φ |
|---|---|---|---|---|---|---|---|
| a Thin films were formed on glass slides via drop casting of a concentrated THF solution (0.5 mg ml−1). b Thin films were formed on glass slides by drop casting of a concentrated DCM solution (0.5 mg ml−1). c Quantum yields were calculated by comparison with the fluorescence observed for fluorescein (F = 91 in NaOH) under identical conditions of irradiation.109 | |||||||
| PhDPP N-Hex | 470 | 529 | 465a | 612a | 59 | 147a | 0.85 (ref. 21) |
| PhOHDPP N-Hex | 481 | 532 | n/a | n/a | 51 | n/a | 0.79 |
| PhOMeDPP N-Hex | 481 | 535 | 494a | 560 (524)a | 55 | 66a | 0.90 |
| PhOTHPDPP N-Hex | 481 | 531 | 498b | 619b | 50 | 121b | 0.84 |
| PhOMeDPP N-2MB | 479 | 534 | 493a | 537a | 55 | 44a | 0.89 |
| PhOHDPP N-2MB | 476 | 533 | 501a | n/a | 57 | n/a | 0.82 |
| PhDPP N-Boc 95 | 490 (ref. 95) | 542 (ref. 95) | n/a | n/a | 52 (ref. 95) | n/a | 0.99 (ref. 95) |
| PhOMeDPP N-Boc | 460 | 517 | 446a | 539a | 57 | 93a | 0.93 |
| PhOBocDPP N-Boc | 436 | 510 | 457a | 545a | 75 | 88a | 0.84 |
For PhDPP N-Hex (ESI† Fig. S35) there is a bathochromic shift in the position of the emission band between solution and solid state, which results from excitonic coupling between neighbouring chromophores3,12 together with a large Stokes shift. The latter hinders exciton mobility but is of interest for luminescent solar concentrators,103–105 as well as biological imaging applications since crossover between excitation and emission wavelengths is avoided.106 The solid state absorption profile is the mirror image of the emission spectrum suggesting that little structural change occurs between ground and excited states. The solid-state emission profile shows an intensity switch between the 0–1 and 0–0 bands compared with solution, with the 0–1 band the most intense. For PhOMeDPP N-2MB the solid state emission spectrum (Fig. 13) is similar to that in solution, albeit with increased vibronic character, as indicated by a growth in the 0–1 band, and this feature is attributed to increased planarization in the excited solid state.46 Given the similarity between the solid state and solution emission spectra then any differences in planarization between the two phases are expected to be small. In contrast, the lack of vibronic structure in the solid-state absorption profile, compared with emission, indicates a large difference in planarization between the ground and excited states. The solution and solid state emission spectra of PhOMeDPP N-Hex are very similar (ESI† Fig. S36). Interestingly, there is a loss in the intensity of the 0–0 band and an increase in intensity of the 0–1 band in the solid state, leading to a bathochromic shift in the emission maxima. A similar result was observed for PhDPP N-Hex suggesting that the chain has an influence over this behaviour and this switch of vibronic band intensity has been observed previously for ThDPP N-alkyl derivatives.107PhOMeDPP N-Hex and N-2MB give similar Stokes shifts in both solid state and solution, with peaks in the solid state spectrum showing a bathochromic shift indicative of stronger intermolecular interactions and excitonic coupling.3 For PhOMeDPP N-Boc the solution emission profile is solvent dependent (ESI† Fig. S37) and the solid emission depends on the depositing solvent. The solid from CH2Cl2 has an emission spectrum with vibronic structure and a shoulder at higher energy. The largest peak is attributed to the 0–1 vibronic transition whilst as a solution in CH2Cl2 this is reversed, and the 0–0 band is larger, suggesting a redistribution of the vibronic energy levels. When grown from THF, there is a single broad band in the solid-state emission spectrum with a slight bathochromic shift compared to that in solution, indicative of stronger intermolecular interactions. The solid-state emission spectrum of a PhOMeDPP N-Boc layer formed from THF shows little vibronic structure, suggesting that the system is more twisted when compared to the solution spectrum. The corresponding absorption spectrum displayed a greater contribution of H-type aggregation than that of a film formed from CH2Cl2. It is known that H-type aggregation leads to a quenching of fluorescence, with J-type resulting in an enhancement108 and this could explain the observed behaviour, with a greater contribution of H-type aggregation in the THF sample leading to reduction in 0–0 band in emission. The presence polymorphs for PhOMeDPP N-Boc will influence the emission properties.
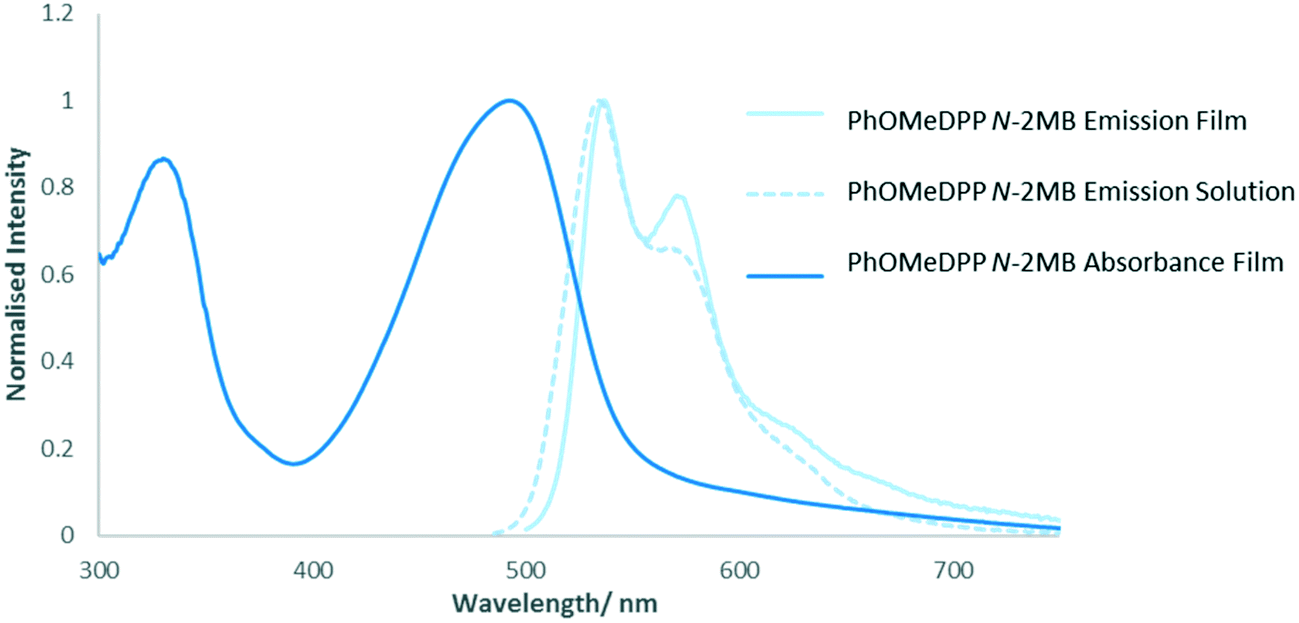 | ||
| Fig. 13 Solution (in THF) and solid-state emission and solid state absorption (normalised to maximum intensity) of PhOMeDPP N-2MB. | ||
The solution and solid-state emission spectra of PhOBocDPP N-Boc are similar to one another (ESI† Fig. S38), albeit with a small bathochromic shift in the solid state attributed to the intermolecular interactions between chromophores. In the solid film, PhOTHPDPP N-Hex possesses a broad featureless emission band that shows a significant bathochromic shift compared with the solution spectrum of the compound, with a large Stokes shift (ESI† Fig. S37), again interesting for biological imaging. This suggests that the excited state structure shows much greater distortion from planarity in the solid state.
There appears to be little correlation between the emission maxima and the DPP-aryl angle, overall displacement and vertical displacement (ESI† Fig. S40–S42), therefore the nature of the unique intermolecular interactions for each species are the biggest contributor to the observed solid state emissive behaviour.
Discussion
PhDPP N-alkyl derivatives possess large dihedral angles compared with other DPPs as a result of steric impediments that generally prevent the close packing observed for their thiophene counterparts.20 This twisting generally results in larger than expected values for molecular displacement in the solid state compared with other DPPs. However, the nature of the chain and aryl substituent also contribute and modulate this displacement. In certain cases, aromatic stacking is not in ideal positions to accommodate the close packing of molecules, suggesting other intermolecular interactions dominate in these instances. This case is observed most evidently for PhOHDPP N-Hex and PhDPP N-Hex, in which the edge to face interaction between the phenyl units and the lactam rings is twisted to accommodate additional intermolecular interactions, specifically hydrogen bonding in the former. This distortion results in a reduced dihedral angle and smaller displacement but also less substantial π overlap between neighbouring molecules. This feature results in these materials having better calculated charge mobility when compared with PhOMeDPP N-Hex. For PhOMeDPP N-Hex the edge to face π interaction is close to perpendicular resulting in larger dihedral angles and large molecular displacement hence low calculated charge mobility as the unit is forced to accommodate this interaction. Similar behaviour is seen for PhOHDPP N-2MB and PhOMeDPP N-2MB, where the edge to face π interactions are close to perpendicular and this interaction is accompanied by increased displacement, larger dihedral angles and reduced calculated mobilities versus analogues in which the interaction is distorted. For N-2MB containing species, aromatic interactions seemingly dominate over hydrogen bonding interactions as was inferred from the increased hydrogen bonding distance versus their PhOHDPP N-Hex analogues. The similarity between the π stacking interactions of PhOHDPP N-2MB and PhOMeDPP N-2MB suggest that these interactions are conserved across this series. The introduction of the hydroxy auxochrome does however modulate this interaction from a phenyl–lactam to phenyl–phenyl interaction. Although the major charge transport pathway is likely to be along π stacks, these systems are, in general, quite distorted. This prevents close packing, resulting in systems with shorter distances to neighbouring chromophores possessing improved theoretical charge mobility regardless of the extent of the π stacking interactions.PhOHDPP N-Hex displays a lamellar 1D chain structure, and is the only compound of those studied here that deviates from a herringbone-type stack.
The size of the end group R′′ on the aryl ring does influence the conformation despite its distant location from the core. PhOHDPP N-Hex has the smallest dihedral angle between the aromatic rings for the N-hexyl series. It is further enforced by analysing the PhOMe analogues in which the interactions are closer to true edge to face and parallel displaced interactions for alkyl and Boc respectively rather than a distorted product of aryl twisting as is the case for others. Although known previously, Boc substitution is a curious case. Preferential formation of the planar structure is ideal for a multitude of applications by providing solubility and substantial π contacts. Substitution of the phenyl ring leads to strong parallel displaced interactions and improved mobility. Polymorph screening of other PhDPP derivatives with this unit could provide other planar systems, where electron mobility would be favourable.
The biggest contributor to the modulation of the optical properties of the materials reported here is their electronic nature rather than structural factors, such as the nature of the alkyl chain.46 The optical properties vary little from case to case because of the electron rich nature of the dominant phenyl substituent. The only substantial change is observed for the Boc substituted variant, where its electron withdrawing nature hypsochromically shifts the absorption and emission spectra, whilst maintaining similar magnitudes of absorptivity and fluorescence. In the solid state, the methoxy and hydroxy auxochromes display predominantly J-type aggregation in their absorption spectra when substituted with alkyl chains, whereas a more oblique stacking case is observed for the t-Boc substituted variant with its tendency to form polymorphs in certain solvents. The introduction of hydrogen bonding modulates solubility and the solid-state absorption profile, as a result of excitonic coupling, whilst also quenching emission in the solid state. The PhOMeDPP N-alkyls show similar behaviour in both solution and the solid state, whereas PhOTHPDPP N-Hex displays similar solid state absorption behaviour to the methoxy and hydroxyl variants but a vast difference in emission maxima. This is also observed for PhDPP N-Hex. Thiophene DPP saw substantial effect of hexyl chain on J aggregation,20 which was not observed to the same extent with the subjects of this paper.
Through simple synthetic transformations the materials produced in this study display favourable optical properties for light harvesting and emitting applications. The twist of the phenyl rings out of the DPP plane, arising from N-alkylation of the lactam core, leads to materials with larger than ideal band gaps for application as donor materials in photovoltaics40 however they have potential as additives for this application,41 especially the chiral derivatives, which could induce a degree of control over the active layer morphology. The emissive properties of the PhOHDPP N-alkyl species in solution suggest potential as sensing materials, because of their ionisable hydroxyl group, whilst the solid-state emission of PhOMeDPP N-Alk derivatives makes them candidates for imaging or lighting applications.
Conclusions
The solid state studies we have performed on three series of PhDPPs reveals that edge-to-face interactions (rather than steric interactions between the N-substituent and the protons attached to the aromatic ring) are responsible for the twisting between the phenyl ring and the DPP core in the solid state. This effect can be overcome when the interaction between the aromatic rings is favoured, as in the stacking seen in PhOMeDPP N-Boc shown here. Therefore, it is possible that stacking of PhDPPs in might be overcome in more extended structures where stacking is favoured.Remote bulky groups at either side or end positions do affect the core conformation through packing effects. Unlike N-benzyl derivatives, in which the π interactions remain largely the same regardless of the 4-substituent, both the 4-phenyl substituent and N-alkyl chain have a competing influence on the crystal packing of the materials in this study. This effect is most evident for the hexyl species with a large variation in their packing with variation of R′. Interestingly, the conformation of the hexyl chain is different in all these structures, showing that the alkyl group can adapt to other packing forces in the solids.
Thin solid films of the materials show emission dominated by local packing effects, that are unique for each compound. The calculated charge mobility of the materials is low, as a result of the twisted conformations of the molecules and their large relative displacement. However, these materials could be useful as light emitters given their large Stokes shift and high emission.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (project EP/M005178/1) and the University of Nottingham for funding (including BNSCF421 and the Beacon of Excellence Propulsion Futures), Dr. Mick Cooper and Mr Ben Pointer-Gleadhill for Mass spectrometry assistance, Mr Shazad Aslam, Mr Kevin Butler and Dr. Adrienne Davis for NMR assistance, Mr Tom Clayton and Mr Mark Guyler for laboratory assistance. DFT calculations were performed using the VUW high-performance computing cluster Rāpoi. The Diamond Light Source Ltd (beamline I19) is thanked for providing beamtime.References
- J. Yang, D. Li, T. Jiang, Y. Gao, B. Li and X. Qu, RSC Adv., 2016, 6, 58434–58442 RSC.
- Y. Cai, W. Si, Q. Tang, P. Liang, C. Zhang, P. Chen, Q. Zhang, W. Huang and X. Dong, Nano Res., 2017, 10, 794–801 CrossRef CAS.
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521–3528 CrossRef.
- S.-S. Li, K.-J. Jiang, F. Zhang, J.-H. Huang, S.-G. Li, M.-G. Chen, L.-M. Yang and Y.-L. Song, Org. Electron., 2014, 15, 1579–1585 CrossRef CAS.
- J. Pan, H. Yin, Y.-Z. Xie, G.-Y. Sun and Z.-M. Su, RSC Adv., 2017, 7, 31800–31806 RSC.
- L. Wang, L. Zhu and D. Cao, New J. Chem., 2015, 39, 7211–7218 RSC.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615–644 CrossRef CAS.
- S. H. Choi, G. H. Kil, N. R. Kim, C. Yoon, J. P. Kim and J. H. Choi, Bull. Korean Chem. Soc., 2010, 31, 3427–3430 CrossRef CAS.
- C. Fu, P. J. Beldon and D. F. Perepichka, Chem. Mater., 2017, 29, 2979–2987 CrossRef CAS.
- J. Mizuguchi and G. Wooden, Ber. Bunsen-Ges. Phys. Chem., 1991, 95, 1264–1274 CrossRef CAS.
- J. Mizuguchi, J. Phys. Chem. A, 2000, 104, 1817–1821 CrossRef CAS.
- W. Herbst, K. Hunger, G. Wilker, H. Ohleier and R. Winter, Ind. Org. Pigment. Prod. Prop. Appl., 2004, 3rd edn, pp. 1–181 Search PubMed.
- O. Wallquist and R. Lenz, Macromol. Symp., 2002, 187, 617–629 CrossRef CAS.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS.
- J. David, M. Weiter, M. Vala, J. Vyňuchal and J. Kučerík, Dyes Pigm., 2011, 89, 137–143 CrossRef CAS.
- S. Luňák, M. Vala, J. Vyňuchal, I. Ouzzane, P. Horáková, P. Možíšková, Z. Eliáš and M. Weiter, Dyes Pigm., 2011, 91, 269–278 CrossRef.
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T. Q. Nguyen, Chem. Mater., 2012, 24, 1699–1709 CrossRef CAS.
- S. Dhar, J. Karothu and D. P. Patil, Chem. Commun., 2015, 51, 97–100 RSC.
- F. Pop, W. Lewis and D. B. Amabilino, CrystEngComm, 2016, 18, 8933–8943 RSC.
- J. Dhar, N. Venkatramaiah, A. Anitha and S. Patil, J. Mater. Chem. C, 2014, 2, 3457–3466 RSC.
- S. Luňák, J. Vyňuchal, M. Vala, L. Havel and R. Hrdina, Dyes Pigm., 2009, 82, 102–108 CrossRef.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS.
- J. Kuwabara, T. Yamagata and T. Kanbara, Tetrahedron, 2010, 66, 3736–3741 CrossRef CAS.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Adv. Mater., 2017, 29, 1600013 CrossRef.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS.
- B. Walker, J. Liu, C. Kim, G. C. Welch, J. K. Park, J. Lin, P. Zalar, C. M. Proctor, J. H. Seo, G. C. Bazan and T.-Q. Nguyen, Energy Environ. Sci., 2013, 6, 952–962 RSC.
- B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS.
- W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS.
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1–12 CrossRef.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198–4207 RSC.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- B. P. Karsten, J. C. Bijleveld and R. A. J. Janssen, Macromol. Rapid Commun., 2010, 31, 1554–1559 CrossRef CAS.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- M. A. Naik, N. Venkatramaiah, C. Kanimozhi and S. Patil, J. Phys. Chem. C, 2012, 116, 26128–26137 CrossRef CAS.
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T. Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543–15552 CrossRef CAS.
- Y. Kim, C. E. Song, A. Cho, J. Kim, Y. Eom, J. Ahn, S.-J. Moon and E. Lim, Mater. Chem. Phys., 2014, 143, 825–829 CrossRef CAS.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS.
- G. S. Thool, K. Narayanaswamy, A. Venkateswararao, S. Naqvi, V. Gupta, S. Chand, V. Vivekananthan, R. R. Koner, V. Krishnan and S. P. Singh, Langmuir, 2016, 32, 4346–4351 CrossRef CAS.
- W. Li, T. Lee, S. J. Oh and C. R. Kagan, ACS Appl. Mater. Interfaces, 2011, 3, 3874–3883 CrossRef CAS.
- Y. Wang, T. Wang, J. Chen, H. Do Kim, P. Gao, B. Wang, R. Iriguchi and H. Ohkita, Dyes Pigm., 2018, 158, 213–218 CrossRef CAS.
- X. fan Liao, J. Wang, S. ying Chen, L. Chen and Y. wang Chen, Chin. J. Polym. Sci., 2016, 34, 491–504 CrossRef.
- Y. Jin, Y. Xu, Y. Liu, L. Wang, H. Jiang, X. Li and D. Cao, Dyes Pigm., 2011, 90, 311–318 CrossRef CAS.
- P. Data, A. Kurowska, S. Pluczyk, P. Zassowski, P. Pander, R. Jedrysiak, M. Czwartosz, L. Otulakowski, J. Suwinski, M. Lapkowski and A. P. Monkman, J. Phys. Chem. C, 2016, 120, 2070–2078 CrossRef CAS.
- M. Grzybowski, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2014, 20, 12493–12501 CrossRef CAS.
- H. Ftouni, F. Bolze and J. F. Nicoud, Dyes Pigm., 2013, 97, 77–83 CrossRef CAS.
- C. Yang, M. Zheng, Y. P. Li, B. L. Zhang, J. F. Li, L. Y. Bu, W. Liu, M. X. Sun, H. C. Zhang, Y. Tao, S. F. Xue and W. J. Yang, J. Mater. Chem. A, 2013, 1, 5172–5178 RSC.
- M. Grzybowski, E. Glodkowska-Mrowka, G. Clermont, M. Blanchard-Desce and D. T. Gryko, Chem. Heterocycl. Compd., 2017, 53, 72–77 CrossRef CAS.
- S. Schutting, S. M. Borisov and I. Klimant, Anal. Chem., 2013, 85, 3271–3279 CrossRef CAS.
- M. Kaur, M. J. Cho and D. H. Choi, Dyes Pigm., 2014, 103, 154–160 CrossRef CAS.
- X. Yang, X. Gong, Y. Li, Z. Liu, B. Gao, G. Zhang, Y. Cui, G. Sun and G. Zhang, Tetrahedron, 2015, 71, 5069–5077 CrossRef CAS.
- S. Schutting, I. Klimant, D. De Beer and S. M. Borisov, Methods Appl. Fluoresc., 2014, 2, 024001 CrossRef.
- D. Aigner, B. Ungerböck, T. Mayr, R. Saf, I. Klimant and S. M. Borisov, J. Mater. Chem. C, 2013, 1, 5685–5693 RSC.
- W. K. Chan, Y. Chen, Z. Peng and L. Yu, J. Am. Chem. Soc., 1993, 115, 11735–11743 CrossRef CAS.
- F. Bruni, M. Sassi, M. Campione, U. Giovanella, R. Ruffo, S. Luzzati, F. Meinardi, L. Beverina and S. Brovelli, Adv. Funct. Mater., 2014, 24, 7410–7419 CrossRef CAS.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y. L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS.
- E. J. MacLean, M. Tremayne, B. M. Kariuki, J. R. A. Cameron, M. A. Roberts and K. D. M. Harris, Cryst. Growth Des., 2009, 9, 853–857 CrossRef CAS.
- J. Mizuguchi, Z. Kristallogr. - New Cryst. Struct., 2003, 218, 134–136 CAS.
- J. Mizuguchi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o469–o471 CrossRef CAS.
- Ciba Specialty Chemicals Corporation, US Pat., 5969154, 1999 Search PubMed.
- F. Pop, J. Humphreys, J. Schwarz, L. Brown, A. Van Den Berg and D. B. Amabilino, New J. Chem., 2019, 43, 5783–5790 RSC.
- R. Beninatto, G. Borsato, O. De Lucchi, F. Fabris, V. Lucchini and E. Zendri, Dyes Pigm., 2013, 96, 679–685 CrossRef CAS.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- R. B. Zerdan, N. T. Shewmon, Y. Zhu, J. P. Mudrick, K. J. Chesney, J. Xue and R. K. Castellano, Adv. Funct. Mater., 2014, 24, 5993–6004 CrossRef CAS.
- T. He, P. Leowanawat, C. Burschka, V. Stepanenko, M. Stolte and F. Würthner, Adv. Mater., 2018, 1804032, 1–8 Search PubMed.
- J. Liu, Y. Zhang, H. Phan, A. Sharenko and P. Moonsin, Adv. Mater., 2013, 25, 3645–3650 CrossRef CAS.
- S. T. Salammal, J. Y. Balandier, J. B. Arlin, Y. Olivier, V. Lemaur, L. Wang, D. Beljonne, J. Cornil, A. R. Kennedy, Y. H. Geerts and B. Chattopadhyay, J. Phys. Chem. C, 2014, 118, 657–669 CrossRef CAS.
- J. Mizuguchi, A. Grubenmann, G. Wooden and G. Rihs, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 696–700 CrossRef.
- S. Ying, M. Chen, Z. Liu, M. Zheng, H. Zhang, S. Xue and W. Yang, J. Mater. Chem. C, 2017, 5, 5994–5998 RSC.
- B. Jiang, C. C. Du, M. J. Li, K. Gao, L. Kou, M. Chen, F. Liu, T. P. Russell and H. Wang, Polym. Chem., 2016, 7, 3311–3324 RSC.
- D. Hablot, P. Retailleau and R. Ziessel, Chem. – Eur. J., 2010, 16, 13346–13351 CrossRef CAS.
- J. Calvo-Castro, M. Warzecha, A. R. Kennedy, C. J. McHugh and A. J. McLean, Cryst. Growth Des., 2014, 14, 4849–4858 CrossRef CAS.
- J. Calvo-Castro, M. Warzecha, I. D. H. Oswald, A. R. Kennedy, G. Morris, A. J. McLean and C. J. McHugh, Cryst. Growth Des., 2016, 16, 1531–1542 CrossRef CAS.
- J. Calvo-Castro, S. Maczka, C. Thomson, G. Morris, A. R. Kennedy and C. J. McHugh, CrystEngComm, 2016, 18, 9382–9390 RSC.
- M. Warzecha, J. Calvo-Castro, A. R. Kennedy, A. N. Macpherson, K. Shankland, N. Shankland, A. J. McLean and C. J. McHugh, Chem. Commun., 2015, 51, 1143–1146 RSC.
- J. Calvo-Castro, G. Morris, A. R. Kennedy and C. J. McHugh, Cryst. Growth Des., 2016, 16, 5385–5393 CrossRef CAS.
- T. Yamagata, J. Kuwabara and T. Kanbara, Eur. J. Org. Chem., 2012, 5282–5290 CrossRef CAS.
- T. Yamagata, J. Kuwabara and T. Kanbara, Tetrahedron Lett., 2010, 51, 1596–1599 CrossRef CAS.
- A. S. Murphy, C. E. Killalea, J. Humphreys, P. A. Hume, M. J. Cliffe, G. J. Murray, E. S. Davies, W. Lewis and D. B. Amabilino, ChemPlusChem, 2019, 84, 1413–1422 CrossRef CAS.
- J. Dhar, D. P. Karothu and S. Patil, Chem. Commun., 2014, 51, 97–100 RSC.
- T. Hasegawa and J. Takeya, Sci. Technol. Adv. Mater., 2009, 10, 024314 CrossRef.
- J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Appl. Phys. Lett., 2007, 90, 102120 CrossRef.
- W. Xu, Y. He, I. Murtaza, D. Zhang, A. Li, Z. Hu, X. Zeng, Y. Guo, Y. Zhu, M. Liu and H. Meng, J. Mater. Chem. C, 2017, 5, 2852–2858 RSC.
- S. M. Ryno, C. Risko and J. L. Brédas, ACS Appl. Mater. Interfaces, 2016, 8, 14053–14062 CrossRef CAS.
- H. Moon, R. Zeis, E. J. Borkent, C. Besnard, A. J. Lovinger, T. Siegrist, C. Kloc and Z. Bao, J. Am. Chem. Soc., 2004, 126, 15322–15323 CrossRef CAS.
- J. Calvo-Castro and C. J. McHugh, J. Mater. Chem. C, 2017, 5, 3993–3998 RSC.
- J. Calvo-Castro, C. J. McHugh and A. J. McLean, Dyes Pigm., 2015, 113, 609–617 CrossRef CAS.
- A. N. Sokolov, S. Atahan-Evrenk, R. Mondal, H. B. Akkerman, R. S. Sánchez-Carrera, S. Granados-Focil, J. Schrier, S. C. B. Mannsfeld, A. P. Zoombelt, Z. Bao and A. Aspuru-Guzik, Nat. Commun., 2011, 2(437), 1–8 Search PubMed.
- B. Baumeier, J. Kirkpatrick and D. Andrienko, Phys. Chem. Chem. Phys., 2010, 12, 11103–11113 RSC.
- E. F. Valeev, V. Coropceanu, D. A. da Silva Filho, S. Salman and J.-L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882–9886 CrossRef CAS.
- T. Jia, S. Huang, H. Bohra and M. Wang, Dyes Pigm., 2019, 165, 223–230 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS.
- S. Wood, J. Wade, M. Shahid, E. Collado-Fregoso, D. D. C. Bradley, J. R. Durrant, M. Heeney and J.-S. Kim, Energy Environ. Sci., 2015, 8, 3222–3232 RSC.
- R. S. Szabadai, J. Roth-Barton, K. P. Ghiggino, J. M. White and D. J. D. Wilson, Aust. J. Chem., 2014, 67, 1330–1337 CrossRef CAS.
- M. Vala, M. Weiter, J. Vyňuchal, P. Toman and S. Luňák, J. Fluoresc., 2008, 18, 1181–1186 CrossRef CAS.
- M. Adachi and S. Nakamura, J. Phys. Chem., 1994, 98, 1796–1801 CrossRef CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS.
- D. Sarkar, P. Das, A. Girigoswami and N. Chattopadhyay, J. Phys. Chem. A, 2008, 112, 9684–9691 CrossRef CAS.
- M. C. Scharber, Adv. Mater., 2016, 28, 1994–2001 CrossRef CAS.
- W. Ma, W. Li, M. Cao, R. Liu, X. Zhao and X. Gong, Org. Electron., 2019, 73, 226–230 CrossRef CAS.
- A. Sanguineti, M. Sassi, R. Turrisi, R. Ruffo, G. Vaccaro, F. Meinardi and L. Beverina, Chem. Commun., 2013, 49, 1618–1620 RSC.
- P. Della Sala, N. Buccheri, A. Sanzone, M. Sassi, P. Neri, C. Talotta, A. Rocco, V. Pinchetti, L. Beverina, S. Brovelli and C. Gaeta, Chem. Commun., 2019, 55, 3160–3163 RSC.
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C. S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 26266–26274 CrossRef CAS.
- G. Wiosna-Salyga, M. Gora, M. Zagorska, P. Toman, B. Luszczynska, J. Pfleger, I. Glowacki, J. Ulanski, J. Mieczkowski and A. Pron, RSC Adv., 2015, 5, 59616–59629 RSC.
- Y. J. Wang, Z. Li, J. Tong, X. Y. Shen, A. Qin, J. Z. Sun and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 3559–3568 RSC.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, other experimental details, separation distances and electronic coupling for unique neighbouring molecules. CCDC 2005924–2005930. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00039j |
| ‡ Current address MOLTECH-Anjou, UMR 6200, CNRS, Univ. Angers, 2bd Lavoisier, 49045 Angers, France. |
| § Current address Chemistry Building, The University of Sydney, Eastern Avenue NSW 2006, Australia. |
| This journal is © The Royal Society of Chemistry 2021 |