DOI:
10.1039/D0CE01579B
(Paper)
CrystEngComm, 2021,
23, 1075-1082
Unveiling the structural features of the host–guest complexes of carboxylated pillar[5]arene with viologen derivatives†
Received
29th October 2020
, Accepted 9th December 2020
First published on 9th December 2020
Abstract
Despite the impressive progress in the study of the aqueous host–guest chemistry of carboxylated pillar[n]arenes, these macrocyclic molecules represent a completely unexplored class of molecular building blocks in the solid state supramolecular chemistry. Here we describe the formation of crystalline host–guest complexes of carboxylated pillar[5]arene with four viologen derivatives, emphasizing its inclusion and self-assembly behavior in the presence of cationic electron-deficient guests. The macrocyclic cavity is prone to slight distortion upon the inclusion of alkyl viologens, but competitive inclusion of ethanol molecules takes place in the case of bulky benzyl viologen. The different distribution of carboxylic versus carboxylate substituents of pillar[5]arene renders self-assembly via carboxylic–carboxylic or carboxylic–carboxylate supramolecular synthons.
Introduction
The supramolecular chemistry of the water-soluble carboxylated pillar[n]arenes (CPAn) has been flourishing since their discovery by Ogoshi in 2010.1 The unique intrinsically rigid and highly symmetric macrocyclic structure decorated with carboxyl groups provides them with outstanding host–guest binding properties in aqueous media.2 The CPAn appear to be relatively non-toxic3 and of tremendous potential application in drug delivery,4 as antitumor agents,5 and supramolecular catalysts6 due to their excellent ability to complex various guest molecules. Despite the impressive progress in the study of their aqueous host–guest and self-assembly properties, these macrocyclic molecules represent a completely unexplored class of molecular building blocks in the solid state supramolecular and coordination chemistry. This is very surprising considering (i) the combination of a rigid electron-rich cavity and flexible carboxylic linkers making them highly promising candidates for the design of solid-state assemblies of various topologies; (ii) the presence of multiple carboxylic groups that can be completely or partially deprotonated, potentially inducing rich binding modes and interesting structures of high dimensionality; (iii) they can act both as multiple hydrogen bond donors and acceptors depending on the degree of deprotonation; (iv) the well-established supramolecular synthons may be used to drive the self-assembly with carboxylic acids. These characteristics may lead to the discovery of the novel class of crystalline materials constructed from the cavity containing macrocyclic building blocks.7
Considering these aspects, we recently began investigating their solid-state supramolecular chemistry and reported the first structural authentication of carboxylic acid substituted pillar[5]arene (CPA5), its basic self-assembly properties and formation of a host–guest complex with active pharmaceutical ingredient tetracaine.8 Our current work is focused on the utilization of CPA5 as a building block in the supramolecular architectures through: 1) exploitation of the properties of its macrocyclic cavity towards guest inclusion, 2) study of the solid-state assembly of CPA5via carboxylic–carboxylic and carboxylic–carboxylate supramolecular synthons. Here we show the formation of host–guest complexes of CPA5 with four viologen derivatives, emphasizing the inclusion and self-assembly behavior of CPA5 with cationic guests in the solid state, Fig. 1. Viologens are commonly used in the supramolecular chemistry to fabricate capsular or threaded host–guest assemblies both in solution and solid state.9 Their cationic electron-deficient nature makes them perfect guests for electron-rich and/or anionic host molecules. Notably, the dimethyl viologen salt (paraquat) was the first guest to reveal the inclusion properties of deca-anionic form of CPA5 in an aqueous solution.1 The molecular recognition of paraquat by larger homologue CPA6 in water has been proposed as a supramolecular treatment of paraquat poisoning.10 It should be emphasized that no crystal structure of a fully anionic form of CPA5 has been reported so far, despite the impressive progress in its solution studies and excellent solubility in water. On the contrary, fully carboxylic functionality of CPA5 renders its low solubility in aqueous solutions, and only the addition of cosolvent (ethanol) as found by us8 entails a significant increase of the solubility of the carboxylic form of CPA5 enabling both solution studies and crystallization of its complexes. The possibility to tune the solubility of CPA5 not only by pH, but by the addition of cosolvent opens up new opportunities in the CPA5 aqueous chemistry. One direction is to integrate the CPA5 macrocycle as a basic supramolecular building block in the crystal engineering using well-studied supramolecular synthons involving carboxylic acid groups.
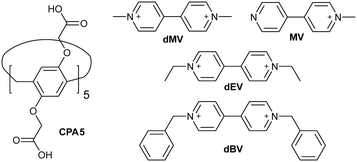 |
| | Fig. 1 Chemical structure of carboxylated pillar[5]arene and four viologen guests used in the host–guest study. | |
Results and discussion
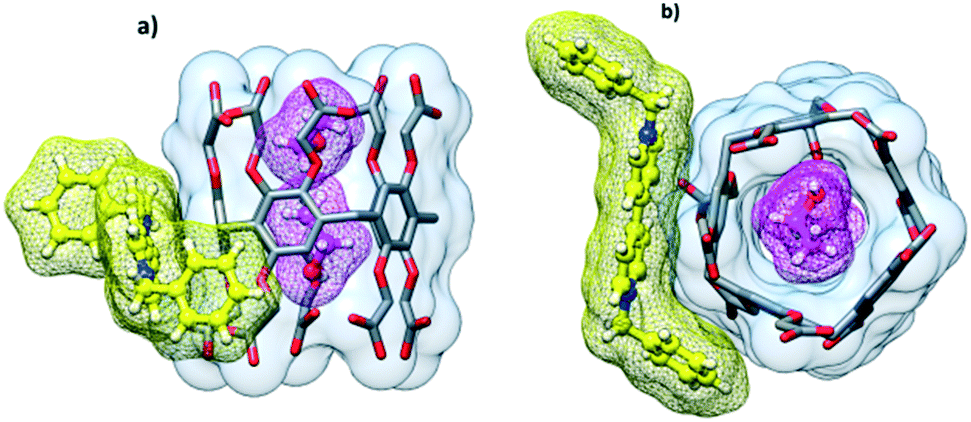
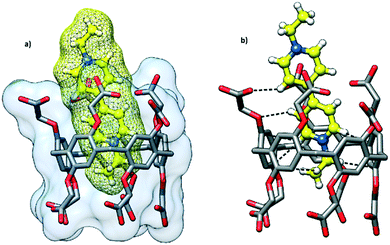
Cocrystallization of dimethyl viologen (N,N′-dimethyl-4,4′-bipyridinium) dichloride, also known as paraquat, with CPA5 from the water–ethanol solution afforded prismatic crystals of the host–guest complex I. The orange color of the crystalline complex was notably different from the solid forms of both host CPA5 and guest dimethyl viologen (dMV) components (white powders). X-ray single crystal analysis revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest inclusion complex of the composition [dMV@CPA5]·5.27(H2O) in the P21/c space group of the monoclinic system. The asymmetric unit comprises one CPA5 molecule, one paraquat cation (modelled as disordered over two positions) included into the host cavity, and water molecules. The dicationic nature of the guest suggests the deprotonation of two of the ten carboxylic substituents of the macrocyclic host to satisfy the electroneutrality of the complex. The dMV penetrates the pentagonal cavity of CPA5 in an asymmetric way with one of the methylpyridinium moieties being encircled by aromatic walls of the macrocycle, while the second methylpyridinium unit is embraced by aliphatic arms of the ‘entrance rim’, Fig. 2a. The depth of the inclusion measured as the distance between the mean plane determined by five methylene carbon atoms of the CPA5 macrocyclic core and centroid of the C–C bond between two pyridinium rings of the dMV is equal to 3.4 Å (3.8 Å for the second position of dMV). The crystals have an orange color suggesting a π–π charge-transfer of the donor–acceptor type between electron-rich aromatic walls of CPA5 and electron-poor pyridinium rings of the dMV guest. One of the phenyl rings of the CPA5 is indeed approximately parallel to the included pyridinium ring of the guest (7° tilt between the mean planes), and the closest donor–acceptor distance is at 3.24 Å (between C3D and C5X). Besides hydrophobic effect, electrostatic attraction and charge transfer, the host–guest complex is stabilized by the C–H⋯π hydrogen bonding between the included pyridinium ring of the dMV and aromatic walls of CPA5 (Fig. 2b); the shortest C⋯π (centroid) distances are 3.21 and 3.16 Å. The second methylpyridinium moiety that protrudes from the cavity is involved in the C–H⋯π and C–H⋯O interactions with the host.
:1 host–guest inclusion complex of the composition [dMV@CPA5]·5.27(H2O) in the P21/c space group of the monoclinic system. The asymmetric unit comprises one CPA5 molecule, one paraquat cation (modelled as disordered over two positions) included into the host cavity, and water molecules. The dicationic nature of the guest suggests the deprotonation of two of the ten carboxylic substituents of the macrocyclic host to satisfy the electroneutrality of the complex. The dMV penetrates the pentagonal cavity of CPA5 in an asymmetric way with one of the methylpyridinium moieties being encircled by aromatic walls of the macrocycle, while the second methylpyridinium unit is embraced by aliphatic arms of the ‘entrance rim’, Fig. 2a. The depth of the inclusion measured as the distance between the mean plane determined by five methylene carbon atoms of the CPA5 macrocyclic core and centroid of the C–C bond between two pyridinium rings of the dMV is equal to 3.4 Å (3.8 Å for the second position of dMV). The crystals have an orange color suggesting a π–π charge-transfer of the donor–acceptor type between electron-rich aromatic walls of CPA5 and electron-poor pyridinium rings of the dMV guest. One of the phenyl rings of the CPA5 is indeed approximately parallel to the included pyridinium ring of the guest (7° tilt between the mean planes), and the closest donor–acceptor distance is at 3.24 Å (between C3D and C5X). Besides hydrophobic effect, electrostatic attraction and charge transfer, the host–guest complex is stabilized by the C–H⋯π hydrogen bonding between the included pyridinium ring of the dMV and aromatic walls of CPA5 (Fig. 2b); the shortest C⋯π (centroid) distances are 3.21 and 3.16 Å. The second methylpyridinium moiety that protrudes from the cavity is involved in the C–H⋯π and C–H⋯O interactions with the host.
 |
| | Fig. 2 The host–guest complex I of CPA5 with dimethyl viologen dMV (paraquat) highlighting the (a) inclusion mode and (b) C–H⋯π and C–H⋯O interactions between the guest and cavity interior. | |
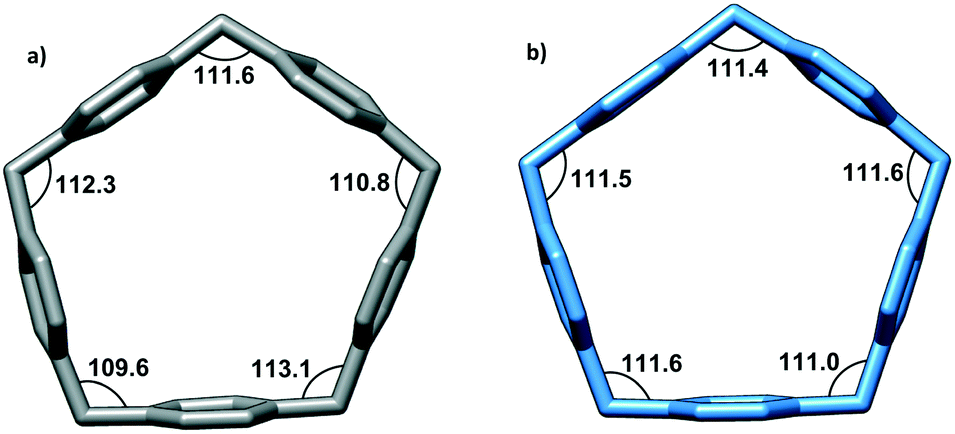
While the shape of the CPA5 macrocyclic core resembles a regular pentagon, the closer look reveals some deviation from regular Euclidean geometry. For a regular pentagon, the interior angles are 108°, while the sp3 hybridization of the bridging methylene groups in pillar[5]arene would favor the bonding angle of 109.28°. The structural strain within the macrocycle is manifested in the expansion of the valence angles of CH2 bridges to ∼111° from the theoretical sp3 value and slight bending and/or tilting of aromatic walls from the center of the cavity as shown from the crystal structure of the carboxylic acid substituted pillar[5]arene, in which the macrocyclic cavity is taken by two ethanol molecules (refcode KOVPES), Fig. 3b.8 Moreover, the inclusion of the bulky paraquat into the CPA5 cavity causes even further distortion of the macrocycle from a regular Euclidean pentagon, Fig. 3a.
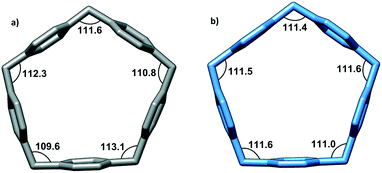 |
| | Fig. 3 The geometry of the CPA5 pentagonal cavity in the host–guest inclusion complexes with (a) dimethyl viologen dMV (paraquat) and (b) ethanol.8 Note the wider range of methylene bonding angles in the dMV@CPA5 complex (109.6–113.1°) than in the EtOH@CPA5 complex (111.0–111.6°), evidencing a more pronounced distortion of the macrocycle from a regular Euclidean pentagon upon inclusion of bulky guest dMV. | |
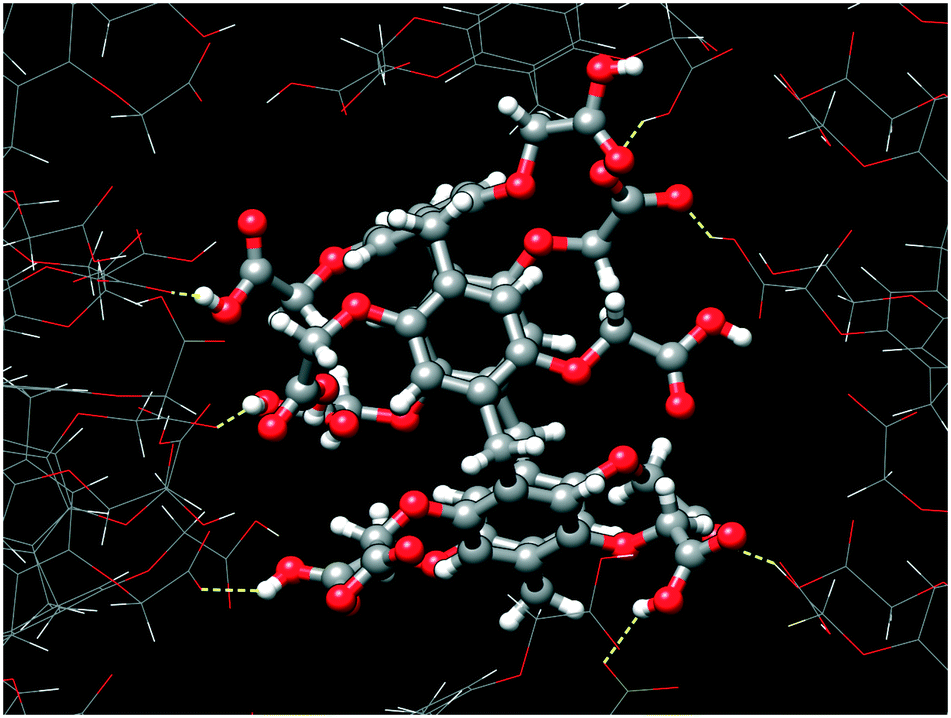
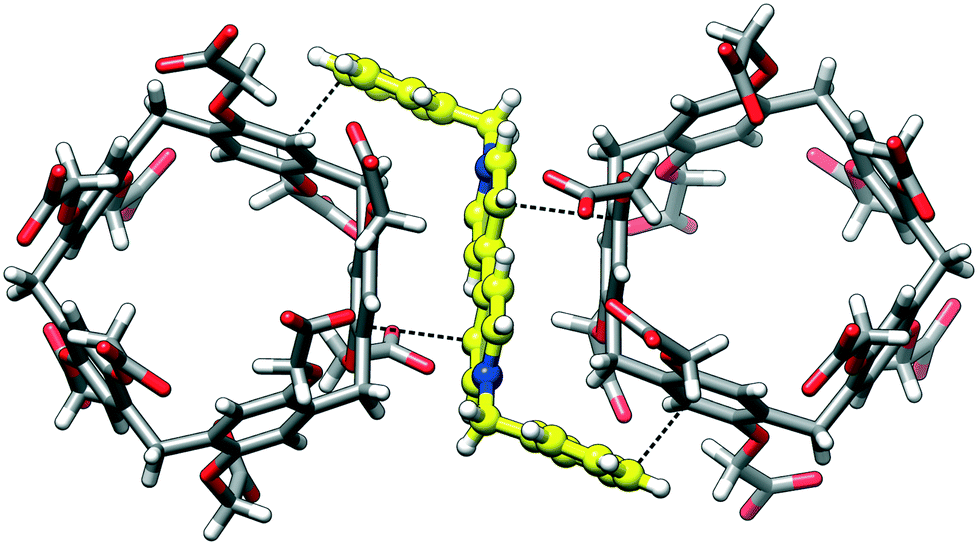
Ten carboxylic/carboxylate groups of CPA5 are involved in extensive O–H⋯O hydrogen bonding (HB) with adjacent CPA5 (Fig. 4) and multiple water molecules in the crystal lattice. Surprisingly, the distance range of the O–H⋯O HB between adjacent CPA5 molecules of 2.45–2.60 Å suggests a very strong HB, which is not observed for the previously reported CPA5 inclusion complex with ethanol in which all of the carboxylic functions of the macrocycle are protonated.8 Moreover, there are no carboxylic–carboxylic cyclic dimers typical for the self-assembly of fully protonated CPA5, suggesting strong preference of carboxylic⋯carboxylate single HB assembly of CPA5 comprising both anionic and neutral carboxylic functionalities. The shortest HB of 2.446(4) Å is a single HB realized between carboxylic and carboxylate groups related by an inversion center, O5D–(1/2 H5D1)⋯(1/2 H5D1)3–O5D3, [symmetry code: (3) –x + 1, −y + 1, −z]. The 50% occupancy of the H5D1 atom was imposed as a consequence of the formation of the hydrogen bonded carboxylic–carboxylate linear dimer over an inversion center. Besides obvious disorder of the position of the hydrogen atom, there is also a crystallographically imposed disorder of carboxylic and carboxylate groups over an inversion center, which was not accounted during refinement due to their similar geometry. This type of disorder was assumed based on statistical data extracted from the CSD on the distance distribution between two O atoms involved in the intermolecular HB for carboxylic–carboxylic and carboxylic–carboxylate dimer structures with low R-factors (R ≤ 0.05).11 The average hydrogen-bonded distance between two O atoms in carboxylic–carboxylate dimers is very short (2.54 ± 0.06 Å). Otherwise, the average distance distribution between two O atoms in the single carboxylic–carboxylic HB dimers is 2.66 ± 0.05 Å. Surprisingly, the shortest HB distances between two O atoms of carboxylic–carboxylate dimers responsible for the self-assembly of adjacent CPA5 of 2.446(4) and 2.469(3) Å (O3A–H3A⋯O3D) are even shorter than the average distance distribution for carboxylic–carboxylate synthons in the CSD. Such very short hydrogen bonds in the carboxylic–carboxylate dimers are rather typical for the intramolecular HB in the mono-anions of dicarboxylic acids (oxalic, malonic, and maleic) and phthalic acid; the average distance distribution is 2.43 ± 0.04 Å.11
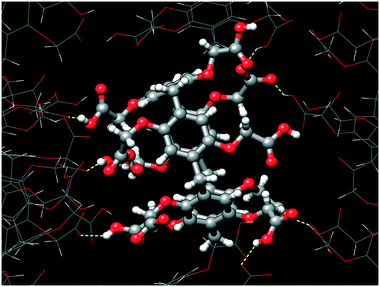 |
| | Fig. 4 The hydrogen bonds generated by the CPA5 molecule with adjacent CPA5 in the crystal lattice of dMV@CPA5. The distance range of O–H⋯O of 2.45–2.60 Å suggests a very strong hydrogen bonding characteristic for a charge-assisted carboxylic–carboxylate synthon. Note the absence of carboxylic–carboxylic association. | |
The short donor–acceptor distances are a characteristic for short strong hydrogen bonds (less than 2.5 Å for the O–H⋯O HB).12 These interactions are believed to be partly covalent in nature based on various experimental and theoretical techniques, such as infrared and NMR spectroscopies, X-ray and neutron diffraction.13 The potential energy surface of very short HBs approach the shape of symmetric single-well potential with a reduced barrier for proton migration rather than the double-well potential of the typical HB.14 Since the CPA5 solid state supramolecular chemistry is still in the flower bud, it is difficult to say if an unusually short carboxylic–carboxylate HB is typical and dominant for the assembly of CPA5 with mixed anionic and neutral carboxylic functionalities. Obviously, it would depend on the presence of competitive alternative HB motifs, for example the pyridyl nitrogen as a HB acceptor, and the number of deprotonated versus neutral carboxylic groups. Besides the short carboxylic–carboxylate HB guiding the assembly, we noted several carboxylic–water HBs and the absence of the carboxylic–carboxylic HB. The feature of the assembly of CPA5 with mixed anionic and neutral carboxylic functionalities is the network of carboxylic–carboxylate HB CPA5 in contrast to the chain-like assembly of the neutral form of CPA5 guided by the cyclic carboxylic–carboxylic HB supramolecular synthons.8
The cocrystallization of ethyl viologen (N,N′-diethyl-4,4′-bipyridinium)dibromide (dEV) with CPA5 from the water–ethanol solution afforded yellow prismatic crystals of host–guest complex II. X-ray single crystal analysis revealed a 1:1 host–guest inclusion complex of the composition [dEV@CPA5]·6.5H2O in the P21/n space group of the monoclinic system, Fig. 5a. The asymmetric unit comprises one CPA5 molecule, one ethyl viologen dication included into the host cavity, and water molecules. The complex is similar to the previously described dMV@CPA5, however, there are some notable differences. The inclusion of the bipyridinium core of the guest is shallower than that in the dMV@CPA5 complex; the depth of the inclusion is 4.1 Å, comparable to 3.4 and 3.8 Å (two disordered positions of dMV) in the dMV@CPA5 complex. The ethylpyridinium moiety immersed in the cavity is held via C–H⋯π interactions; the shortest C⋯π (centroid) distance is at 3.31 Å, Fig. 5b. The closest donor–acceptor distance is at 3.23 Å (C3C⋯C5P) involving edge-to-edge orientation of the donor (CPA5) and acceptor (included pyridinium moiety) π systems responsible for the yellow color of the host–guest crystals. The second ethylpyridinium moiety protrudes from the cavity interacting via the C–H⋯O hydrogen bonding with the carboxyl groups of host CPA5 and adjacent CPA5 in the crystal lattice. The distortion of the CPA5 pentagonal cavity from a regular pentagon due to the inclusion of dEV is similar to the case of dMV@CPA5 host–guest complex I.
 |
| | Fig. 5 Host–guest complex II of CPA5 with diethyl viologen dEV highlighting the (a) inclusion mode and (b) C–H⋯π and C–H⋯O interactions between the guest and cavity interior. | |
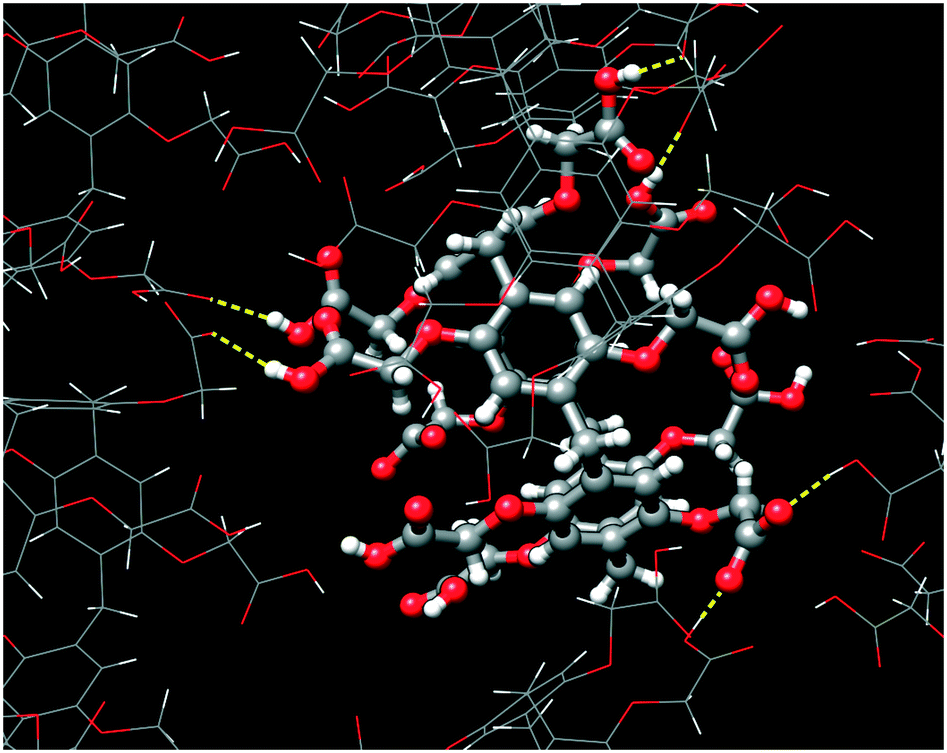
The self-assembly of adjacent CPA5 macrocycles is featured by realization of both carboxylic–carboxylate and carboxylic–carboxylic types of HB, Fig. 6. The two short distances between pairs of carboxyl partners involved in the intermolecular HB are 2.475(5) and 2.572(4) Å, evidencing the carboxylic–carboxylate synthon. The two long distances of 2.649(4) and 2.669(4) Å are typical for carboxylic–carboxylic dimers. These carboxylic–carboxylic dimers involve a single HB linking the two units. No cyclic carboxylic–carboxylic dimers have been identified in the dEV@CPA5 assembly. CPA5 are also involved in the extensive carboxylic–water HB.
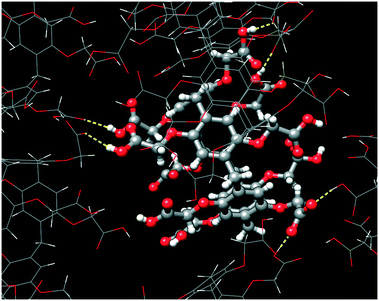 |
| | Fig. 6 The hydrogen bonds generated by a CPA5 molecule with adjacent CPA5 in the crystal lattice of dEV@CPA5. The distance range of O–H⋯O of 2.47–2.67 Å suggests the realization of both carboxylic–carboxylate and carboxylic–carboxylic supramolecular synthons. | |
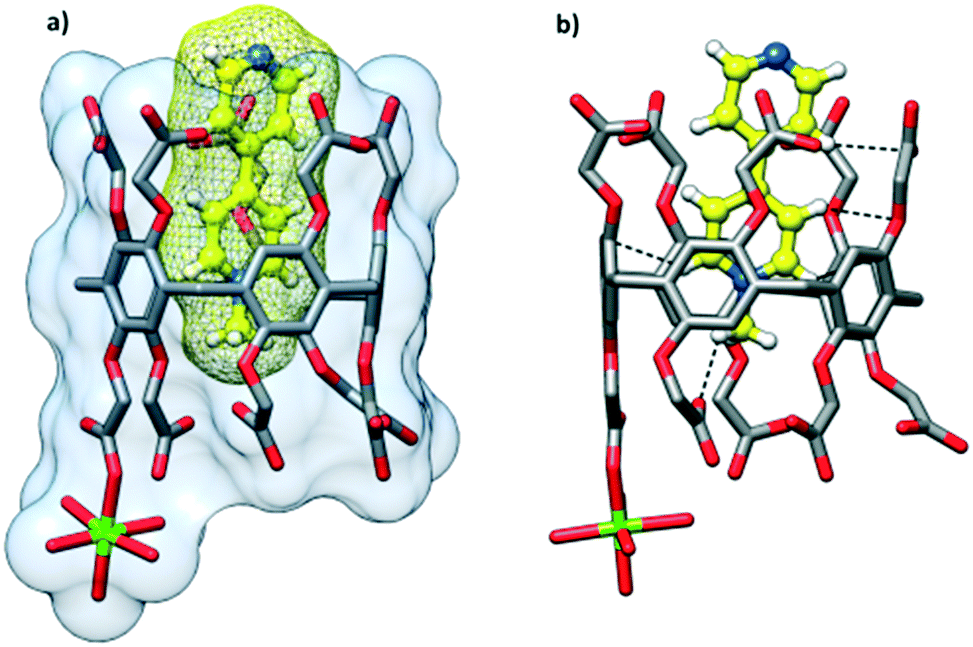
The cocrystallization of CPA5 with 1-methyl-4,4′-bipyridinium (MV) iodide resulted in yellow prismatic crystals of host–guest complex III. The asymmetric unit comprises one CPA5, one guest MV included into the host macrocyclic cavity, one magnesium cation coordinated to CPA5, and 11.5 water molecules, Fig. 7. The presence of a magnesium cation as a component of the host–guest crystalline complex was rather unexpected, as the magnesium chloride has been used by us as an additive in all crystallization experiments with CPA5 to produce better quality single crystals. The magnesium ion was not present in the crystal lattice of the CPA5 complexes with dMV and dEV. The reason for the simultaneous guest inclusion and coordination of magnesium to CPA5 carboxyl groups may be related to the monocationic type of guest MV instead of dicationic dMV and dEV without magnesium interference. However, additional structural studies on the coordination mode of magnesium (as well as other metal cations) with CPA5 and mixed coordination/host–guest complexes are necessary to give more insight into the problem. No crystal structure on the coordination complexes of CPA5 has been reported so far, which is surprising considering the tremendous interest in the incorporation of CPAn's into metal–organic frameworks, for example, for drug loading and release.15 The crystal structure was solved and refined in monoclinic space group I2/a. The methylpyridinium moiety of the guest penetrates the central cavity of the host in a manner similar to dMV@CPA5, while the pyridine part of the guest is embraced by aliphatic substituents of the ‘entrance’ rim with pyridine nitrogen protruding from the host rim. The depth of the inclusion is 3.7 Å. The main difference between monocationic guest MV and dicationic dMV besides charge is the presence of pyridine nitrogen in the MV guest; the excellent acceptor of HB was realized in the carboxylic acid–pyridine heterosynthon.16 Thus, we expected the competitive participation of MV as an acceptor in the HB network between adjacent CPA5 hosts. Indeed, single crystal X-ray analysis has shown that the pyridine nitrogen of the included guest act as the acceptor of HB from one of the carboxylic groups of the symmetry-equivalent CPA5 (O–H⋯N distance 2.591 Å) altering exclusive O–H⋯O HB assembly observed in the dMV@CPA5 and dEV@CPA5 complexes, Fig. 8.
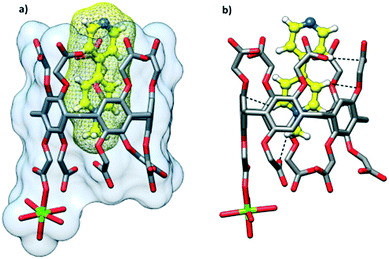 |
| | Fig. 7 Host–guest complex III of CPA5 with methyl viologen MV highlighting the (a) inclusion mode of MV and coordination mode of magnesium cation (in green); (b) C–H⋯π and C–H⋯O interactions between the guest and cavity interior. | |
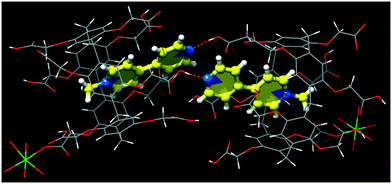 |
| | Fig. 8 The hydrogen bonding between carboxylic group of CPA5 and N-pyridine nitrogen of the guest from symmetry-equivalent MV@CPA5. | |
Magnesium cation is coordinated directly to three carboxyl groups (two carboxylate and one carboxylic) of three adjacent CPA5 in the monodentate mode, in other words, three of the ten carboxyl substituents of each CPA5 are coordinated to magnesium ions (Mg–O distances are in the range of 2.00–2.15 Å). Interestingly, the coordination is realized only on one of the rims of CPA5 – the ‘free’ rim opposite to the ‘entrance’ rim occupied with the pyridine moiety of the guest. The magnesium cation is in the typical octahedral coordination with three aqua ligands complementing the coordination sphere in addition to three carboxyl oxygen atoms of CPA5, Fig. 9a. Monodentately bound carboxylates stabilize themselves further via HB in the first coordination sphere between one carboxylic ligand (donor) and one carboxylate ligand (HB acceptor) (2.509 Å), and between one aqua ligand (HB donor) and one carboxylate (2.679 Å). There is also additional HB to second-sphere functionalities. Such coordination mode leads to the 1-D coordination polymer consisting of double-strand of CPA5, Fig. 9b. Besides coordination through an Mg cation, adjacent CPA5 are assembled via O–H⋯O HB; two short distances between oxygen atoms of 2.480 and 2.509 Å imply carboxylic–carboxylate association; one long distance 2.724 Å corresponds to the carboxylic–carboxylic supramolecular synthon.
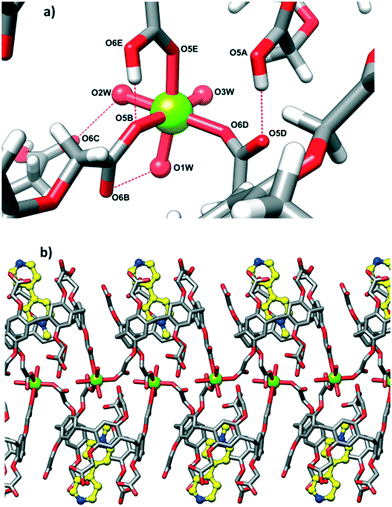 |
| | Fig. 9 (a) The coordination environment of a magnesium cation in complex III. Two carboxylate, one carboxylic and three aqua ligands constitute the octahedral coordination of Mg2+. Such coordination mode is stabilized by additional hydrogen bonding in the first coordination sphere between carboxylic and carboxylate ligands (O–H O distance of 2.509 Å) and between water and carboxylate (O–H O distance of 2.679 Å). (b) The 1-D double-strand coordination polymer is formed by the coordination of Mg2+ to three CPA5 molecules. | |
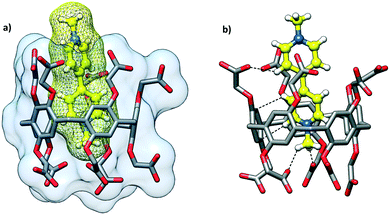
The cocrystallization of CPA5 with benzyl viologen (N,N′-dibenzyl-4,4′-bipyridinium) (dBV) chloride resulted in red plate-like crystals of host–guest complex IV. X-ray single crystal analysis revealed a host–guest complex of the exclusion type of 2:1 stoichiometry in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit comprises one CPA5 molecule, two ethanol molecules occupying the host cavity, half of the benzyl viologen dBV dication lying on the inversion center outside of the host cavity, (Fig. 10) and water molecules. The crystal structure is characterized by a high degree of disorder; eight of the ten carboxyl substituents of CPA5, ethanol molecules inside the cavity, benzyl viologen dication and water molecules were modeled as disordered. Nevertheless, the quality of the structure and appropriate modeling of disorder were sufficient to recognize the host–guest chemistry and assembly mode of the complex in the solid state. The macrocyclic cavity is filled with two ethanol molecules as in EtOH@CPA5 which was reported by us previously.8 Obviously, the inclusion of the bulky dBV would require the significant distortion of the CPA5 cavity and thus was energetically unfavorable.
space group. The asymmetric unit comprises one CPA5 molecule, two ethanol molecules occupying the host cavity, half of the benzyl viologen dBV dication lying on the inversion center outside of the host cavity, (Fig. 10) and water molecules. The crystal structure is characterized by a high degree of disorder; eight of the ten carboxyl substituents of CPA5, ethanol molecules inside the cavity, benzyl viologen dication and water molecules were modeled as disordered. Nevertheless, the quality of the structure and appropriate modeling of disorder were sufficient to recognize the host–guest chemistry and assembly mode of the complex in the solid state. The macrocyclic cavity is filled with two ethanol molecules as in EtOH@CPA5 which was reported by us previously.8 Obviously, the inclusion of the bulky dBV would require the significant distortion of the CPA5 cavity and thus was energetically unfavorable.
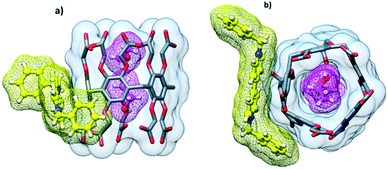 |
| | Fig. 10 Host–guest complex IV of CPA5 with dibenzyl viologen dBV. The macrocyclic cavity is occupied by two ethanol molecules (in magenta), while dibenzyl viologen (in yellow) is complexed outside the cavity: (a) side view and (b) top view of the complex. The asymmetric unit contains only half of the dBV dication lying on the inversion center; the entire dBV is shown here for the appropriate illustration of the host–guest chemistry. | |
Despite the lack of the inclusion complexation between dBV and CPA5, the deep red color of crystals suggests charge transfer between the electron-rich walls of CPA5 and electron-poor viologen moiety. Indeed, close inspection of the surroundings of dBV in the crystal lattice revealed its sandwiching between outer walls of two CPA5 macrocycles, Fig. 11. The aromatic walls of CPA5 are approximately parallel to the inserted viologen core of the dBV (6° tilt between mean planes); the closest donor–acceptor distance is at 3.29 Å (C3A⋯C4X). The 2:1 stoichiometry between CPA5 and dicationic dBV implies that only one of the ten carboxyl substituents of CPA5 is in the anionic carboxylate form. The self-assembly mode of adjacent CPA5 also resembles that of EtOH@CPA5 in a fully carboxylic form, Fig. 12.8 The only two ordered carboxylic groups are engaged in the HB cyclic dimer with O–H⋯O distances of 2.622 and 2.667 Å. The conformation of CPA5 resembles a regular pentagon as in the EtOH@CPA5 complex in contrast to inclusion complexes dMV@CPA5 and dEV@CPA5 with distorted cavities.
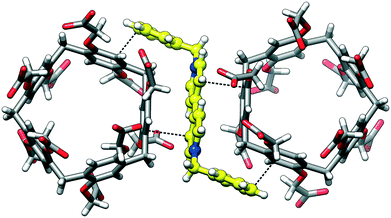 |
| | Fig. 11 The sandwiching of dibenzyl viologen dBV between outer walls of two CPA5 molecules in complex IV explaining the deep red color of crystals typical for the charge-transfer of the donor–acceptor type. The electron-deficient viologen moiety of the guest is almost parallel to electron-rich aromatic walls of CPA5 (6° tilt between the mean planes); the closest donor–acceptor distance is at 3.29 Å (shown as dashed black line between viologen and aromatic carbons of CPA5). The complexation mode is also stabilized by C–H⋯π host–guest interactions. | |
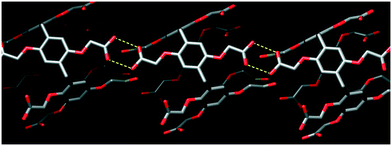 |
| | Fig. 12 The self-assembly between adjacent CPA5via the carboxylic–carboxylic cyclic supramolecular synthon in host–guest complex IV of CPA5 with dibenzyl viologen. The identification of other hydrogen bonding types was not attempted due to the heavy disorder of eight of the ten carboxylic substituents of the CPA5 (only one position for each of the disordered groups is depicted). | |
Conclusions
CPA5 have been shown to be a versatile building block for the host–guest assemblies with viologen derivatives both of inclusion and exclusion types. In all complexes, the charge-transfer between the electron-rich macrocyclic cavity and electron-deficient viologen moiety of the guest contributed to the complexation, Fig. 13. The different distribution of carboxylic versus carboxylate substituents of CPA5 renders self-assembly via carboxylic–carboxylic or carboxylic–carboxylate supramolecular synthons. It appears from the studied complexes that very short HB with O–H⋯O distances less than 2.50 Å are typical for the self-assembly of CPA5 with mixed carboxylic and carboxylate functionalities. Further studies will deal with the possibility to disrupt these preferable types of CPA5 association through targeted heterosynthon formation (for example carboxylic acid–pyridine) by the introduction of multiple or multivalent guest partners. The presence of ten carboxylic/carboxylate functional groups provides both opportunities and challenges in the future design of self-assembled architectures based on the CPA5 building block. On the one hand, it affords the possibility to participate in various intermolecular interactions in addition to the inherent cavity features, thus there is a strong temptation to utilize multivalent CPA5 for the supramolecular fabrication of multicomponent crystalline materials. On the other hand, gaining control over multiple/competing interactions when utilizing CPA5 might be a challenge for crystal engineers. The design strategies based on the use of bulky organic cations17 and/or directing metal ions,18 templating effect of solvent,19 and outer surface interactions20 lend some insight into the targeted self-assembly of cavity-containing macrocyclic hosts. These strategies have been demonstrated to be successfully implemented in many examples with water-soluble p-sulfonatocalix[n]arene21 and cucurbit[n]uril22 architectures, and may be proved useful in gaining some level of control in constructing CPA5 complex assemblies. It is clear, however, that much work remains to be done to study the self-assembly and crystallization of CPA5 complexes and, in particular, to reveal and understand the host–guest and coordination solid-state chemistry of CPA5. Our future work will focus on the further utilization of CPA5 as a building block in the supramolecular architectures while mapping out their assembly properties in the solid-sate.
 |
| | Fig. 13 Optical microscopy images of the host–guest crystals of CPA5 with dimethyl viologen (a), diethyl viologen (b), methyl viologen (c) and dibenzyl viologen (d). The charge transfer of the donor–acceptor type between electron-rich CPA5 cavity and electron-poor viologen guests is responsible for the crystal colour. Note that the intense red colour is a characteristic for the sandwiching of the dibenzyl viologen guest between exo-walls of two CPA5 molecules. | |
Experimental
Materials
Dimethyl, methyl, diethyl and dibenzyl viologens were purchased from Sigma-Aldrich. Carboxylic acid substituted pillar[5]arene was synthesized according to a literature procedure.23
Crystallization conditions
Complex I.
1 mg of CPA5 and 8.5 mg of MgCl2 were dissolved in 1.5 ml of a 1:1 water–ethanol mixture under gentle heating. The solution of 0.8 mg of dimethyl viologen (1,1′-dimethyl-4,4′-bipyridinium) dichloride, also known as paraquat, in 1 ml of a 1:1 water–ethanol mixture was slowly added to the solution of CPA5. The diffraction-quality crystals of complex I dMV@CPA5 grew after several days.
Complex II.
0.9 mg of CPA5 and 4.8 mg of MgCl2 were dissolved in 1.5 ml of a 1:1 water–ethanol mixture under gentle heating. The solution of 0.6 mg of diethyl viologen (1,1′-diethyl-4,4′-bipyridinium) dibromide in 0.5 ml of a 1:1 water–ethanol mixture was slowly added to the solution of CPA5. The diffraction-quality crystals of complex II dEV@CPA5 grew after several days.
Complex III.
1 mg of CPA5 and 8.7 mg of MgCl2 were dissolved in 1.5 ml of a 1:1 water–ethanol mixture under gentle heating. The solution of 1.1 mg of methyl viologen (1-methyl-4,4′-bipyridinium) iodide in 0.5 ml of a 1:1 water–ethanol mixture was slowly added to the solution of CPA5. The diffraction-quality crystals of complex III MV@Mg(CPA5) grew after several days.
Complex IV.
1.2 mg of CPA5 and 7.2 mg of MgCl2 were dissolved in 1.5 ml of a 1:1 water–ethanol mixture under gentle heating. The solution of 0.5 mg of dibenzyl viologen (1,1′-dibenzyl-4,4′-bipyridinium) dichloride in 0.5 ml of a 1:1 water–ethanol mixture was slowly added to the solution of CPA5. The diffraction-quality crystals of complex IV 2(EtOH)@(CPA5)(dBV) grew after several days.
Crystallography
The crystals were embedded in the inert perfluoropolyalkylether (viscosity 1800cSt; ABCR GmbH) and mounted using Hampton Research Cryoloops. The crystals were flash cooled to 100.0(1) K in a nitrogen gas stream and kept at this temperature during the experiments. The X-ray data were collected on a SuperNova Agilent diffractometer using CuKα radiation (λ = 1.54184 Å). The data were processed with CrysAlisPro.24 The structures were solved by direct methods and refined using SHELXL25 under WinGX.26 The figures were prepared using Chimera.27
Crystal data for I.
(C55H48O30)·(C12H14N2)·5.27(H2O), Mr = 1470.1, orange blocks, monoclinic, space group P21/c, a = 21.4549(4), b = 12.5308(1), c = 26.2757(4) Å, β = 111.934(2)°, V = 6552.8(2) Å3, Z = 4, ρcalc = 1.49 g cm−3, μ(CuKα) = 0.79 mm−1, θmax = 72.11°, 45391 reflections measured, 12768 unique, 1159 parameters, R = 0.069 and wR = 0.199 (R = 0.094 and wR = 0.219 for all data). GooF = 1.06. CCDC 2040337.
Crystal data for II.
(C55H48O30)·(C14H18N2)·6.5(H2O), Mr = 1520.3, yellow blocks, monoclinic, space group P21/n, a = 21.8515(1), b = 12.3750(6), c = 27.435(2) Å, β = 112.883(9)°, V = 6834.9(9) Å3, Z = 4, ρcalc = 1.48 g cm−3, μ(CuKα) = 1.04 mm−1, θmax = 67.80°, 43895 reflections measured, 12194 unique, 975 parameters, R = 0.072 and wR = 0.180 (R = 0.123 and wR = 0.204 for all data). GooF = 0.88. CCDC 2040338.
Crystal data for III.
[Mg(C55H47O30)·3(H2O)](C11H11N2)·8.5(H2O), Mr = 1590.6, yellow blocks, monoclinic, space group I2/a, a = 35.5280(3), b = 12.9487(1), c = 33.1933(3) Å, β = 105.019(1)°, V = 14748.7(2) Å3, Z = 8, ρcalc = 1.43 g cm−3, μ(CuKα) = 1.12 mm−1, θmax = 66.60°, 30850 reflections measured, 12912 unique, 1064 parameters, R = 0.099 and wR = 0.283 (R = 0.105 and wR = 0.289 for all data). GooF = 0.97. CCDC 2040339.
Crystal data for IV.
(C55H49O30)·0.5(C24H22N2)·2(C2H5OH)·3 (H2O), Mr = 1505.3, red plates, triclinic, space group P, a = 12.9186(5), b = 13.6147(6), c = 21.7390(8) Å, α = 89.925(3), β = 81.101(3), γ = 76.021°, V = 3663.0(3) Å3, Z = 2, ρcalc = 1.37 g cm−3, μ(CuKα) = 0.94 mm−1, θmax = 69.71°, 29147 reflections measured, 13627 unique, 1132 parameters, R = 0.106 and wR = 0.284 (R = 0.120 and wR = 0.297 for all data). GooF = 0.99. CCDC 2040336.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was funded by the National Science Centre of Poland (grant PRELUDIUM 14 nr 2017/27/N/ST5/01931).
Notes and references
- T. Ogoshi, M. Hashizume, T. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46, 3708 RSC.
- T. Xiao, W. Zhong, L. Xu, X. Q. Sun, X. Y. Hu and L. Wang, Org. Biomol. Chem., 2019, 17, 1336 RSC; N. Song, T. Kakuta, T. Yamagishi, Y.-W. Yang and T. Ogoshi, Chem, 2018, 4, 2029 Search PubMed; P. J. Cragg, Isr. J. Chem., 2018, 58, 1194 CrossRef CAS; Y. Liu, F. Zhou, F. Yang and Da Ma, Org. Biomol. Chem., 2019, 17, 5106 RSC; T. Kakuta, T. Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656 CrossRef; H. Zhang, Z. Liu and Y. Zhao, Chem. Soc. Rev., 2018, 47, 5491 RSC; S. Dasgupta and P. S. Mukherjee, Org. Biomol. Chem., 2017, 15, 762 RSC; N. L. Strutt, H. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47(8), 2631 CrossRef.
- X. Y. Hu, K. Jia, Y. Cao, Y. Li, S. Qin, F. Zhou, C. Lin, D. Zhang and L. Wang, Chem. – Eur. J., 2015, 21, 1208 CrossRef CAS; X. Wu, Y. Li, C. Lin, X. Y. Hu and L. Wang, Chem. Commun., 2015, 51, 6832 RSC; L. Gao, B. Zheng, W. Chen and C. A. Schalley, Chem. Commun., 2015, 51, 14901 RSC; Y. L. Sun, Y. W. Yang, D. X. Chen, G. Wang, Y. Zhou, C. Y. Wang and J. F. Stoddart, Small, 2013, 9, 3224 Search PubMed.
- G. Sun, Z. He, M. Hao, M. Zuo, Z. Xu, X. Y. Hu, J. J. Zhu and L. Wang, J. Mater. Chem. B, 2019, 7, 3944 RSC; G. Sun, Z. He, M. Hao, Z. Xu, X.-Y. Hu, J.-J. Zhu and L. Wang, Chem. Commun., 2019, 55, 10892 RSC; B. Li, Z. Meng, Q. Li, X. Huang, Z. Kang, H. Dong, J. Chen, J. Sun, Y. Dong, J. Li, X. Jia, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2017, 8, 4458 RSC; G. Ping, Y. Wang, L. Shen, Y. Wang, X. Hu, J. Chen, B. Hu, L. Cui, Q. Meng and C. Li, Chem. Commun., 2017, 53, 7381 RSC; N. J. Wheate, K.-A. Dickson, R. R. Kim, A. Nematollahi, R. B. Macquart, V. Kayser, G. Yu, W. B. Church and D. J. Marsh, J. Pharm. Sci., 2016, 105, 3615 CrossRef CAS.
- J. Chen, H. Ni, Z. Meng, J. Wang, X. Huang, Y. Dong, C. Sun, Y. Zhang, L. Cui, J. Li, X. Jia, Q. Meng and C. Li, Nat. Commun., 2019, 10, 3546 CrossRef.
- S. Kosiorek, N. Rad and V. Sashuk, ChemCatChem, 2020, 12, 2776 CrossRef CAS.
- Y. Zhou, K. Jie, R. Zhao and F. Huang, Adv. Mater., 2020, 32, 1904824 CrossRef CAS.
- O. Danylyuk and V. Sashuk, CrystEngComm, 2015, 17, 719 RSC.
- A. Swartjes, P. B. White, M. Lammertink, J. A. A. W. Elemans and R. J. M. Nolte, Angew. Chem., Int. Ed., 2020, 59, 2 CrossRef CAS; N. K. Beyeh, H. H. Jo, I. Kolesnichenko, F. Pan, E. Kalenius, E. V. Anslyn, R. H. A. Ras and K. Rissanen, J. Org. Chem., 2017, 82, 5198 CrossRef; K. Moon and E. Kaifer, Org. Lett., 2004, 6(2), 185 CrossRef.
- G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489 CrossRef CAS.
- L. D'Ascenzo and P. Auffinger, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 164 CrossRef.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1994, 116, 909 CrossRef CAS;
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- L. H. R. Dos Santos, B. L. Rodrigues, Y. M. Idemori and N. G. Fernandes, J. Mol. Struct., 2012, 1014, 102 CrossRef CAS; L. Sobczyk, S. J. Grabowski and T. J. Krygowski, Chem. Rev., 2005, 105(10), 3513 CrossRef.
- L. K. Saunders, H. Nowell, L. E. Hatcher, H. J. Shepherd, S. J. Teat, D. R. Allan, P. R. Raithby and C. C. Wilson, CrystEngComm, 2019, 21, 5249 RSC; C. L. Perrin, Acc. Chem. Res., 2010, 43, 1550 CrossRef CAS.
- H. Zhang, Z. Liu, F. Xin and Y. Zhao, Coord. Chem. Rev., 2020, 420, 213425 CrossRef CAS; K. Yang, K. Yang, S. Chao, J. Wen, Y. Pei and Z. Pei, Chem. Commun., 2018, 54, 9817 RSC; L.-L. Tan, H. Li, Y.-C. Qiu, D.-X. Chen, X. Wang, R.-Y. Pan, Y. Wang, S. X.-A. Zhang, B. Wang and Y.-W. Yang, Chem. Sci., 2015, 6, 1640 RSC.
- T. R. Shattock, K. K. Arora and M. J. Zaworotko, Cryst. Growth Des., 2008, 8(12), 4533 CrossRef CAS.
- I. Ling, Y. Alias, B. W. Skelton and C. L. Raston, Cryst. Growth Des., 2012, 12, 1564 CrossRef CAS.
- R. E. Fairbairn, S. J. Teat, I. Ling and J. Dalgarno, CrystEngComm, 2019, 21, 6659 RSC; P. P. Cholewa and S. J. Dalgarno, CrystEngComm, 2014, 16, 3655 RSC; I. Ling, K. S. Iyer, C. S. Bond, A. N. Sobolev, Y. Aliasa and C. L. Raston, CrystEngComm, 2012, 14, 8541 RSC.
- S. Kennedy, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2012, 12, 679 CrossRef CAS; S. Kennedy, I. E. Dodgson, C. M. Beavers, S. J. Teat and S. J. Dalgarno, Cryst. Growth Des., 2012, 12, 688 CrossRef; S. Kennedy and S. J. Dalgarno, Chem. Commun., 2009, 5275 RSC.
- Y. Huang, R.-H. Gao, M. Liu, L.-X. Chen, X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, K. Chen and Z. Tao, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202002666; X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, S.-F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386 CrossRef CAS.
- S. L. Loh, A. N. Sobolev, S. H. Lim and I. Ling, CrystEngComm, 2020, 22, 52 RSC; I. Ling, H. Kumari, M. Mirzamani, A. N. Sobolev, C. J. Garvey, J. L. Atwood and C. L. Raston, Chem. Commun., 2018, 54, 10824 RSC; I. Ling, A. N. Sobolev and C. L. Raston, CrystEngComm, 2015, 17, 1526 RSC.
- L.-X. Chen, M. Liu, Y.-Q. Zhang, Q.-J. Zhu, J.-X. Liu, B.-X. Zhu and Z. Tao, Chem. Commun., 2019, 55, 14271 RSC; L. Mei, F.-Z. Li, J.-H. Lan, C.-Z. Wang, C. Xu, H. Deng, Q.-Y. Wu, K.-Q. Hu, L. Wang, Z.-F. Chai, J. Chen, J. K. Gibson and W.-Q. Shi, Nat. Commun., 2019, 10, 1532 CrossRef CAS.
- T. Ogoshi, S. Kanai, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130(15), 5022 CrossRef CAS; C. Li, X. Shu, J. Li, S. Che, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, J. Org. Chem., 2011, 76(20), 8458 CrossRef.
-
Agilent Technologies, CrysAlisPro, Version 1.171.38.46 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25(13), 1605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format. CCDC 2040336–2040339. For crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01579b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sandra
Kosiorek
,
Volodymyr
Sashuk
,
Sandra
Kosiorek
,
Volodymyr
Sashuk