
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concerning the selection of crystallization modifiers for non-hydrogen bonded systems: the case of benzophenone†
Simon N.
Black
 ,
Adrian
Hutchinson
and
Roger J.
Davey
*
,
Adrian
Hutchinson
and
Roger J.
Davey
*
Department of Chemical Engineering and Analytical Science, School of Engineering, University of Manchester, M13PL, UK. E-mail: roger.davey@manchester.ac.uk
First published on 8th January 2021
Abstract
A design strategy for the selection of crystal growth modifiers for non-polar crystals is proposed and its application demonstrated for the case of benzophenone. The strongest intermolecular interaction in the crystal structure of the stable form of benzophenone combines two strong phenyl–phenyl approaches, which embody screw symmetry along the x-axis. Two of the nine additives selected to disrupt this interaction were successful in modifying the morphology and decreasing crystal growth rates of benzophenone crystals. This efficacy is discussed and interpreted in the light of the crystal structures of the additives, which display a great variety of phenyl⋯phenyl approaches and associated symmetries. As part of this study we made use of the idea of ‘favourable connections’, based on a simple geometric method, to reveal ‘symthons’: symmetry-forming phenyl–phenyl approaches.
Introduction
The design and selection of additives suitable for modifying and controlling the crystallization of a desired solute have been well studied over the last few decades. Thus in the context of habit modification, inhibition of crystal growth and control of polymorphic form the notion of ‘tailor-made additives’ has become ubiquitous.1–4 For polar molecules whose crystals utilize significantly directional inter-molecular connections, such as H-bonds, the methodology for additive selection, based on a knowledge of molecular packing in relevant crystal faces, has been widely applied. Thus, imposter molecules are chosen which can simultaneously enter a growing surface and disrupt the extension of a chosen directional interaction thereby selectively inhibiting growth of the face.5 However, for weakly polar molecules and in the absence of H-bonds, no such coherent strategy has been articulated and clearly, a different procedure is needed. One much explored example, typical of this class of materials, is the alkanes, where crystallization modifiers are an important component of diesel fuels.6 Here it is apparent that successful outcomes employ the strongest whole-molecule to whole-molecule interactions with additives designed to disrupt or terminate chains of the multiple intermolecular connections7,8 involved. A related method was reported for α p-aminobenzoic acid where inhibitors were selected which were capable of binding as whole molecules to aromatic stacks and hence inhibiting growth along the crystal's needle axis.9In exploring the potential of this technique in other weakly polar systems we chose benzophenone (BZP) as a target for study because it cannot form hydrogen bonds with itself, but does contain distinct functional groups – two phenyl rings and a carbonyl group. Hence, crystal structures of this polymorphic material contain close approaches (e.g. phenyl⋯phenyl) of different functional groups but no continuous H-bonded motif that may be targeted by a tailor-made additive. This demonstrates the challenge for additive design with molecules of this type and we define a strategy in which we first identify the strongest potential BZP⋯BZP intermolecular interactions based on the molecule and its crystal structures. This analysis was performed qualitatively by visualizing and counting “favourable connections”, judged based on the distance, between non-hydrogen atoms. Secondly, potential additives were selected, as whole molecules, to exploit or disrupt these strong interactions.
It is worth commenting on the vital role that these favourable connections play. Comparison of the measured sublimation and melting enthalpies of BZP (95.1 kJ mol−1 (ref. 10) and 16.9 kJ mol−1 (ref. 11)) indicate that in the transition from vapour, via liquid, to crystal, the majority of the enthalpy change is associated with molecular condensation. Just ∼20% of the sublimation (lattice) energy is used to direct the intermolecular contacts that give rise to crystallographic symmetry and lead to crystallization and this takes place via these favourable connections.
As part of this study, a brief description of the morphology of BZP was undertaken leading to an experimental investigation of the effectiveness of the selected additives as habit modifiers. This qualitative assessment was underpinned by measurement of growth rates of specific crystallographic directions. Comparison of these results with the predictions highlighted some unexpected features. The crystal structures of additives, where available, were consulted for further insights. Some general comments are made on different ways of representing and evaluating interactions involving phenyl groups and conjugated substituents.
Previous studies of BZP
BZP has two known polymorphic crystal structures. The morphology of the stable form is described in Groth12 and the references cited therein. He defined the orthorhombic point group and assigned the unit cell axes such that c < a < b. This assignment has been perpetuated in studies of crystal growth rates,13 crystal structure,14,15 habit modification by solvents10 and colour change by dye incorporation.16 There are nine crystal structure determinations of this orthorhombic form of BZP in the CSD,17 all in space group P212121. While BPHENO01 (ref. 14) and BPHENO10 (ref. 15) adopt the axis assignment from Groth,12 in BPHENO12 (ref. 18) the axes are assigned differently with a < b < c (a = 7.7378 Å, b = 10.2421 Å and c = 12.0395 Å). This assignment has been adopted in the other six more recent structure determinations16,19,20 as given in the CSD. In accordance with the space group P212121 and Z' = 1, a single crystal comprises only one of the two mirror image conformations. Indeed, a recent study,20 determined the absolute configurations of individual crystals by X-ray diffraction and confirmed them by circular dichroism.Throughout the current work, we have used BPHENO12 for the stable form of BZP. In this structure determination, all hydrogen atom positions were assigned rather than determined experimentally, and all the C–H bond lengths are 0.950 Å.
The preparation of a metastable, monoclinic, polymorph of BZP from supercooled melts was first reported in 1871 (ref. 12) and there are three crystal structure determinations in the CSD: BPHENO02,21 BPHENO03 (ref. 21) and BPHENO11.22 The space group is C2/c with the same axis settings and here we used BPHENO11 for this metastable form.
Using this structural data for the two forms we now proceed to examine the conformations of BZP and the nature of the strongest BZP interactions.
The BZP molecule
BZP (Scheme 1) contains two phenyl rings linked by a central carbonyl group. It follows that conformations of BZP are compromises between the unfavourable overlap of two ortho hydrogens and the favourable, extended conjugation through the carbonyl group. The conformation of BZP in BPHENO12 is shown in Fig. 1. The two phenyl rings are denoted A1 and A2 at their centroids. The carbonyl group, the neighbouring phenyl carbons, and the centroids, lie in the mean plane, shown in yellow in Fig. 1. The two phenyl rings are twisted out of this plane with torsion angles of 31.2° and 31.0°, close to the most common angles for 7742 similar molecules.23 This suggests that this is the minimum energy conformation, but that other conformations may be accessed to optimize packing energies (e.g. as in the metastable form, BPHENO11, see below). We may thus consider the BPHENO12 conformation to comprise two overlapping conjugated systems each with eight heavy atoms. BZP is oriented relative to the crystallographic axes as shown (Fig. 1), with the mean plane nearly perpendicular to the x axis, the long axis of the molecule close to the y axis, and the carbonyl groups almost parallel to the z axis. Hence the phenyl rings “face” approximately along the x axis.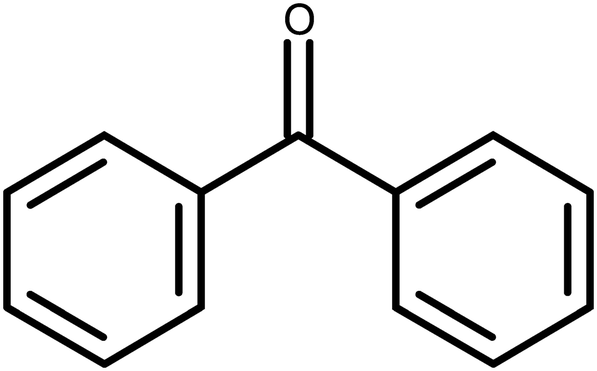 | ||
| Scheme 1 The BZP molecule. | ||
 | ||
| Fig. 1 BZP in BPHENO12, viewed along x (above) and z (below). | ||
Favourable connections
Each BZP molecule has four phenyl faces and eight accessible phenyl edges. Intermolecular interactions between aromatic molecules are often conveniently categorized as “face⋯face” phenyl⋯phenyl approaches (as in graphite) or “edge⋯face” approaches (as in benzene). More generally, intermolecular interactions may also be analysed as sets of atom⋯atom connections – the basis for many computational methods for calculating intermolecular interaction energies. Here we utilize the concepts of intermolecular interactions, functional group approaches, and atom⋯atom connections in BZP by visualizing selected atom⋯atom connections.Connections between carbon atoms were defined as “favourable” for interatomic separations <4.70 Å. While this is significantly larger than the sum of the van der Waals radii for C⋯C connections (3.40 Å), energy calculations using the UNI function in Mercury show (see S1 and Fig. S1.1†) that even at this separation C⋯C connections are indeed more attractive24 than all C⋯H and H⋯H connections. This conclusion, although surprising, clarifies the relevance of connections at distances beyond (>0.5 Å) the sum of the VDW radii24 and supports the insights revealed here in the search for “favourable connections”. For simplicity, the same distance criterion was used here to define all favourable heavy atom⋯heavy atom separations and based on the above discussion, the weaker C⋯H and H⋯H connections were ignored. Fig. S2.1 and Table S2.1† underline the validity of this geometric approach, for the BZP/additive systems studied here, by comparing the number of favourable contacts with the equivalent interaction energies. In what follows, strong interactions between benzophenone molecules are identified by the number of such favourable connections (NFC), grouped as phenyl⋯phenyl, phenyl⋯carbonyl or carbonyl⋯carbonyl, and further split according to which phenyl rings they connect. This allows an assessment of how phenyl and carbonyl groups within BZP contribute to each intermolecular interaction. This analysis was complemented by use of the “Aromatic Analyser”25 supplied with the Mercury software (see S3† for full details).
This method identifies up to 50 favourable atom⋯atom connections for each BZP⋯BZP intermolecular interaction. No repulsive C⋯C connections (separations <3.40 Å) were found.26 The large number of C⋯C connections captures the diffuse nature of phenyl⋯phenyl approaches, recognizing that, unlike hydrogen bonds, they are not “point contacts”. The method is illustrated below in detail for the two strongest BZP⋯BZP interactions originating from its two polymorphic forms.
BZP(1): the strongest intermolecular interaction in BPHENO12
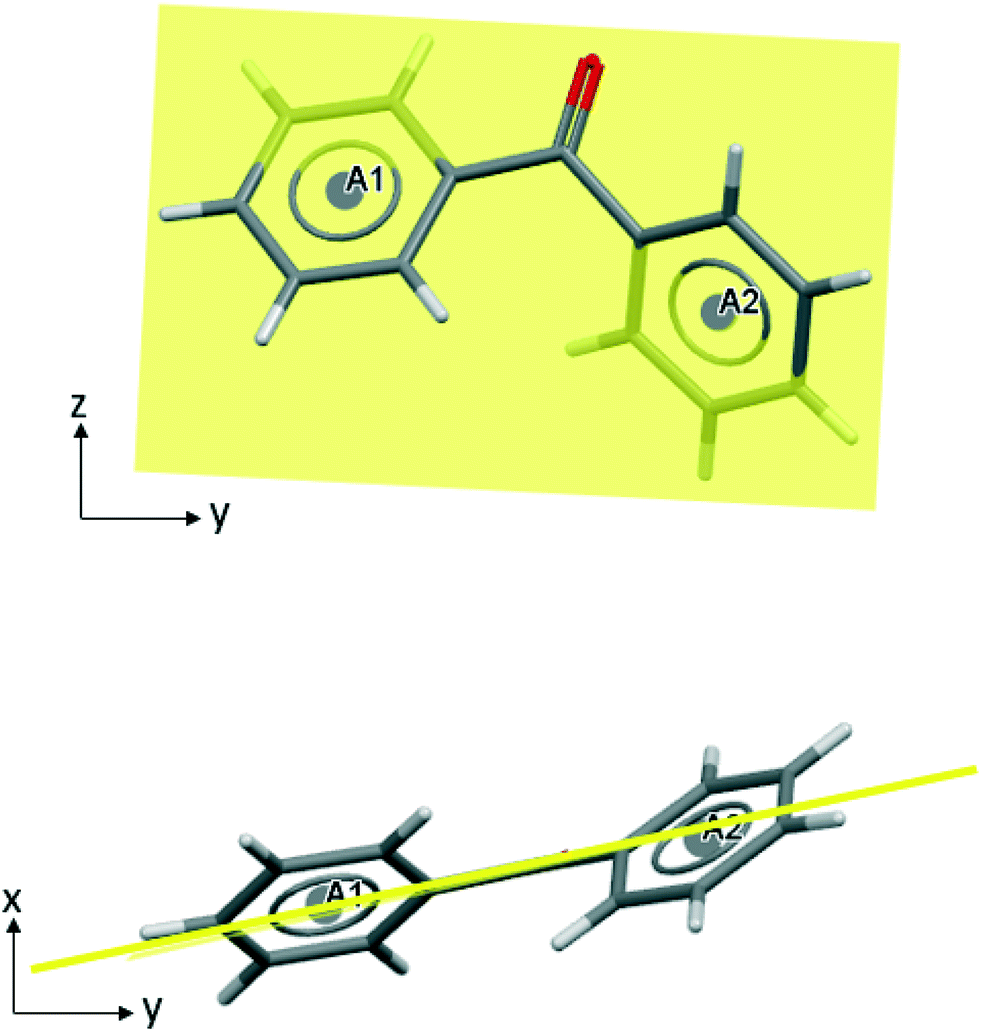
In the stable polymorph, BPHENO12, the intermolecular interaction that contains the highest number of these favourable connections is referred to here as BZP(1) and is shown in Fig. 2.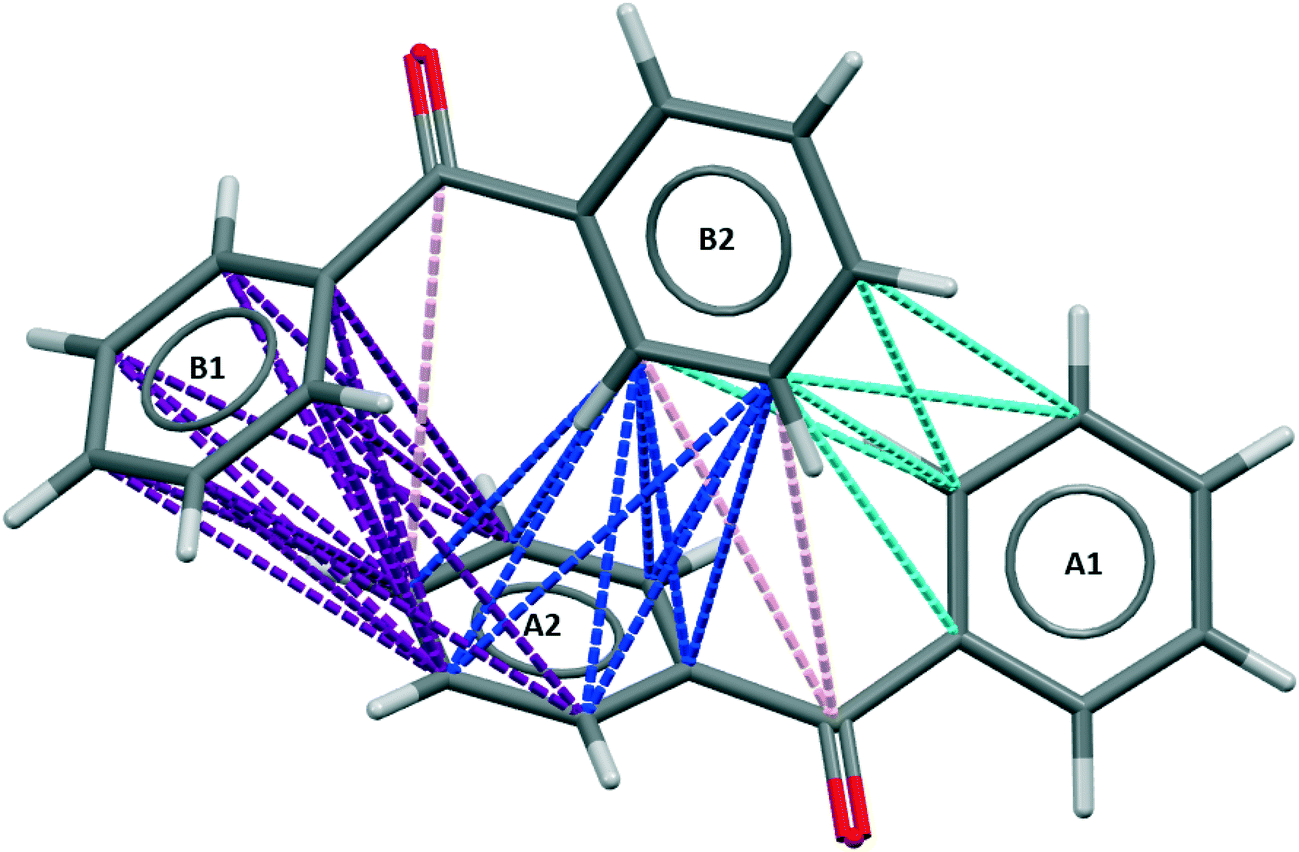 | ||
| Fig. 2 BZP(1) – the strongest intermolecular interaction in BPHENO12. The aromatics rings are designated A1, A2 (lower molecule) and B1, B2 (upper molecule). See text for further explanation. | ||
For this dimer, in which molecules are related by the 2-fold screw axis along x, there are a total of 37 favourable connections.
This analysis identifies the A2⋯B1 approach as the strongest phenyl–phenyl approach in this crystal structure, with four carbon atoms from phenyl ring A2 connected to all six carbon atoms in phenyl ring B1. The 18 favourable connections in this phenyl⋯phenyl approach are coloured purple in Fig. 2. This is consistent with the Aromatic Analyser, which adds that the relative orientation of these phenyl rings is 39° (see S3† for further details). This approach is neither “face⋯face” nor “edge⋯face” but could be described as “skew”.
In the A2⋯B2 approach, two carbon atoms from phenyl ring B2 are connected to all six carbon atoms in phenyl ring A2 with the 10 favourable connections, coloured blue in Fig. 2. The angle between the planes is 84°. This is a typical “edge⋯face” approach, similar to those in benzene. The A1⋯B2 approach is designated as “weak” (defined here as having fewer than 10 favourable connections), coloured cyan in Fig. 2. The remaining three favorable connections, which are coloured pink in Fig. 2, are from two phenyl⋯carbonyl approaches that play a minor role in this interaction.
This interaction embodies the 2-fold screw axis in the x direction with every second molecule in the resulting homochiral chain related by the a repeat of 7.74 Å. There are no other BZP⋯BZP interactions in BPHENO12 with NFC > 25. Further information on weaker interactions is given in section S4 of the ESI.†
BZP(2): the strongest intermolecular interaction in BPHENO11
The strongest intermolecular interaction in the centrosymmetric, metastable polymorph, BPHENO11, involves the two mirror image conformers related by an inversion centre, as shown in Fig. 3.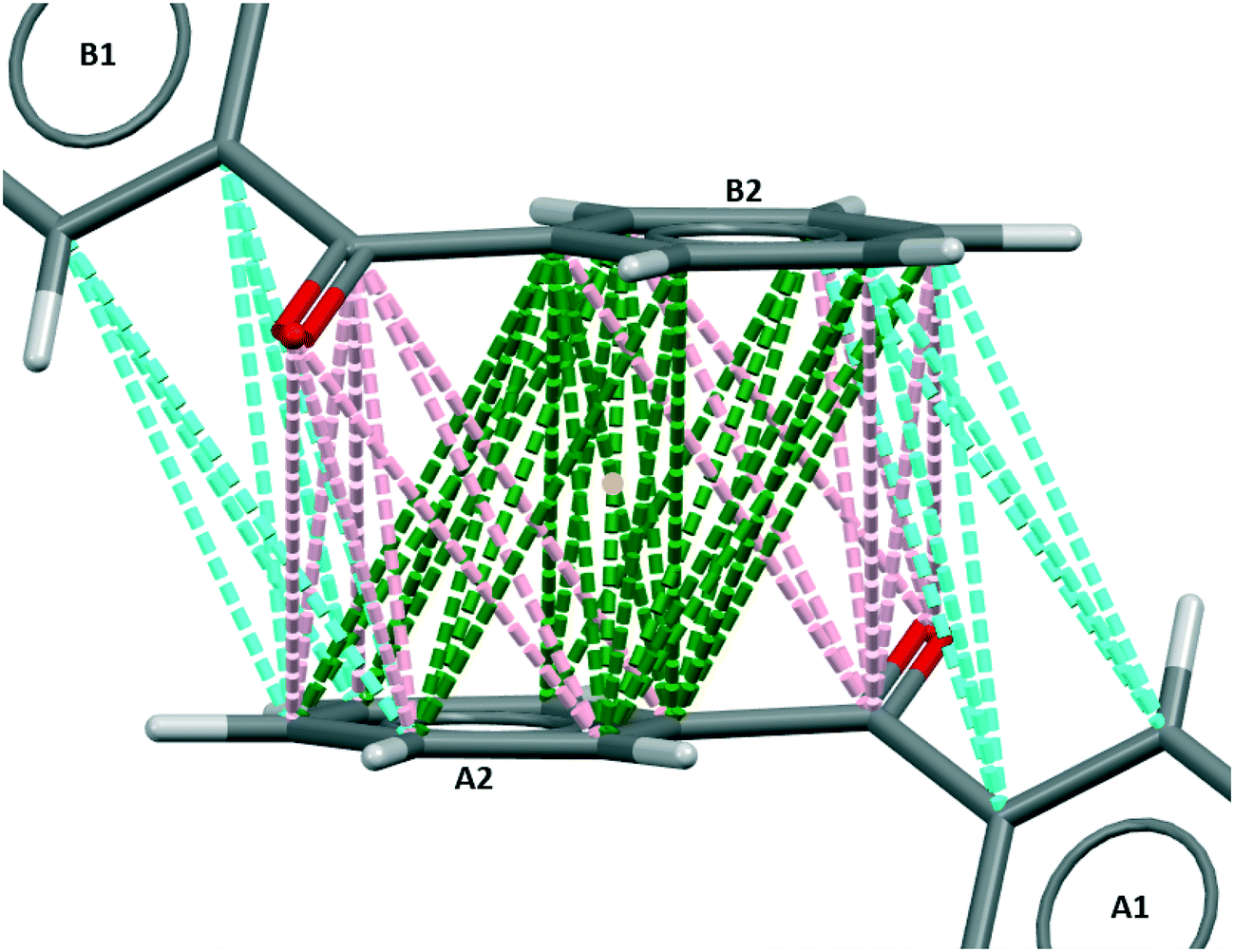 | ||
| Fig. 3 BZP(2) – the strongest intermolecular interaction in BPHENO11. The yellow dot identifies the inversion centre. See text for further explanation. | ||
The strongest approach is the face⋯face approach of A2 and B2 phenyl groups, with 22 favourable connections, involving all twelve carbon atoms and shown in green in Fig. 3. This is supplemented by two carbonyl⋯phenyl approaches, each with 8 favourable connections, shown in pink in Fig. 3. There are also weaker A1⋯B2 and A2⋯B1 approaches, each with 6 favourable connections, shown in cyan in Fig. 3.
The face⋯face interplanar separation is 3.72 Å – significantly larger than is typically taken for π–π stacking. The torsion angles are 30.7° (similar to BPHENO12) and 44.3°. Mogul analysis23 indicates that the larger torsion is less common in molecules of this type. This distortion, combined with the larger interplanar spacing, suggests that this interaction is a compromise between optimizing torsion angles and optimizing the face⋯face approach.
The approaches in the two intermolecular interactions BZP(1) and BZP(2) are summarized and compared in Table 1 for the two polymorphs.
| Polymorph | BPHENO12 | BPHENO11 | ||
|---|---|---|---|---|
| Orthorhombic, stable | Monoclinic, metastable | |||
| Strongest interaction | BZP(1) | BZP(2) | ||
| Approach | N FC | Colour/type | N FC | Colour/type |
| A2⋯B2 | 10 | Blue/edge⋯face | 22 | Green/face⋯face |
| A2⋯B1 | 18 | Purple/skew | 6 | Cyan |
| A1⋯B2 | 6 | Cyan | 6 | Cyan |
| A2⋯carbonyl | 1 | Pink | 8 | Cyan |
| B2⋯carbonyl | 2 | Cyan | 8 | Cyan |
| Total | 37 | 2-Fold screw axis | 50 | Inversion |
Additive design
In considering the choice of additives to modify the crystal habit/growth kinetics of the stable polymorph of BZP it would be accepted procedure to consider the molecular packing in the targeted growth directions and choose BZP-like molecules capable of disrupting the related interactions. However, the choice of such inhibitor molecules must also be informed by the previous discussion above concerning BZP–BZP interactions, since to form strong interactions with BZP molecules during growth this whole molecule interaction must be mimicked in order for additives to compete with a large excess of other BZP molecules in solution. With this in mind, it is clear that the two strongest interactions in Table 1 are the benchmark against which additives must compete and this enables us to define certain selection criteria:1. BZP(1), the strongest molecular interaction in BPHENO12 (Fig. 2) includes close approaches of both phenyl rings in each molecule. This suggest that additives should also contain two phenyl rings linked by a single intermediate atom.
2. BZP(2), the strongest interaction in BPHENO11 (Fig. 3) includes a “face-to-face” approach between two phenyl rings, supported by multiple favourable connections with the conjugated carbonyl groups. This suggests that the number of favourable connections (NFC) could be further increased by adding other planar, conjugated substituents such as halides, amino or nitro groups to the phenyl ring.
This leads to the prediction that adding substituents to the phenyl rings will give efficacious additives, but removing or replacing atoms in the carbonyl group will not. In considering how such potential additives might interact with the growing crystal, we next explore the morphology of the stable form of BZP.
Crystal morphology & additive design
The experimental morphology of the stable polymorph of BZP is compared with the BFDH prediction in Fig. 4. Both images are drawn in the same crystallographic orientation, with the x axis close to vertical and the z axis perpendicular to the orange, {002}, face. In what follows, all Miller indices have been adjusted, where necessary, to refer to the unit cell of BPHENO12 in which a < b < c.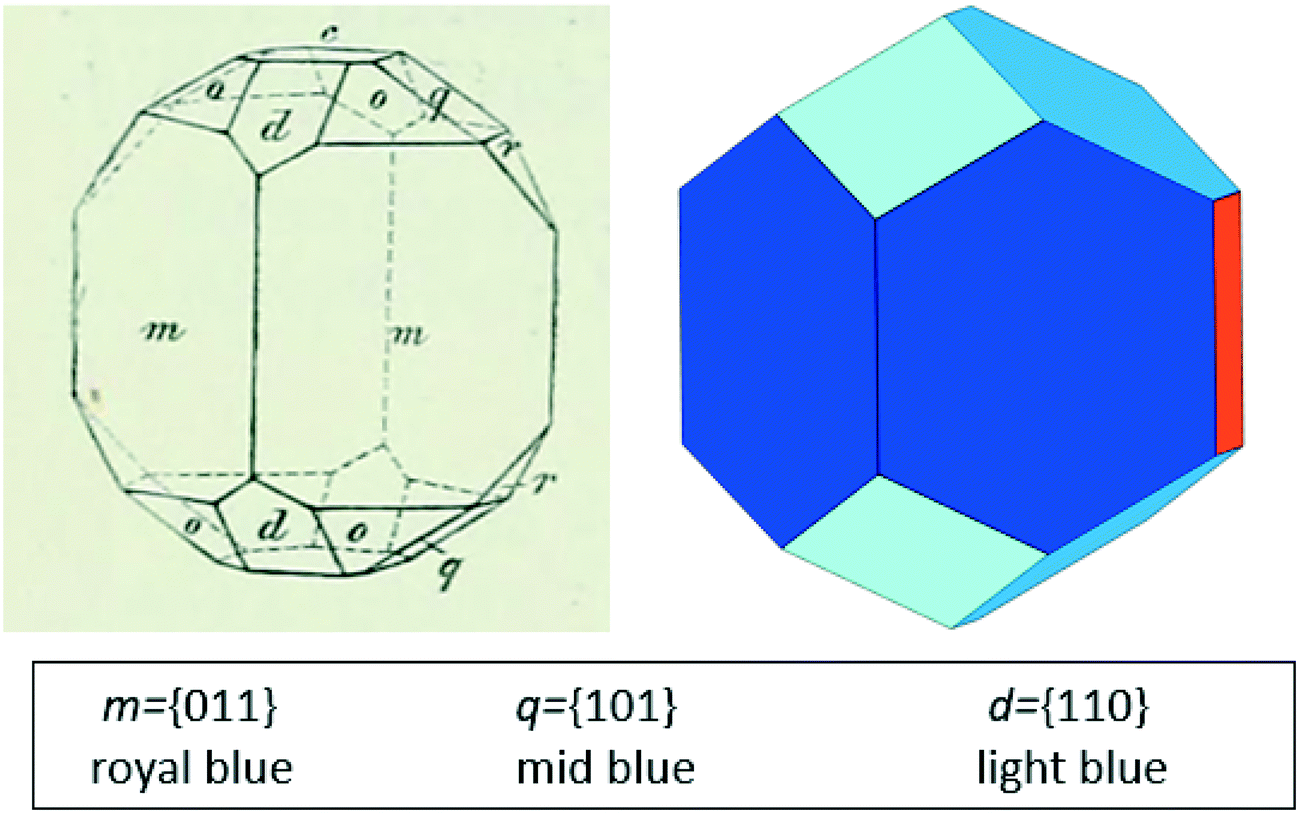 | ||
| Fig. 4 Experimental (left, Groth12) and predicted, BDFH (right) morphologies of BZP. Key identifies the Groth indexing and our colour coding. See Table 2 and text for further details. | ||
The experimental morphology shown in Fig. 4 (left) was measured before X-ray crystallography, relying instead on accurate optical measurements of the angles between crystal faces. This enabled the determination of the point group symmetry (“rhombisch bissphenoidisch”), the relative repeat ratios for the three crystallographic axes dimensions, and Miller indices for each face, all of which are consistent with later X-ray determination of the point group (222) and the unit cell dimensions. Groth12 labels the faces in drawings such as Fig. 4 (left) with lower case letters rather than Miller indices, to clarify which faces are related by symmetry. Faces at the back of the image are not labelled. He clarifies that in this point group the four {−111} faces and the four {111} faces are not symmetry-related despite all being labelled (o). Fig. 4 (right) shows the BFDH predicted morphology, prepared using Mercury where, inspired by Groth,12 we have assigned symmetry-equivalent faces the same colour manually. Table 2 gives further details of how the two morphologies compare, showing the relationship between Groth's letters, the BFDH colours and the Miller indices.
Groth summarises a range of morphologies from earlier reports, in which {002}, {102}. {111} and {1−11} are not always present but in which the largest four faces (in agreement with BDFH) are {011}. The morphology of crystals grown from 2-propanol in the absence of additives in this study was determined by examination of crystals mounted on a single-crystal X-ray diffractometer.27 The results are also included in Table 2. This morphology is consistent with Fig. 4 and with Roberts et al.10 The four {011} faces are the most important with eight {110} and {101} faces usually present to cap the ends of crystals and other faces occasionally present.
Combining this morphological data with the above analysis of BZP interactions, it becomes apparent that additives, which interfere with growth in the direction of the strongest intermolecular interaction, will slow down growth along the x axis. If this is their strongest effect, the result will be more ‘plate-like’ crystals, possibly showing large {200} faces.
Additive selection
Commercially available additives were selected, based on the above discussion. All contained two phenyl rings, at least one of which was unsubstituted, linked by one central atom. The molecular structures of these additives are summarized in Table 3. The initial expectation, based on the above discussion, was that the six additives containing a carbonyl group would be more effective than the three additives in which it has been replaced.| Molecule | Abbreviation | Phenyl groups linked by | Difference in number of heavy atoms | Position of substituent | Melting point27 (°C) |
|---|---|---|---|---|---|
| Benzophenone | BZP | >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
— | — | 49 |
| Diphenylamine | DPA | >NH2 | −1 | — | 53 |
| Diphenylmethane | DPM | >CH2 | −1 | — | 25 |
| Diphenylethene | DPE | >CCH2 |
0 | — | 7 |
| 4-Aminobenzophenone | 4ABZP | >CO |
+1 | Para | 123 |
| 4-Methylbenzophenone | 4MBZP | >CO |
+1 | Para | 55 |
| 4-Nitrobenzophenone | 4NBZP | >CO |
+3 | Para | 138 |
| 4-Fluorobenzophenone | 4FBZP | >CO |
+1 | Para | 47 |
| 3-Chlorobenzophenone | 3ClBZP | >CO |
+1 | Meta | 83 |
| 2-Methylbenzophenone | 2MBZP | >CO |
0 | Ortho | −18 |
Experimental data
In order to explore the impact of the chosen additives on the crystallization of BZP, three different experimental programmes were pursued, all carried out at 15 °C. All experimental work was carried out on the stable form (orthorhombic, BPHENO12) of BZP. Additive concentrations are given as mole% of total dissolved solids. Firstly, the solubility of BZP in IPA was determined gravimetrically in the absence and presence of the chosen additives. This information is vital to enable definition of supersaturation (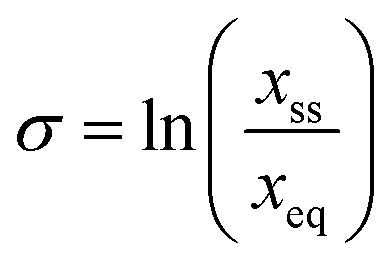 with x in mole fraction). Secondly, a morphology screen was performed to test the qualitative impact of the additives on the crystal shape of BZP. Crystals were first grown in pure solutions at six different supersaturations (0.03. 0.038, 0.05, 0.1, 0.135 and 0.20) to test the impact of supersaturation. Seed crystals were then prepared by slow cooling of solutions in the presence of 10% of each additive. Selected crystals from each experiment were analysed in a single-crystal X-ray diffractometer to determine the relative orientations of the dominant faces and the location of the x-axis.27 The extent of additive uptake was measured by HPLC on crystals grown in unseeded experiments at a supersaturation of 0.3 in the presence of 10% of each additive. These samples were also analysed by powder X-ray diffraction (PXRD) to check for changes in unit cell dimensions.
with x in mole fraction). Secondly, a morphology screen was performed to test the qualitative impact of the additives on the crystal shape of BZP. Crystals were first grown in pure solutions at six different supersaturations (0.03. 0.038, 0.05, 0.1, 0.135 and 0.20) to test the impact of supersaturation. Seed crystals were then prepared by slow cooling of solutions in the presence of 10% of each additive. Selected crystals from each experiment were analysed in a single-crystal X-ray diffractometer to determine the relative orientations of the dominant faces and the location of the x-axis.27 The extent of additive uptake was measured by HPLC on crystals grown in unseeded experiments at a supersaturation of 0.3 in the presence of 10% of each additive. These samples were also analysed by powder X-ray diffraction (PXRD) to check for changes in unit cell dimensions.
Finally, the crystal growth rates of the length and width dimensions (the [200] and [011] directions) of single seed crystals (100 × 100μm) were measured in a microscope growth cell as a function of both additive concentration and supersaturation.4
Further details of the solubility determinations, morphology screen, HPLC/PXRD and growth rate measurements are given in sections S5, S6, S7 and S8 respectively of the ESI.†
Solubility
The approximate (heat capacity, Cp unaccounted for) ideal solubility of BZP was calculated from DSC data11 (Tm = 48 °C, ΔHm = 16.9 kJ mol−1) as 0.46 mole fraction at 15 °C. This equates to 2.6 g/g 2-propanol, which is much larger than the measured value of 0.17 g/g. This is consistent with a reluctance of the phenyl rings to mix with this non-aromatic solvent. The effect of all but one of the additives at 0.1 mole fraction (relative to BZP) is to increase the solubility of BZP to between 0.19 and 0.24 g/g (see S5† for full data). These data are consistent with the introduction of additional phenyl rings into the solvent. 4NBZP did not dissolve in 2-propanol at the required level – hence it was excluded from all further investigations.Crystal morphologies
Supersaturation had no major impact on the crystal morphology of BZP. Fig. 5 shows the effect of the various additives. In all cases the crystallographic a axis (i.e. the x direction) is indicated and the crystals all lie on large {011} facets. | ||
| Fig. 5 Morphology screen, showing direction of a axis and additives coded as per Table 3. Longest dimension of all crystals 1–1.5 mm. | ||
Overall, it is evident that of all the additives only 4ABZP and 4MBZP have a significant observable effect, both giving rods bounded by {011} side faces and elongated in the x direction. This is the opposite effect to the {200} plates predicted above. All the other additives had negligible impact on the morphology. In order to shed further light on these results, additive uptake and growth rate data for 4ABZP and 4MBZP were examined and compared to DPM, one of the non-habit-modifying additives.
Additive incorporation
Crystals grown in the presence of DPM contained no additive, while those grown in the presence of 4MBZP and 4ABZP showed significant additive uptake: in the case of 4MBZP 2.1% (which reduced to 1.4% on washing) and for 4ABZP 9%, both before and after washing. The latter is similar to the level present in solution (10%), implying indiscriminate incorporation in the crystal structure. DSC data on these samples showed no significant change in melting point, PXRD showed small peak shifts to lower 2θ values and no new peaks. Taken together, these results are consistent with the formation of a partial solid solution of 4ABZP in BPHENO12.Face growth rates
The effect of 10% of 4ABZP, 4MBZP and DPM on the length ([200]) and width ([011]) growth rates of BZP are shown in Fig. 6 upper and lower respectively.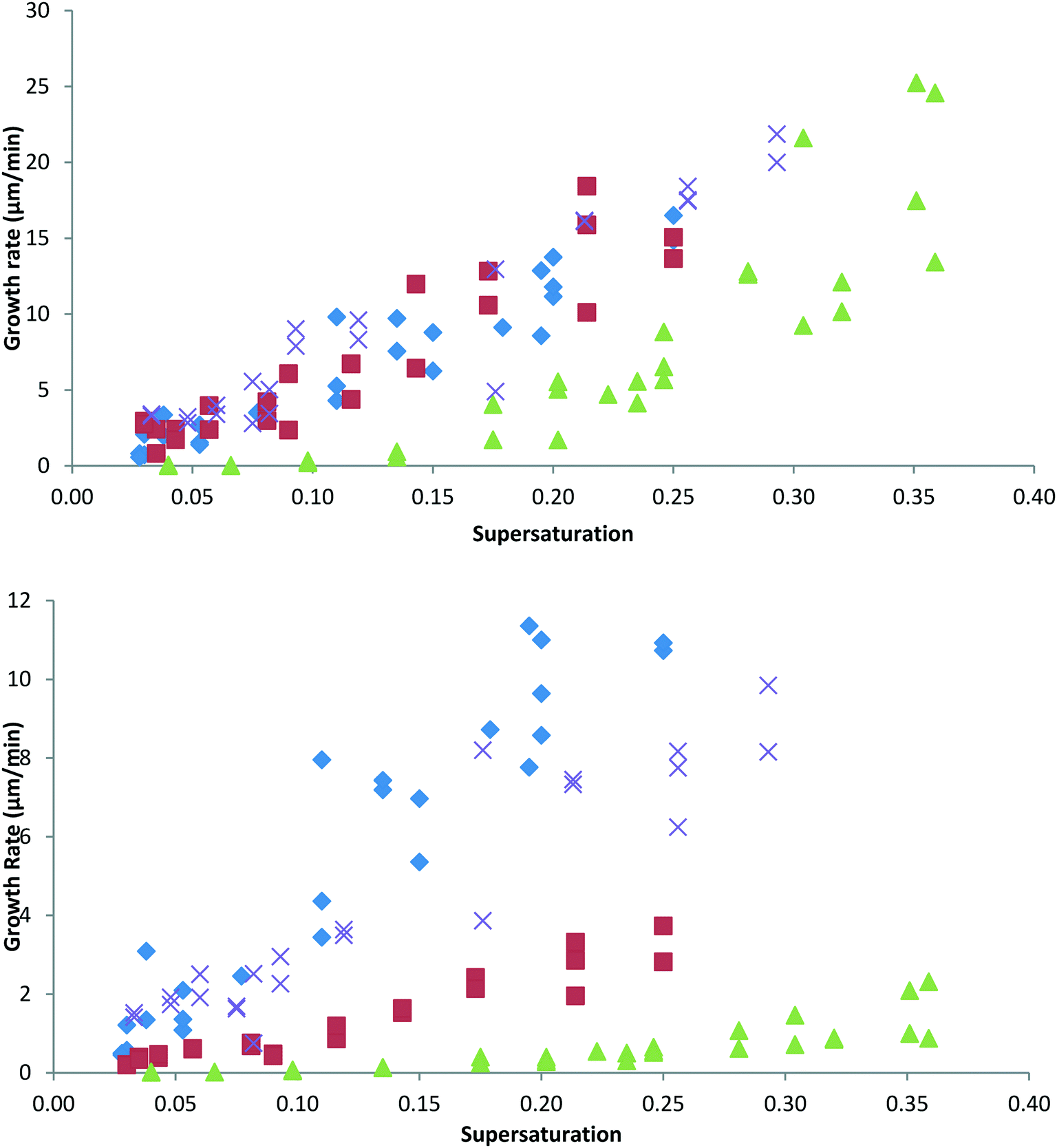 | ||
| Fig. 6 The linear growth rates of BZP crystals in the presence of additives: upper the effect of the selected additives on the length ([200], x-axis) growth rate of BZP and lower the effect of the selected additives on the width ([011]) growth rates of BZP. Blue diamond pure, red square 4ABZP, green triangle 4MBZP, cross DPM. | ||
It is clear that, as predicted, for a molecule in which the carbonyl group has been replaced and as expected on the basis of morphology and additive uptake, DPM has no significant effect on the growth kinetics of BZP. It is also worth noting that the morphological changes are not necessarily a good indication of the underlying kinetic effects. The morphologies suggest that both additives only affect the width, however this is not the whole story – while 4ABZP does indeed reduce only the width growth rate, 4MBZP reduces both length and width growth rates, giving rise to ‘dead zones’ in both directions at low supersaturations.28 These observations are thus qualitatively consistent with the morphological observations of Fig. 5, but not with the expectation that the additives would affect the [200] selectively. Specifically, and unexpectedly 4MBZP and 4ABZP affect width growth rates, more strongly than [200] growth rates.
Summarising these data and ranking the additives in order of decreasing efficacy we note the following. 4MBZP creates dead zones, has the larger effect on growth rates in both directions, yields rods and is concentrated at crystal surfaces. 4ABZP affects only width growth rates, gives rods and forms a solid solution. DPM has little effect on growth rates or morphology and no incorporation was detected. The other additives DPA, DPE, 4FBZP, 3ClBZP, 2MBZP had no effect on morphology and hence were not tested in the kinetic measurements.
Discussion
We now consider these results in the light of potential additive-BZP interactions. An important component of this analysis is to examine the interactions within the crystal structures of the additives themselves. It was of particular interest to see if the additives were capable of “mimicking” the two strongest interactions in the BZP polymorphs. Accordingly, the strongest interactions in each of these six crystal structures are summarized in Table 4. Comparison with BZP(1) and BZP(2) shows that none of these mimic the symmetry of either BZP(1) or BZP(2). Fig. 7 displays these interactions with additional crystal structure data given in S9.†| CSD reference | Strongest interaction | Torsion 1, ° | Torsion 2, ° | N FC |
|
Symmetry |
|---|---|---|---|---|---|---|
| BPHENO12 | BZP(1) | 31.2 | 31.0 | 37 | n/a | 2-Fold screw |
| BPHENO11 | BZP(2) | 30.7 | 44.3 | 50 | n/a | Inversion |
| FEVNAV01 | 4MBZP(1) | 32.2 | 39.5 | 30 | 30 | Translation, a = 5.70 |
| VOFVAN21 | 4ABZP(1) | 17.8 | 48.5 | 33 | 33 | Translation, b = 5.47 |
| BOJVIH | 4FBZP(1) | 30.7 | 28.0 | 43 | 43 | Glide |
| FEVNAV02 | 4MBZP(2) | 32.2 | 33.2 | 47 | 47 | 3-Fold screw |
| VOFVAN23 | 4ABZP(2) | 12.1 | 37.3 | 81 | 79 | Translation, b = 3.87 |
| BOJVON | 4NBZP(1) | 36.4 | 21.5 | 88 | 77 | Translation, a = 3.92 |

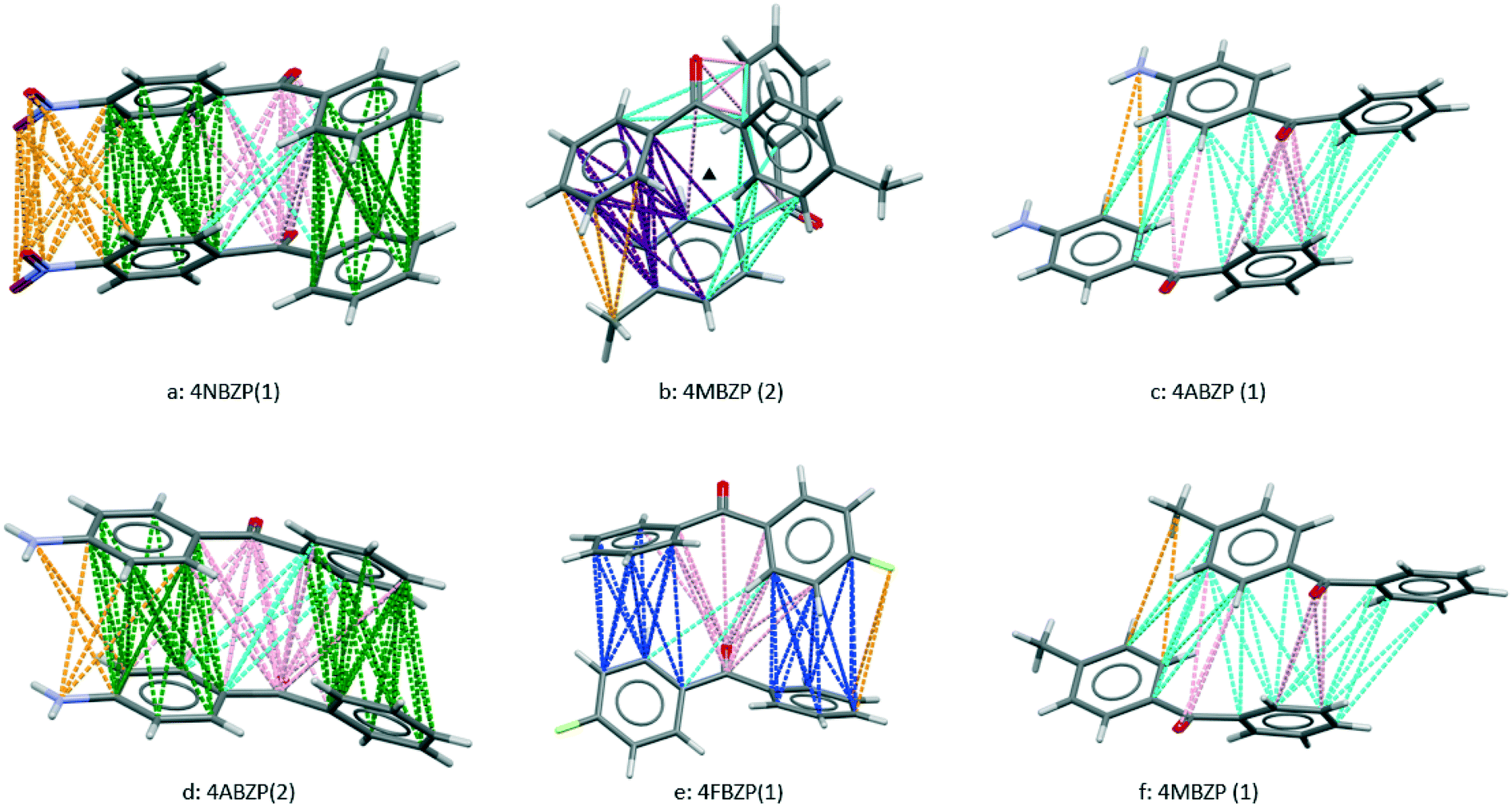 | ||
| Fig. 7 Strong interactions in crystal structures of additives. As in Fig. 2 and 3, colours denote the type of approach, with para-substituent approaches in orange. Further details are given in Tables 4 and 5 and the text. | ||
The range of torsion angles of para-substituted BZP molecules in Table 4 is consistent with the expectation above that other conformations may be accessed to optimize packing energies. The additives in which the carbonyl group is replaced show more extreme conformational change. In DPM the torsion angles are 72° and 64° and in DPA (Z′ = 8) torsion angles are in ranges 6 to −6°and 46 to −46°.22 In both cases, freedom from the constraint of sharing conjugation across a carbonyl group results in different molecular shapes, incompatible with BZP. This accounts for the inefficacy of these two additives as crystal growth modifiers. In the case of other ineffective additives, no crystal structures were found for DPE, 3ClBZP or 2MBZP.
The strongest interactions in each of the six available additive crystal structures are shown in Fig. 7. Table 4 gives their respective NFC values determined by inspection from Fig. 7 (see also S10†). Table 4 also contains summaries of the two strongest BZP⋯BZP interactions, taken from Table 1, for comparison. Like BZP, the two most active additives, 4MBZP and 4ABZP each have two polymorphs, and the strongest interactions in each are labelled, in the second column of Table 4, as (1) and (2) respectively.
4FBZP (Fig. 7e) contains disorder between para-fluorine and para-hydrogen atoms. This raises the possibility of a partial solid solution between BZP and 4FBZP without habit modification, a feature not explored further in this work. In 4NBZP (Fig. 7a), not investigated by virtue of its low solubility in IPA, two “green” face⋯face approaches embody translation symmetry giving a π–π stack (cf. in BZP(2), as shown in Fig. 3, there is a face⋯face approach which embodies inversion symmetry). This is facilitated by changes in the torsion angles. The approaches are supplemented by favourable connections with and between both the carbonyl groups and the para substituent. This gives the interaction with highest NFC. This is consistent with the low solubility of 4NBZP and suggests that it could be effective if tested at lower concentrations or using an alternative solvent.
Having established (see above) the correlation between NFC and interaction energies, the relative strengths of additive⋯BZP interaction were then evaluated using their NFC values in Table 4. For example, in the 4ABZP⋯4ABZP interaction in Fig. 7d the upper amino group takes part in two favorable connections. Removing this amino group leaves a potential BZP⋯4BAZP interaction with 2 fewer favourable connections, so  = 81–2 = 79, as shown in the sixth column in Table 4. Extending this to the other additives then gives a predicted order of binding efficiency to BZP as 4ABZP > 4NBZP ≫ 4MBZP > 4FBZP. Given that 4ABZP(2) and 4NBZP(1) are not relevant to the experimental study, (4NBZP was not evaluated experimentally and 4ABZP(2) is only accessible at elevated pressures29) this leaves a revised order of 4MBZP > 4FBZP> 4ABZP. From the orientation of BZP in BPHENO12 shown in Fig. 1, this is also the predicted order of binding in the x direction. Thus, 4MBZP is predicted to be the best inhibitor of growth in the x direction, consistent with the experimental data in Fig. 5 and 6.
= 81–2 = 79, as shown in the sixth column in Table 4. Extending this to the other additives then gives a predicted order of binding efficiency to BZP as 4ABZP > 4NBZP ≫ 4MBZP > 4FBZP. Given that 4ABZP(2) and 4NBZP(1) are not relevant to the experimental study, (4NBZP was not evaluated experimentally and 4ABZP(2) is only accessible at elevated pressures29) this leaves a revised order of 4MBZP > 4FBZP> 4ABZP. From the orientation of BZP in BPHENO12 shown in Fig. 1, this is also the predicted order of binding in the x direction. Thus, 4MBZP is predicted to be the best inhibitor of growth in the x direction, consistent with the experimental data in Fig. 5 and 6.
As far as width growth along BZP [011] is concerned the activity of 4ABZP and 4MBZP may be understood by reference to Fig. 8 which emphasizes the short, unfavorable, para H⋯H separation (2.896 Å) between central and right hand side molecules, suggesting that substitution here, on either phenyl ring, would be highly unfavorable. This is evident if we consider replacing the central BZP molecule in Fig. 8 with either additive such that the para-substituent is on the right. The methyl or amino group sits exactly where the crystal structure requires a favourable C⋯C connection of 3.632 Å, blocking the addition of the next molecule in this chain.
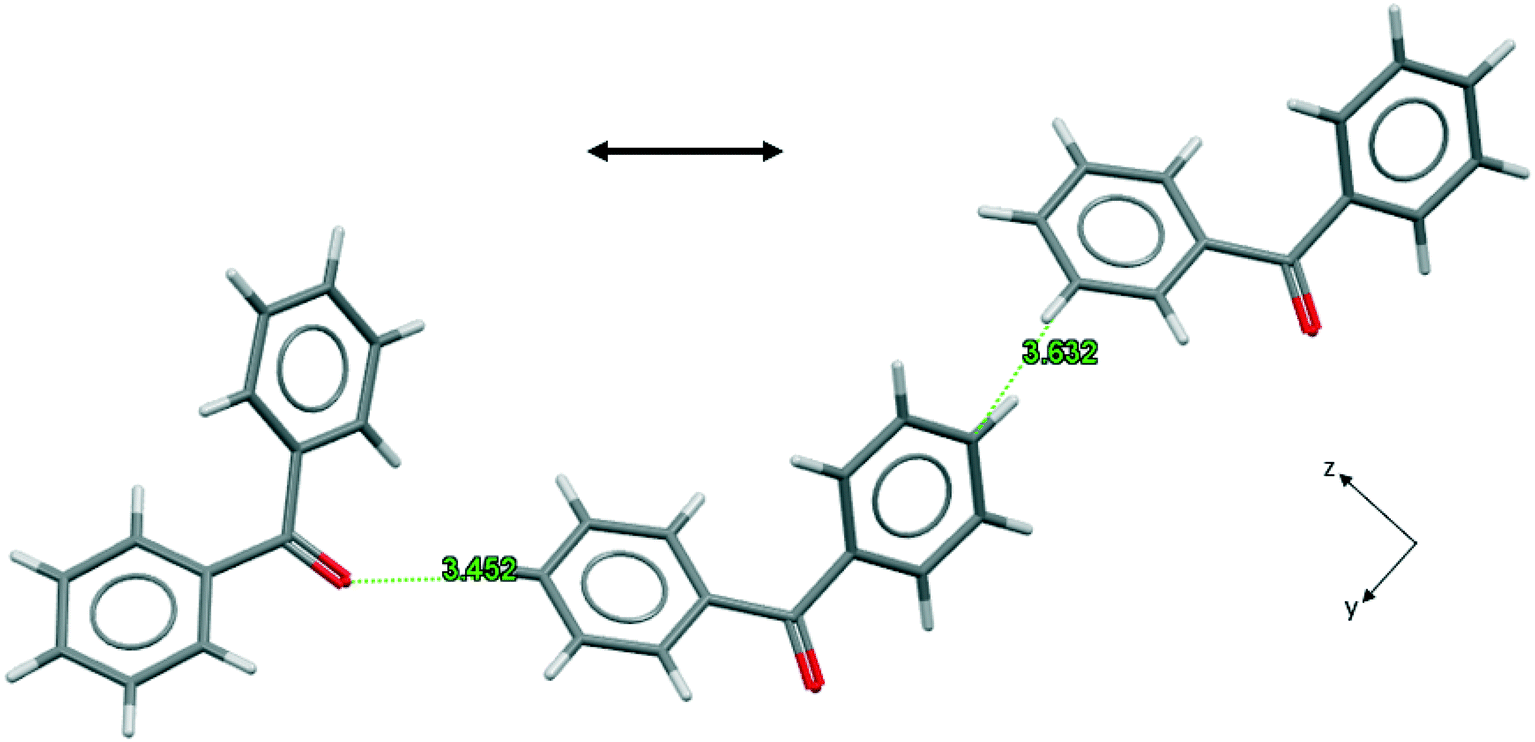 | ||
| Fig. 8 Favourable connections in BPHENO12 involving the para carbon atom, viewed parallel to the x axis. The double-headed arrow shows the directions of width growth. | ||
Now consider the effect of substituting the additive molecules such that the substituents are attached to the phenyl group on the left. In the case of 4ABZP, the amine group is now close to a carbonyl oxygen and favourably oriented for a C–N–H⋯O hydrogen bond. Such a hydrogen bond is present in the crystal structure of 4ABZP (VOFVAN21) with a C⋯O separation of 3.71 Å. The C⋯O separation above is 3.45 Å, so incorporation of the amine group requires some distortion of the crystal structure and/or the hydrogen bond. The existing interaction between these two molecules is relatively weak (NFC = 6, BZP(10); see Fig. S4.7 in the ESI†) compared to the potential benefit of introducing the only hydrogen bond into this crystal structure. This is consistent with the formation of a partial solid solution and further implies that 4ABZP will be incorporated primarily through {011} growth sectors. In the case of 4MBZP, when the methyl substituent is on the left, the incorporation of 4MBZP is much less favourable, as no hydrogen bonds are created to mitigate the distortion introduced by the methyl group.
In summary, 4MBZP may hinder growth at both ends of this chain. 4ABZP may hinder growth to the right and be incorporated to the left. These observations are consistent with the observations of reduced growth rate and high levels of incorporation of 4ABZP, and of surface rejection and greater reduction in growth rate with 4MBZP. This suggests that that a BZP derivative with conjugated para functionality that is larger than the methyl group and contains no hydrogen bond donors will be a more effective habit modifier with less incorporation. 4NBZP would fall into this category.
Conclusions
In this work we have shown how Groth's rich compilation of crystallographic data, pre-X-ray crystallography, remains relevant and useful today. Its availability as an online resource12 is a potential boon to experimentalists. We suspect that translation into English from the original German would enhance its potential usage even further. Groth recognised the value of identifying which crystal faces are related by symmetry. This is illustrated here by the manual colouring of facets in the BFDH morphology (Fig. 4). As the {111} faces in orthorhombic BZP show, faces that are predicted to have the same shape, size and surface area are often but not always related by symmetry. Over 100 years on from Groth's labelling scheme, an automated identification of symmetry-related faces would be a useful addition to the Mercury software. Leading on from this we note that assignment of crystallographic axes is a matter of fashion, and fashions may overlap. For the stable polymorph of BZP, the early assignment, initiated by Groth in 1919 persisted until at least 2015. This is a reminder to identify the unit cell when quoting Miller indices.A key element of this work has been to consider and compare the strength of intermolecular interactions between weakly polar molecules through the idea of “favourable connections”. This is a simple, geometric way of visualizing and comparing intermolecular interactions in a method that requires only one parameter (a limiting interatomic distance) and is easy to implement within the “Mercury” software. The method further identifies contributions from the close approaches of different functional groups. Three types of phenyl⋯phenyl approach were identified; “face⋯face”, “edge⋯face” and “skew”. These are differentiated by the relative orientations of the planes, which are parallel in face⋯face approaches, >45° in edge⋯face approaches and between 0° and 45° in skew approaches. In edge⋯face approaches only two or three of the carbon atoms in one phenyl ring (the “edge”) connect favourably to 5 or 6 of the carbon atoms in the other phenyl ring (the “face”), as in benzene. A molecule like BZP, containing two non-coplanar phenyl rings, has many options for phenyl⋯phenyl approaches with the strongest intermolecular interactions including two or more.
Visualizing multiple connections and approaches in this way reveals (see Fig. 5) how elements of crystallographic symmetry are formed by phenyl⋯phenyl approaches and embodied in intermolecular interactions that are the building blocks of molecular crystals. In synthetic chemistry and crystal engineering,30 the term “synthon” is used to describe building blocks for complex molecules and crystals. Here, by analogy, we propose the additional term “symthon” for phenyl⋯phenyl approaches that form crystallographic symmetry. Thus, inspection of Fig. 7 and Tables 4 and 5 suggests that different types of phenyl⋯phenyl approach form different symmetries and symthons. Face⋯face approaches form inversion or short translational (“π–π stacking”) symmetries. These approaches are strengthened by co-planar substitutions on the phenyl rings. In contrast, 2-fold screw axes are formed by edge⋯face approaches. Graphite and benzene are archetypes of these two classes. Further work is necessary to explore the generality of these observations beyond the 8 structures contained in Table 5.
| CSD reference | Strongest interaction | Fig. | Strongest phenyl⋯phenyl approaches (NFC) | Symthon symmetry |
|---|---|---|---|---|
| YOPTUU02 | “graphitic” | S11.1† | Face⋯face (29) | Inversion |
| BOJVON | 4NBZP(1) | 7a | Face⋯face (26) + face⋯face(25) | Translation |
| VOFVAN23 | 4ABZP(2) | 7d | Face⋯face (25) + face⋯face(25) | Translation |
| BPHENO11 | BZP(2) | 3 | Face⋯face (22) | Inversion |
| FEVNAV02 | 4MBZP(2) | 7b | Skew (21) | 3-Fold screw |
| BPHENO12 | BZP(1) | 2 | Skew (18) + edge⋯face (10) | 2-Fold screw |
| BPHENO11 | BZP(3) | S4.1† | Skew (17) | 2-Fold screw |
| BOJVIH | 4FBZP(1) | 7e | Edge⋯face (14) + edge⋯face (13) | Glide |
| BPHENO12 | BZP(4) | S4.2† | Edge⋯face (12) | 2-Fold screw |
| BENZEN | Benzene “T” | S11.1† | Edge⋯face (11) | 2-Fold screw |
In terms of its contribution to the design and selection of crystallization modifiers, this work has set out a strategic methodology involving a whole molecule paradigm applicable to weakly polar molecules and demonstrated its utility in the case of BZP. This differs from the conventional design of tailor-made additives for polar molecules in which one targets a chain of strong H-bonds and selects a molecule that is essentially the host with a donor or acceptor missing or derivatised. Here the chains in question exist by virtue of numerous intermolecular (dispersive) connections and a different strategy is required, in which more emphasis has to be placed on the ability of the additive to compete with the solute for surface sites. Thus, when we choose our additive it is not enough simply to add a steric or polar substituent; we also have to be concerned that it can have sufficient contacts with the host that it binds to the surface. In our case, this meant selecting on the basis that the additive can replicate the contacts that BZP likes. For example, an additive with only a single phenyl ring would not be expected to work because it has no chance of competing with other BZP molecules. Actually, habit modification of BZP grown from pure toluene as a solvent has been suggested previously10 and an investigation of BZP morphology as a function of toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IPA solvent ratio, while outside the scope of this work, would complement both studies.
:IPA solvent ratio, while outside the scope of this work, would complement both studies.
The “favourable connections” method enables us to understand and visualize key strong interactions arising from multiple atomic non-bonded contacts in both BZP and additive crystal structures. These insights help to account for the efficacy of 4MBZP and 4ABZP as habit and growth kinetic modifiers, and the ineffectiveness of other additives.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
AH thanks Syngenta for financial support and for helpful discussions with Dr. N. George.References
- Z. Berkovitch-Yellin, J. Van Mil, L. Addadi, M. Idelson, M. Lahav and L. Leiserowitz, J. Am. Chem. Soc., 1985, 107, 3111–3122 CrossRef CAS.
- I. Weissbuch, M. Lahav and L. Leiserowitz, Cryst. Growth Des., 2003, 3, 125–150 CrossRef CAS.
- R. J. Davey, N. Blagden, S. Righini, H. Alison and E. S. Ferrari, J. Phys. Chem. B, 2002, 106, 1954–1959 CrossRef CAS.
- R. Dowling, R. J. Davey, R. A. Curtis, G. Han, S. K. Poornachary, P. S. Chow and R. B. H. Tan, Chem. Commun., 2010, 46, 5924–5926 RSC.
- C. Scott and S. Black, Org. Process Res. Dev., 2005, 9, 890–893 CrossRef CAS.
- ‘From Molecules to Crystallisers: An Introduction to Crystallisation’ Oxford Chemistry Primers No 86, Oxford University Press, ed. R. J. Davey and J. Garside, 2000, Cf., ch. 9 Search PubMed.
- B. Simon, A. Grassi and R. Boistelle, J. Cryst. Growth, 1974, 26, 90–96 CrossRef CAS.
- D. H. M. Beiny, J. W. Mullin and K. Lewtas, J. Cryst. Growth, 1990, 102, 801–806 CrossRef CAS.
- J. F. B. Black, A. J. Cruz-Cabeza, R. J. Davey, R. D. Willacy and A. Yeoh, Cryst. Growth Des., 2018, 18, 7518–7525 CrossRef CAS.
- K. J. Roberts, J. N. Sherwood, C. S. Yoon and R. J. Docherty, Chem. Mater., 1994, 6, 1099–1102 CrossRef CAS . Note: BZP crystals with a different morphology from that shown in Fig. 4 were obtained from concentrated solutions of BZP in toluene at elevated temperatures. These crystallisation conditions are different to those adopted here for studying additives (although similar to those for making the metastable monoclinic form); so are not discussed further here.
- J.-C. Bradley, A. Williams and A. Lang, Jean-Claude Bradley Open Melting Point Dataset, Figshare Dataset, 2014, DOI:10.6084/m9.figshare.1031637.
- P. Groth, Chemische Kristallographie: Teil V, W. Engelman, Leipzig, 1919, pp. 88–102, available on-line at https://archive.org/details/chemischekrystal05grotuoft/page/n4 Search PubMed.
- J. B. Morris and R. F. Strickland-Constable, Trans. Faraday Soc., 1954, 50, 1378–1393 RSC.
- G. M. Lobanova, Kristallografiya, 1968, 13, 984 CAS.
- E. B. Fleischer, N. Sung and S. Hawkinson, J. Phys. Chem., 1968, 72, 4311–4312 CrossRef CAS.
- H. Yadav, N. Sinha, N. Tyagi and B. Kumar, Cryst. Growth Des., 2015, 15, 4908–4917 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- J. Moncol and P. Coppens, CSD Communication, 2004 Search PubMed.
- A. M. Reilly, D. A. Wann, M. J. Gutmann, M. Jura, C. A. Morrison and D. W. H. Rankin, J. Appl. Crystallogr., 2013, 46, 656–662 CrossRef CAS.
- A. Matsumoto, S. Tsuchiya, Y. Hagiwara, K. Ishikawa, H. Koshima, T. Asahi and K. Soai, Chem. Lett., 2016, 45, 526–528 CrossRef CAS.
- H. Kutzke, H. Klapper, R. B. Hammond and K. J. Roberts, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 486–496 CrossRef.
- J. Bernstein, A. Ellern and J.-O. Henck, CSD Communication, 2002 Search PubMed.
- S. N. Black, C. L. Woon and R. J. Davey, Cryst. Growth Des., 2018, 18, 6918–6928 CrossRef CAS.
- A. Gavezzotti, Acc. Chem. Res., 1994, 27, 309–314 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J, P. T. A. Cottrell, P. Galek, E. McCabe, M. Pidcock, G. P. Platings, J. S. Shields, M. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 396–406 CrossRef CAS.
- A. Hutchison, The effect of additives on the growth of Benzophenone, PhD thesis, University of Manchester, UK, 2014 Search PubMed.
- Y. Liu, J. F. B. Black, K. F. Boon, A. J. Cruz-Cabeza, R. J. Davey, R. J. Dowling, N. George, A. Hutchinson and R. Montis, Cryst. Growth Des., 2019, 19, 4579–4587 CrossRef CAS.
- E. Marelli, N. Casati, F. Gozzo, P. Macchi, P. Simoncic and A. Sironie, CrystEngComm, 2011, 13, 6845–6849 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce01547d |
| This journal is © The Royal Society of Chemistry 2021 |