
Photo-modulated supramolecular self-assembly of ortho-nitrobenzyl ester-based alkynylplatinum(II) 2,6-bis(N-alkylbenzimidazol-2′-yl)pyridine complexes†
Andy Shun-Hoi
Cheung
,
Michael Ho-Yeung
Chan
 ,
Charlotte
Po
,
Eugene Yau-Hin
Hong
and
Vivian Wing-Wah
Yam
*
,
Charlotte
Po
,
Eugene Yau-Hin
Hong
and
Vivian Wing-Wah
Yam
*
Institute of Molecular Functional Materials and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk
First published on 29th November 2021
Abstract
The enhanced supramolecular self-assembly behaviors of photo-caged platinum(II) complexes have been triggered by applying light as the external stimulus. Distinct morphological transformation of the nanoaggregates has been observed in the photo-caged complexes before and after UV irradiation.
Owing to the intriguing spectroscopic properties and high propensity to form non-covalent metal–metal interactions,1–25 platinum(II) polypyridine complexes have attracted a lot of attention.1–25 Successful construction of highly-ordered supramolecular assemblies has been a result of a delicate balance and interplay of a variety of non-covalent interactions, such as π–π stacking, hydrophobic–hydrophobic interactions, hydrogen bonding etc.26,27 In contrast, the use of metal–metal interactions in enhancing molecular association and supramolecular assembly has been relatively much less explored and it is only until recent years that there is a growing interest in this area of study.10–25 The modulation of Pt⋯Pt interactions by external stimuli, such as a change of solvation,11,19,21 temperature,12,20 pH13,22 and addition of polyelectrolytes,14 has been reported and platinum(II) systems are shown to exhibit drastic spectroscopic changes and morphological responses upon a change in the micro-environment.
The utilization of light as an external stimulus to trigger the self-assembly and to perturb the molecular associations of platinum(II) complexes, modulated by Pt⋯Pt interactions, has been relatively less investigated.24,25,28–31ortho-Nitrobenzyl (o-NB) derivatives have received enormous interests owing to their distinct absorption profiles in the UV region and the high yield of deprotection in various solvents.30–33 Photochemical reaction of o-NB derivatives has been found to alter the hydrophobic–hydrophilic balance and the solubility of the compounds in the system, rendering them as photodegradable networks,34 hydrogels,35 photo-responsive delivery and release systems,36 and photoresists.37 However, most of the studies on the utilization of o-NB derivatives as the photo-removable protecting group have been confined to the organic systems, for photoresists in materials fabrication and phototriggers and photocontrolled drug release and activated therapy in the biological systems,30–37 with less emphasis on the modulation of supramolecular order and assemblies. It is envisaged that the incorporation of the o-NB photoremovable protecting group (PPG) to the platinum(II) system could lead to the construction of new photo-activatable organometallic assemblies. Herein, we report the design and synthesis of photoactivatable ortho-nitrobenzyl ester-based alkynylplatinum(II) 2,6-bis(N-decylbenzimidazol-2′-yl)pyridine (bzimpy) complexes and their uncaged reference control complexes (Scheme 1) as well as their photophysical and self-assembly properties.
 | ||
Scheme 1 Molecular structures of alkynylplatinum(II) bzimpy complexes, [Pt{bzimpy(C10H21)2}{C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C6H4–(COOCH2–C6H4–NO2-2)-4}]PF6 (1a), [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOH)-4}]PF6 (1b), [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOCH2–C6H4–NO2-2)-2}]PF6 (2a) and [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOH)-2}]PF6 (2b). C–C6H4–(COOCH2–C6H4–NO2-2)-4}]PF6 (1a), [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOH)-4}]PF6 (1b), [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOCH2–C6H4–NO2-2)-2}]PF6 (2a) and [Pt{bzimpy(C10H21)2}{CC–C6H4–(COOH)-2}]PF6 (2b). | ||
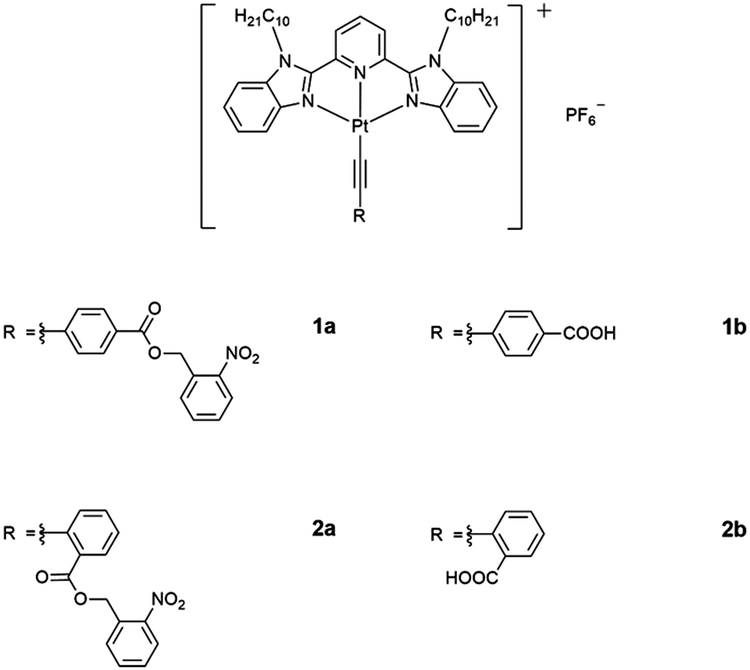
The electronic absorption spectra of 1a–2b in DMSO display intense high-energy absorption bands at ca. 290–370 nm with molar extinction coefficients in the order of 104 dm3 mol−1 cm−1 and less intense low-energy absorption bands at ca. 410–470 nm with molar extinction coefficients in the order of 103–104 dm3 mol−1 cm−1. The high-energy absorption bands are assigned as a mixture of intraligand (IL) [π→π*] transitions of the bzimpy ligands and the alkynyl ligands, while the low-energy absorption bands are assigned as an admixture of the metal-to-ligand charge transfer (MLCT) [dπ(Pt)→π*(bzimpy)] and ligand-to-ligand charge transfer (LLCT) [π(alkynyl) →π*(bzimpy)] transitions (Table S1 and Fig. S1, ESI†). Upon increasing the water content in DMSO solutions of 1a and 2a, a drop in the absorbance at 290–330 nm, 340–380 nm and 420–440 nm, together with a growth of a low-energy tail at ca. 480 nm is observed. The drop in the high-energy absorption bands has revealed the hypochromic phenomenon in the ligand-centered and MLCT absorptions, suggestive of formation of aggregate species with close proximity of π-conjugated aromatic systems (Fig. 1a and Fig. S2, ESI†).38 The growth of the low-energy absorption tail at λ ≈ 480 nm is tentatively assigned as the metal–metal-to-ligand charge transfer (MMLCT) transition. This has further been confirmed by red shift of the low-energy emission band of the corresponding solvent-dependent emission studies (vide infra). To gain more insights into the effect of change of solvation on the assembly process, corresponding emission studies have been performed for 1a and 2a in the DMSO–water mixed-solvent systems (Fig. 1b and Fig. S3–S5, ESI†). Upon addition of water to a DMSO solution of 1a and 2a, a low-energy emission band emerges at 650–680 nm with enhancement in luminescence. In addition to the decrease in the emission band at 550–570 nm, typical of the monomeric emission of the alkynylplatinum(II) bzimpy complexes, the growth of the low-energy emission band is found to be red-shifted upon increasing the water content, suggesting an increase in the extent of the Pt⋯Pt and/or π–π stacking interactions. It is believed that the Pt⋯Pt interaction is more significant in 1a than that in 2a as revealed by the red shift in the emission band maximum of 1a (ca. 680 nm for 1a and ca. 650 nm for 2a). The stronger directional Pt⋯Pt interactions could result in the formation of highly-ordered one-dimensional nanoaggregates in 1a while the relatively weaker Pt⋯Pt interactions in 2a could only lead to the formation of the spherical-like nanoaggregates as revealed by the TEM experiments (vide infra). Upon increasing the temperature from 298 to 348 K, the emission bands of 1a in DMSO–water mixtures (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80, v/v) are found to be temperature-responsive and the emission shows a drop in intensity (Fig. S6, ESI†). In addition to the increase in the non-radiative decay at elevated temperatures, which could lead to the decrease in the emission intensity, the emission bands are also found to be blue-shifted (Fig. S7, ESI†). This observation may be attributed to the disruption of the Pt⋯Pt interactions at high temperatures, leading to the disassembly of the platinum(II) complexes. Therefore, the emergence of the low-energy emission bands has been ascribed to supramolecular assembly via intermolecular Pt⋯Pt and π–π stacking interactions that leads to emission of 3MMLCT excited state origin upon reducing solvation.
:80, v/v) are found to be temperature-responsive and the emission shows a drop in intensity (Fig. S6, ESI†). In addition to the increase in the non-radiative decay at elevated temperatures, which could lead to the decrease in the emission intensity, the emission bands are also found to be blue-shifted (Fig. S7, ESI†). This observation may be attributed to the disruption of the Pt⋯Pt interactions at high temperatures, leading to the disassembly of the platinum(II) complexes. Therefore, the emergence of the low-energy emission bands has been ascribed to supramolecular assembly via intermolecular Pt⋯Pt and π–π stacking interactions that leads to emission of 3MMLCT excited state origin upon reducing solvation.
 | ||
| Fig. 1 (a) UV-Vis absorption and (b) emission traces of 1a in DMSO at 298 K upon increasing the water content from 0 to 90%. | ||
Nanorods with diameters and lengths of ca. 80 nm and 400–800 nm are observed for 1a while irregular spherical aggregates with diameters of ca. 200 nm are observed for 2a (Fig. 2). The steric demand of the o-NB moiety on the alkynyl ligand is believed to alter the overall molecular packing and results in the formation of aggregates with different morphologies. In addition, the crystalline nature of the nanorods prepared from 1a has been determined by SAED experiment (Fig. 2c and d). A pair of arcs with a d-spacing of 3.41 Å along the long axis has been revealed, attributing to the possible involvement of the intermolecular Pt⋯Pt and π–π stacking interactions in the aggregates. These observations are in line with the solvent-dependent emission studies in which 1a is found to have a stronger Pt⋯Pt interactions, which may assist the formation of the nanorods. The hydrodynamic diameters of 1a and 2a (in 50% DMSO–water mixtures) are determined to be ∼136 and ∼146 nm respectively (Table S2, ESI†).
 | ||
| Fig. 2 TEM images of (a) 1a and (b) 2a prepared from a 50% DMSO–water mixture. (c) Magnified TEM image of 1a and (d) the corresponding SAED pattern. | ||
Upon photo-irradiation of the photocaged complexes at ca. 300 nm in DMSO solutions, similar to the observations in UV-vis absorption studies in the DMSO–water mixed-solvent, a drop in the absorbance at 300–330 nm and 350–380 nm, together with a slight growth of the low-energy tail at ca. 470 nm is observed (Fig. 3 and Fig. S8, ESI†). The drop in the high-energy absorption bands has suggested the consumption of the photocaged complex and revealed the hypochromic phenomenon in the ligand-centered absorptions, ascribed to the formation of aggregate species and the close proximity of the aromatic chromophores. With reference to the literature,32,33,36 the o-NB moiety could be cleaved by UV irradiation to give the corresponding benzoic acid moiety. The change in the hydrophobic–hydrophilic balance of the complex and the enhancement in planarization are believed to enhance the molecular association, leading to a slight growth of the low-energy absorption tail at ca. 470 nm in 1a. The uncaged reference controls (1b and 2b) have been synthesized for comparisons while detailed investigations are conducted for 1b. Upon increasing the temperature from 298 to 348 K, the aromatic signals of 1b in DMSO-D6 are found to show a slight downfield shift, suggesting the presence of substantial π–π stacking interactions (Fig. S9, ESI†).19 Upon cooling the DMSO solution of 1b from 348 to 298 K, a decrease in the MLCT absorption band and an increase in the low-energy absorption tail at ca. 470 nm are observed (Fig. S10, ESI†). The low-energy absorption tail is tentatively assigned as the MMLCT transition, facilitated by the intermolecular Pt⋯Pt and π–π stacking interactions in the supramolecular assembly state. Long rod-like aggregates with diameters and lengths of ca. 100 nm and up to 1–2 mm are observed for 1b (Fig. S11, ESI†). The corresponding SAED experiment indicates the crystalline nature of the rod-like nanostructures and reveals a pair of arcs with a d-spacing of about 3.50 Å parallel to the long axis of the nanorod, attributed to the presence of Pt⋯Pt and/or π–π stacking interactions. The powder X-ray diffraction (XRD) pattern reveals d-spacings of 21.63, 10.93, 7.07 and 5.46 Å in a ratio of ca. 4:3:2:1, characteristic of the formation of lamellar packing (Fig. S12, ESI†). It is also found to exhibit a diffraction peak at 2θ = 25.56° (d-spacing = 3.51 Å), indicative of the presence of Pt⋯Pt and/or π–π stacking interactions. Together with the temperature-dependent UV-vis absorption study of the uncaged reference control complex, 1b in DMSO solution, the low-energy absorption tail at 470 nm that emerges upon photo-irradiation in 1a is assigned as the MMLCT transition.
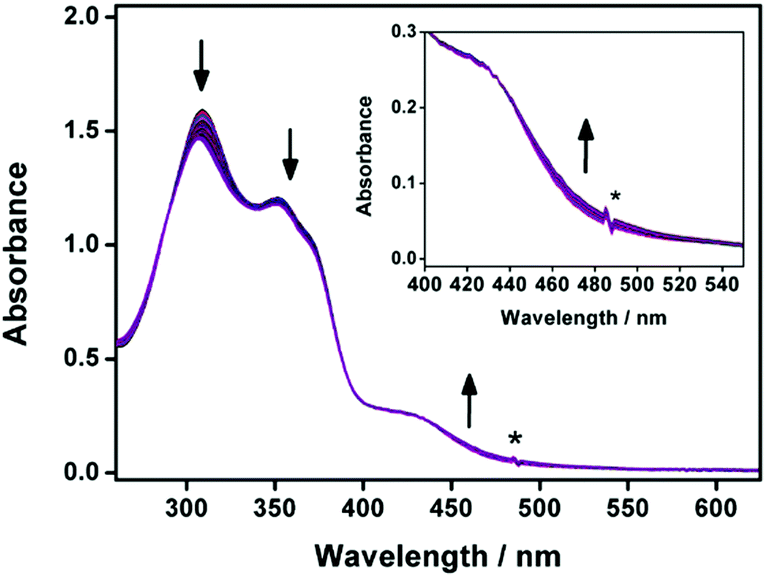 | ||
| Fig. 3 UV-Vis absorption spectral traces of 1a in DMSO solution at 298 K upon prolonged UV irradiation at 310 nm for 20 hours. Inset: Magnified UV-vis absorption spectral traces in the low-energy absorption region. The asterisk denotes an instrumental artifact. The direction of arrows indicates the direction of spectral changes upon UV irradiation. | ||
After UV irradiation, apart from the [M − PF6]+ molecular ion peak of 1a and 2a, a new set of signals has been observed at m/z 931, which corresponds to the photo-uncaged carboxylic acid product by the loss of a [–CH2–C6H4–NO2] fragment, in the high-resolution ESI† experiments (Fig. S13 and S14, ESI†). These suggest that upon UV irradiation, photoactivation of the photocaged platinum(II) complex occurs to afford the photo-uncaged platinum(II) complex. Interestingly, before UV irradiation, no well-defined nanostructures are observed in the TEM image, in line with the absence of scattering signals in the DLS experiment of 1a in DMSO. After UV irradiation, rod-like nanostructures are observed with diameters and lengths of ca. 100 nm and up to 1 mm respectively, which are comparable to the nanorods observed for the uncaged reference control complex, 1b (Fig. 4). These observations have illustrated that the removal of the ortho-nitrobenzyl ester group by photo-irradiation has led to an enhancement of the amphiphilic character and promotion of a better molecular alignment of the amphiphilic alkynylplatinum(II) bzimpy complexes to afford well-defined self-assembled nanostructures, resembling those observed in the uncaged reference control complex 1b.
 | ||
| Fig. 4 TEM images of 1a prepared from a DMSO solution (a) before and (b) after UV irradiation. | ||
Morphological transformation from ill-defined objects to sheet-like nano-layers is also observed in 2a with the formation of the sheet-like aggregates that closely resemble that prepared by the uncaged reference control (Fig. S15 and S16, ESI†). The observed difference in the morphologies of the aggregates for 1b (rod-like aggregates) and 2b (sheet-like aggregates) could probably be attributed to the different molecular packing, as one would expect the position of hydrophilic carboxylic acid moiety would lead to steric hindrance of different extents. Similar observations regarding the effect of positional difference on the formation of aggregates with different morphologies were observed in the related triazine-containing alkynylplatinum(II) terpyridine systems.39 For the photocaged complexes 1a and 2a, the floppy nature of the ester linkage as well as the steric hindrance of the o-NB moiety would probably result in the monomeric form of molecules in pure DMSO solution.
1a has been further analyzed by 1H NMR spectroscopy before and after UV irradiation in DMSO-D6 (Fig. S17–S20, ESI†). The 1H NMR spectrum of 1a after prolonged UV irradiation for about 20 hours only gives one dominant species. From the 1H NMR change, approximately 16.1% of 1a has undergone photocleavage reaction, resulting in the decrease in the integral of the [–CH2–C6H4–NO2] moiety. In addition, there is an upfield shift of the pyridine and benzimidazole protons after UV irradiation (Fig. S19, ESI†). Upon increasing the temperature from 298 to 348 K, the aromatic proton signals show a significant downfield shift (Fig. S20, ESI†). These observations suggest the presence of substantial π–π stacking interactions between the bzimpy ligand, probably serving as one of the driving forces to enhance the molecular associations upon photolysis. Based on the above-mentioned experiments, the o-NB-based alkynylplatinum(II) bzimpy complexes would undergo photolysis upon UV irradiation. The original well-solvated complexes have enhanced the overall amphiphilicity, leading to the formation of ordered nanoaggregates.
In conclusion, based on the above-mentioned experiments, the o-NB-based alkynylplatinum(II) bzimpy complexes would undergo photolysis upon UV irradiation. The current study has illustrated that the removal of the ortho-nitrobenzyl ester group by photo-irradiation has led to an enhancement of the amphiphilic character and promotion of a better molecular alignment of the amphiphilic alkynylplatinum(II) bzimpy complexes to afford well-defined self-assembled nanostructures, which may provide insights into the rational molecular design of stimuli-responsive materials with functional properties.
V. W.-W. Y. acknowledges UGC funding administered by The University of Hong Kong (HKU) for supporting the electrospray ionization quadrupole time-of-flight mass spectrometry facilities under the support for Interdisciplinary Research in Chemical Science, the University Development Fund and the Dr Hui Wai Haan Fund of The University of Hong Kong for funding the Bruker D8 ADVANCE Powder X-ray Diffractometer. This work was supported by a General Research Fund (GRF) grant from the Research Grants Council of the Hong Kong Special Administrative Region, People's Republic of China (HKU17309220) and the CAS-Croucher Funding Scheme for Joint Laboratory on Molecular Functional Materials for Electronics, Switching and Sensing. A. S.-H. C. acknowledges the receipt of a postgraduate studentship and a university postgraduate fellowship from the University of Hong Kong. Dr Kam-Hung Low is gratefully acknowledged for his assistance in collecting PXRD data. We also thank Mr Frankie Yu-Fee Chan at the Electron Microscope Unit of The University of Hong Kong for his helpful technical assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839 CrossRef CAS PubMed.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28(8), 1529 CrossRef CAS.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446 CrossRef CAS.
- V. M. Miskowski, V. H. Houlding, C. M. Che and Y. Wang, Inorg. Chem., 1993, 32, 2518 CrossRef CAS.
- J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34(18), 4591 CrossRef CAS.
- W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS.
- C. A. Daws, C. L. Exstrom, J. R. Sowa and K. R. Mann, Chem. Mater., 1997, 9(1), 363 CrossRef CAS.
- C. E. Buss and K. R. Mann, J. Am. Chem. Soc., 2002, 124(6), 1031 CrossRef CAS PubMed.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS PubMed.
- K. Wang, M.-a. Haga, H. Monjushiro, M. Akiba and Y. Sasaki, Inorg. Chem., 2000, 39(18), 4022 CrossRef CAS PubMed.
- V. W. W. Yam, K. M. C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124(23), 6506 CrossRef CAS PubMed.
- V. W. W. Yam, K. H. Y. Chan, K. M. C. Wong and B. W. K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169 CrossRef CAS PubMed.
- K. M. C. Wong, W. S. Tang, X. X. Lu, N. Zhu and V. W. W. Yam, Inorg. Chem., 2005, 44(5), 1492 CrossRef CAS PubMed.
- C. Yu, K. M. C. Wong, K. H. Y. Chan and V. W. W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791 CrossRef CAS PubMed.
- A. Y. Y. Tam, W. H. Lam, K. M. C. Wong, N. Zhu and V. W. W. Yam, Chem. – Eur. J., 2008, 14(15), 4562 CrossRef CAS PubMed.
- A. Y. Y. Tam, K. M. C. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed.
- M. C. L. Yeung, K. M. C. Wong, T. K. T. Tsang and V. W. W. Yam, Chem. Commun., 2010, 46, 7709 RSC.
- M. C. L. Yeung and V. W. W. Yam, Chem. Sci., 2013, 4(7), 2928 RSC.
- C. Po, A. Y. Y. Tam, K. M. C. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2011, 133(31), 12136 CrossRef CAS PubMed.
- C. Po, A. Y. Y. Tam and V. W. W. Yam, Chem. Sci., 2014, 5(7), 2688 RSC.
- A. F. F. Cheung, E. Y. H. Hong and V. W. W. Yam, Chem. – Eur. J., 2018, 24(6), 1383 CrossRef CAS PubMed.
- M. H. Y. Chan, S. Y. L. Leung and V. W. W. Yam, J. Am. Chem. Soc., 2019, 141(31), 12312 CrossRef CAS PubMed.
- A. S. Y. Law, L. C. C. Lee, M. C. L. Yeung, K. K. W. Lo and V. W. W. Yam, J. Am. Chem. Soc., 2019, 141(46), 18570 CrossRef CAS PubMed.
- S. Fang, S. Y. L. Leung, Y. Li and V. W. W. Yam, Chem. – Eur. J., 2018, 24(58), 15596 CrossRef CAS PubMed.
- S. Fang, M. H. Y. Chan and V. W. W. Yam, Mater. Chem. Front., 2021, 5(5), 2409 RSC.
- J. M. Lehn, Science, 2002, 295(5564), 2400 CrossRef CAS PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105(4), 1491 CrossRef CAS PubMed.
- J. A. Barltrop, P. J. Plant and P. Schofield, Chem. Commun., 1966, 822 RSC.
- Z. Gao, Y. Han and F. Wang, Nat. Commun., 2018, 9(1), 3977 CrossRef PubMed.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1(7), 441 CrossRef PubMed.
- Y. V. Il'ichev, M. A. Schwörer and J. Wirz, J. Am. Chem. Soc., 2004, 126(14), 4581 CrossRef PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45(4), 1723 CrossRef CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113(1), 119 CrossRef PubMed.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromolecules, 2007, 40(10), 3589 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324(5923), 59 CrossRef CAS PubMed.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39(13), 4633 CrossRef CAS.
- P. G. Taylor, J. K. Lee, A. A. Zakhidov, M. Chatzichristidi, H. H. Fong, J. A. DeFranco, G. G. Malliaras and C. K. Ober, Adv. Mater., 2009, 21(22), 2314 CrossRef CAS.
- L. Brunsveld, E. W. Meijer, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2001, 123(33), 7978 CrossRef CAS PubMed.
- H. L.-K. Fu, S. Y.-L. Leung and V. W.-W. Yam, Chem. Commun., 2017, 53(82), 11349 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05754e |
| This journal is © The Royal Society of Chemistry 2021 |