
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New approaches to ondansetron and alosetron inspire a versatile, flow photochemical method for indole synthesis†‡
Wei
Sun
*a,
William A. T.
Raimbach
a,
Luke D.
Elliott
b,
Kevin I.
Booker-Milburn
 b and
David C.
Harrowven
*a
b and
David C.
Harrowven
*a
aChemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: dch2@soton.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 26th November 2021
Abstract
An oxidative photocyclisation of N-arylenaminones to indoles is described, that mirrors the Fischer indole synthesis but uses anilines in place of arylhydrazines. Its value is exemplified with new approaches to the WHO-listed APIs ondansetron and alosetron.
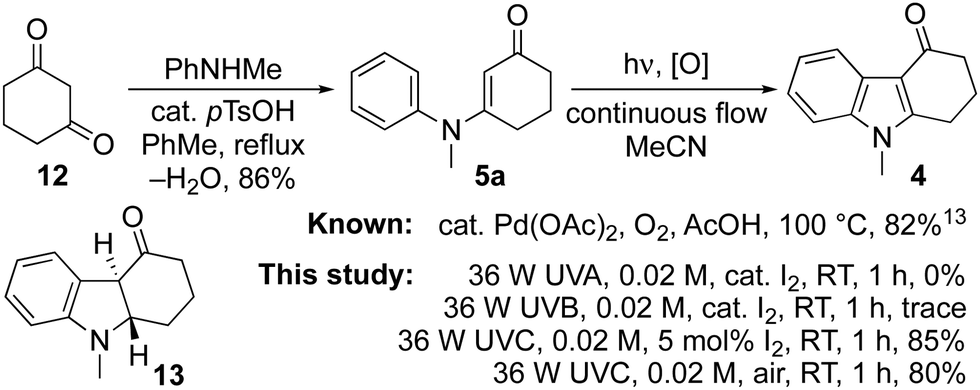
Tetrahydrocarbazolones and related heterocyclic ring systems are privileged structures in medicinal chemistry with a number of active pharmaceutical ingredients bearing such motifs.1 For example, alosetron 1 is a 5-HT3 antagonist used in the management of chronic irritable bowel syndrome (IBS),2 while ondansetron 2 is a WHO listed essential medicine used to treat nausea in cancer and COVID-19 patients.3,4 The pandemic has given impetus to calls for the on-shoring of the manufacture of such essential medicines and in that context, we sought to develop a synthesis of the tetrahydrocarbazolone core 4 of ondansetron 2 that was cheaper and more benign than the published strategies (Fig. 1).5–20 The approach we envisioned (Scheme 1) used the known condensation of bulk chemicals N-methylaniline and 1,3-cyclohexadione 12, followed by photocyclization of the resulting adduct 5a under oxidative conditions.21 Herein we describe our realisation of that goal leading to the development of a general method for the synthesis of indoles that mirrors the Fischer indole synthesis but uses anilines in place of arylhydrazines.6
 | ||
| Fig. 1 Approaches to the tetrahydrocarbazolone core of ondansetron. | ||
 | ||
| Scheme 1 Approaches to the tetrahydrocarbazolone core of ondansetron. | ||
An attractive feature of the strategy was its potential to effect the synthesis without recourse to stoichiometric reagents, metal catalysts or harsh reaction conditions. It also offered high atom-economy with a reduced and more benign waste-stream compared to the current industrial synthesis involving a palladium catalysed cyclisation of aryl bromide 5d (R = Me).13 The conversion of adduct 5a to ondansetron precursor 4 has been described previously using catalytic palladium acetate in acetic acid at 100 °C under an oxygen atmosphere.9,20 Its cyclization to dihydroindole 13 has also been reported to proceed in benzene using a 400 W medium pressure mercury lamp, with subsequent oxidation to 4 accomplished using DDQ or Mn(OAc)3.10
Thus, our initial focus was to establish conditions to achieve the direct conversion of 5a to 4, photochemically under flow. A viable method was established when a 0.02 M solution of 5a in acetonitrile containing 5 mol% iodine underwent conversion to 4 in 85% yield following irradiation with a 36 W Philips UVC lamp for 1 h.22–24 Analogous reactions with UVA and UVB lamps had proven ineffective, largely returning the starting material with trace by-products. Similarly, the reaction was slowed when the concentration of iodine was increased, suggesting that it competed for photons with other components in the reaction mixture.25 Pleasingly, air was also effective as an oxidant when introduced as bubbles into the flow stream. Though crude product mixtures were not as clean as those attained with catalytic iodine, the isolated yield of 4 was comparable.
Buoyed by this success, we next sought to apply the method in the preparation of alosetron precursor 14 (Scheme 2).2,26 We were disappointed to find that precursor 16 failed to give the anticipated product 14 on irradiation with UVC light under the aforementioned oxidative cyclization sequences, delivering instead an intractable product mixture. The failure was traced to a photochemical scission of cyclic amide 14,23 as evidenced by the formation of indole 15 as the primary product when methanolic solutions of 14 or 16 were irradiated with UVC. Pleasingly, the analogous Boc protected precursor 17 gave the reaction smoothly to deliver alosetron precursor 14 in 63% yield after deprotection with TFA.
 | ||
| Scheme 2 A formal synthesis of alosetron 1. | ||
Development of the procedure into a general method for the conversion of N-arylenaminones to indoles became our next goal. While similar 6π-photocyclization reactions have been described for the synthesis of indolines, these are characterised by poor productivity, low energy efficiency and limited substrate scope.10,27 Recent advances in photocatalysis have allowed cyclisations to be performed using blue light but the need for long irradiation times and expensive iridium catalysts limits its appeal.28 The scope of the reaction has been shown to be good for carbamate protected N-arylenaminones under UVA irradiation,29 but for API precursors 4 and 14 this would require subsequent deprotection, indoline oxidation and N-methylation steps. Thus, the UVC-induced oxidative cyclisation procedure had the potential to provide a complimentary method for substrates bearing other nitrogen substituents.
To that end, we first targeted tetrahydrocarbazolones 18b–n bearing substituents on nitrogen and the cyclohexene ring (Table 1). Pleasingly, each proceeded smoothly in yields of ∼70%. Notably, substrates bearing an N-benzyl substituent required shorter residence times than those with N-methyl and N-ethyl residues. Though secondary enaminone 5a (R = R′ = H) failed to give tetrahydrocarbazolone 3, that product could be accessed from the N-Boc-indole 18c, which was delivered in high yield when using catalytic I2 as the oxidant.
| † 3 can be formed in high yield by Boc deprotection of 18c, as detailed in the ESI |
|---|

|
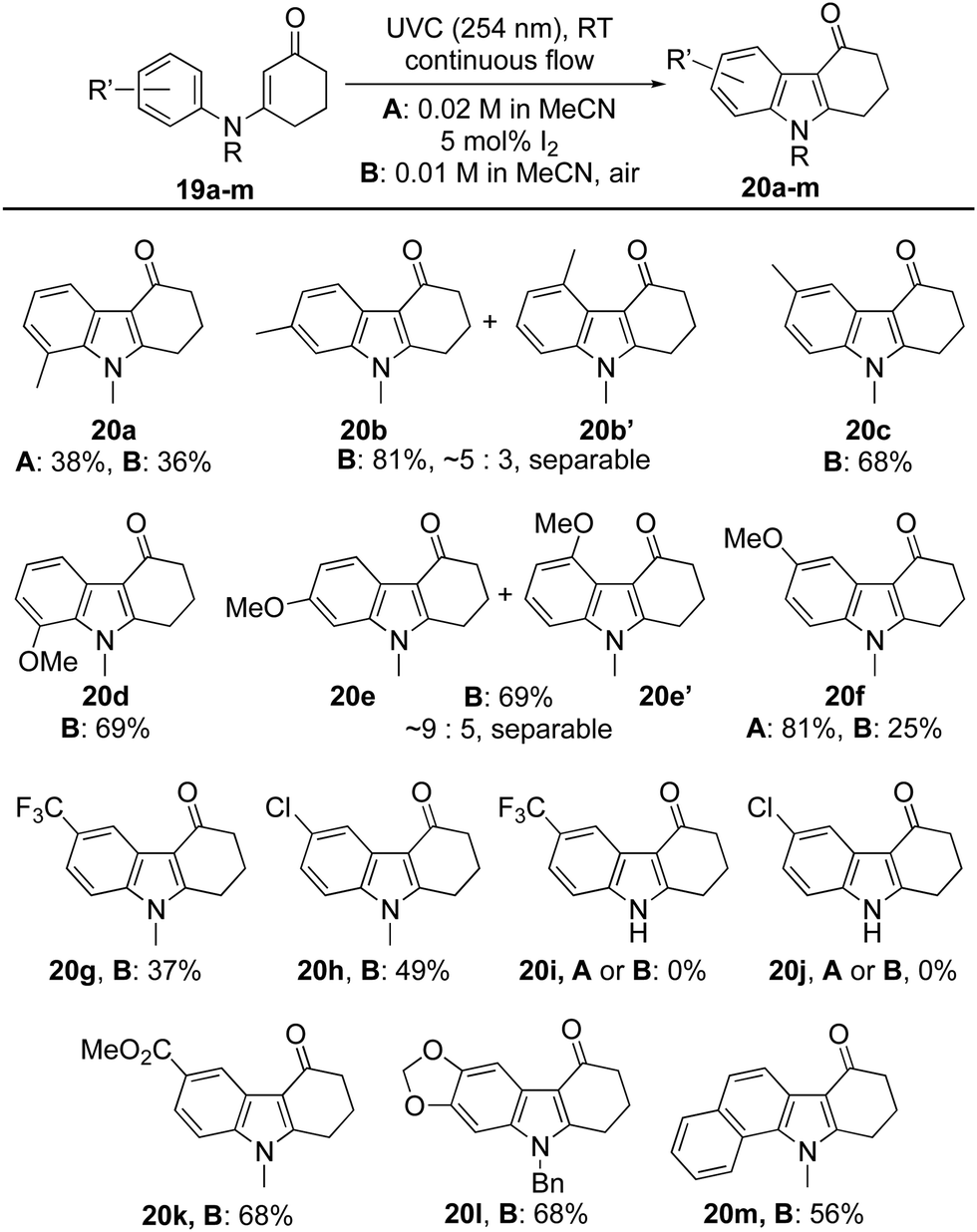
Attention next turned to the influence of aromatic substituents on the course of the reaction (Table 2). The ortho-, meta- and para-tolyl and anisole derivatives 19a–f were each prepared and converted into indoles 20a–f by the aforementioned protocols. In most cases, reactions proceeded well using air as the oxidant, with hydrolysis of the starting material accounting for much of the outstanding mass balance. Indeed, this proved especially significant for the ortho-tolyl derivative 19a → 20a, leading to a modest yield. In this case, switching the oxidant to catalytic I2 had little impact on the efficiency of the reaction, in stark contrast to the ortho-anisole derivative 19d where the switch elevated the yield of indole 20d from 25% to 81%. Competitive hydrolysis also contributed to the modest 37% yield attained for indole 20g while for the para-chloro analogue 20h (49%), other side reactions reduced the yield significantly. The reaction failed to give indoles 20i or 20j from secondary enaminones 19i and 19j (R = H) but was extended successfully to the fused polycycles 20l and 20m.
|
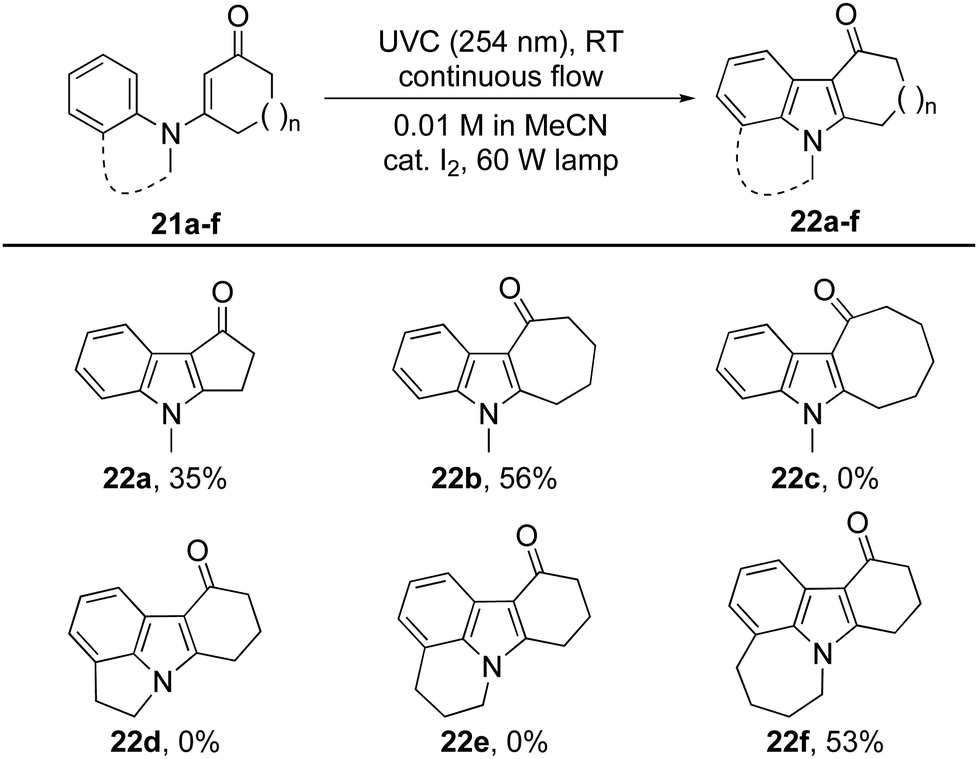
Changing the size of the cyclic enone had a dramatic effect on the reactions’ efficiency (Table 3).29 Indeed, for cyclopenten-one 21a (n = 0) only traces of the expected indole 22a were observed using the aforementioned procedures. Optimization studies were rewarded with the production of indole 22a in 35% isolated yield when cat. I2 was used in conjunction with a 60 W lamp. Under these conditions, cyclisation was able to compete with hydrolysis of the starting material. The same conditions also allowed cycloheptenone 21b to be transformed into indole 22b in 56% yield, but proved ineffective with cyclooctenone 21c, which returned N-methylaniline as the major product.
|
N-Arylenaminones derived from bicyclic amines were also investigated (Table 3). While the dihydroindole and dihydro-quinoline derivatives failed to give indoles 22d and 22e, the corresponding tetrahydrobenzoazepine was successfully converted into tetracyclic indole 22f. These observations indicate that the size of the ring fused to the N-arylenaminone determines whether a reactive conformer with good orbital overlap can be achieved.24
Initial attempts to induce cyclisations of acyclic N-aryl-enaminones (Table 4) were beset by low yields and the recovery of starting material.30 A breakthrough came with the observation that reactions could be induced under conditions of high dilution. Thus, at 0.002 M in acetonitrile using air as the oxidant, we were able to realize each of the oxidative cyclisation reactions of enamines 23a–l to indoles 24a–l. A notable anomaly was enamine 23m (R = H, R1 = Me, R2 = R3 = CO2Me), which was returned under these conditions but gave indole 24m in 63% when acetic acid was added to the reaction mixture. We presume that acetic acid catalyses the proton transfer step, 25 → 26, that follows conrotatory 6π-electrocyclisation of 23via its singlet excited state (Scheme 3). Another point of note is that the substitution pattern for indole 24e mirrors that of the nonsteroidal anti-inflammatory and analgesic, pravadoline.31
| †Required the addition of AcOH, as detailed in the ESI |
|---|

|
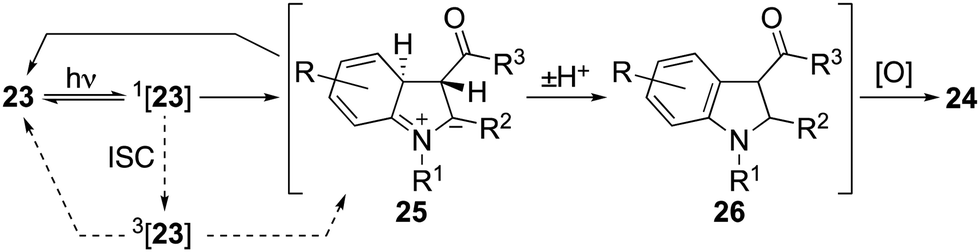 | ||
| Scheme 3 Mechanistic course of the reaction. | ||
Finally, the method has been extended to N-arylenaminones where the amine is bound to the α-carbon of the enone, and bond formation occurs to its β-carbon (Table 5).32 As with the aforementioned examples, the cyclohexanone derivatives 27a–c each gave the corresponding indoles 28a–c in good yield after a residence time of 1 h, while the yield for cyclisation of the acyclic enone 27d to indole 28d was modest.

|
In conclusion, we have developed a cheap, efficient and atom economic route to indoles that has wide applicability. Strategically, the method mirrors the Fischer indole synthesis but replaces the arylhydrazine component with an aniline which are generally cheaper, less toxic and more widely available. The method allows products such as the ondansetron precursor 4 to be synthesized on a gram per hour basis using a standard laboratory set-up (see ESI‡). Efforts to scale the reaction further are currently under investigation along with extensions to other APIs and fused heteroaromatic ring systems.
Wei Sun and William Raimbach performed all of the reported experiments and contributed equally in respect of the practical work. Kevin Booker-Milburn, Luke Elliott and David Harrowven conceived the project and acquired the funding to support it. David Harrowven was the primary supervisor.
We gratefully acknowledge financial support from EPSRC [EP/P013341/1, EP/L003325/1 and EP/K039466/1] and the European Regional Development Fund [ERDF Interreg Va programme (Project 121)].
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B. Chen, P. J. Kling, K. A. Kunkel, J. P. Springer and J. Hirshfieldt, J. Med. Chem., 1988, 31, 2235 CrossRef CAS PubMed; (b) P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2017, 56, 7971 CrossRef CAS PubMed.
- J. W. F. Whitehead, K. Mills, I. H. Coates, A. W. Oxford and P. C. North, US Pat., 1993, 5196534 Search PubMed.
- http://www.who.int/publications/i/item/WHOMVPEMPIAU2019http://.06 (accessed 6.10.2021).
- I. H. Coates, J. A. Bell, D. C. Humber and G. B. Ewan, UK Pat., 1985, 2153821. See also http://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-1 (accessed 6.10.2021) Search PubMed.
- F. Khan, M. Fatima, M. Shirzaei, Y. Vo, M. Amarasiri, M. G. Banwell, C. Ma, J. S. Ward and M. G. Gardiner, Org. Lett., 2019, 21, 6342 CrossRef CAS.
- D.-Q. Xu, J. Wu, S.-P. Luo, J.-X. Zhang, J.-Y. Wu, X.-H. Du and Z.-Y. Xu, Green Chem., 2009, 11, 1239 RSC.
- J. Wang, M. Wang, K. Chen, S. Zha, C. Song and J. Zhu, Org. Lett., 2016, 18, 1178 CrossRef CAS.
- X. Yao, X. Shan and L. Zu, Org. Lett., 2018, 20, 6498 CrossRef CAS.
- (a) S. Würtz, S. Rakshit, J. J. Neumann, T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 7230 CrossRef; (b) J. J. Neumann, S. Rakshit, T. Dröge, S. Würtz and F. Glorius, Chem. – Eur. J., 2011, 17, 7298 CrossRef CAS; (c) W. Bi, X. Yun, Y. Fan, X. Qi, Y. Du and J. Huang, Synlett, 2010, 2899 CAS.
- C. Tietcheu, C. Garcia, D. Gardette, D. Dugat and J.-C. Gramain, J. Heterocycl. Chem., 2002, 39, 965 CrossRef CAS.
- L. V. Kudzma, Synthesis, 2003, 1661 CrossRef CAS.
- B. Knipp, R. Kucznierz, T. Sattelkau and T. Zeibig, US Pat., 2007, 112054 Search PubMed.
- A. Osuka, Y. Mori and H. Suzuki, Chem. Lett., 1982, 2031 CrossRef CAS.
- (a) U. S. Sørensen and E. Pombo-Villar, Helv. Chim. Acta, 2004, 87, 82 CrossRef; (b) S. Yan, H. Wu, N. Wu and Y. Jiang, Synlett, 2007, 2699 CAS.
- (a) Y. Uozumi, M. Mori and M. Shibasaki, Chem. Commun., 1991, 81 RSC; (b) M. Mori, Y. Uozumi and M. Shibasaki, Heterocycles, 1992, 33, 819 CrossRef CAS.
- (a) T. L. Scott and B. C. G. Söderberg, Tetrahedron, 2003, 59, 6323 CrossRef CAS; (b) D. Janreddy, V. Kavala, J. W. J. Bosco, C.-W. Kuo and C.-F. Yao, Eur. J. Org. Chem., 2011, 2360 CrossRef CAS.
- Y. Ban, K. Yoshida, J. Goto, T. Oishi and E. Takeda (neé Ishigamori), Tetrahedron, 1983, 39, 3657 CrossRef CAS.
- R. A. Bunce and B. Nammalwar, J. Heterocycl. Chem., 2009, 46, 172 CrossRef CAS.
- X. Ban, Y. Pan, Y. Lin, S. Wang, Y. Du and K. Zhao, Org. Biomol. Chem., 2012, 10, 3606 RSC.
- D. Bhattacherjee, S. Ram, A. S. Chauhan, Yamini, Sheetal and P. Das, Chem. – Eur. J., 2019, 25, 5934 CrossRef CAS PubMed.
- D. Szymor-Pietrzak, M. N. Khan, A. Pagès, A. Kumar, N. Depner and D. L. Clive, J. Org. Chem., 2021, 86, 619 CrossRef CAS PubMed.
- (a) D. C. Harrowven, M. Mohamed, T. P. Gonçalves, R. J. Whitby, D. Bolien and H. F. Sneddon, Angew. Chem., Int. Ed., 2012, 51, 4405 CrossRef CAS; (b) M. Mohamed, T. P. Gonçalves, R. J. Whitby, H. F. Sneddon and D. C. Harrowven, Chem. – Eur. J., 2011, 17, 13698 CrossRef CAS; (c) T. P. Gonçalves, M. Mohamed, R. J. Whitby, H. F. Sneddon and D. C. Harrowven, Angew. Chem., Int. Ed., 2015, 54, 4531 CrossRef; (d) D. E. Collin, E. H. Jackman, N. Jouandon, W. Sun, M. E. Light, D. C. Harrowven and B. Linclau, Synthesis, 2021, 1307 CAS.
- M. A. Manning, W. Sun, M. E. Light and D. C. Harrowven, Chem. Commun., 2021, 57, 4556–4559 RSC.
- M. J. Ralph, D. C. Harrowven, S. Gaulier, S. Ng and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2015, 54, 1527 CrossRef CAS.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769 CrossRef CAS.
- (a) B. Miao, S. Li, G. Li and S. Ma, Org. Lett., 2016, 18, 2556 CrossRef CAS; (b) L. Li, X. Zhou, B. Yu and H. Huang, Org. Lett., 2017, 19, 4600 CrossRef CAS PubMed.
- (a) J.-C. Gramain, H.-P. Husson and Y. Troin, J. Org. Chem., 1985, 50, 5517 CrossRef CAS; (b) J.-C. Gramain, Y. Troin and H.-P. Husson, J. Heterocycl. Chem., 1988, 25, 201 CrossRef CAS; (c) N. Benchekroun-Mounir, D. Dugat and J.-C. Gramain, Tetrahedron Lett., 1992, 33, 4001 CrossRef CAS; (d) N. Benchekroun-Mounir, D. Dugat, J.-C. Gramain and H.-P. Husson, J. Org. Chem., 1993, 58, 6457 CrossRef CAS; (e) F. Toda, H. Miyamoto, T. Tamashima, M. Kondo and Y. Ohashi, J. Org. Chem., 1999, 64, 2690 CrossRef CAS PubMed; (f) J. L. Berry, D. R. Dillin and D. G. Watson, Tetrahedron Lett., 1985, 26, 143 CrossRef CAS; (g) D. Dugat, J.-C. Gramain and G. Dauphin, J. Chem. Soc., Perkin Trans. 2, 1990, 605 RSC.
- C.-J. Wu, W.-X. Cao, T. Lei, Z.-H. Li, Q.-Y. Meng, X.-L. Yang, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2017, 53, 8320 RSC.
- S. G. Modha, A. Pöthig, A. Dreuw and T. Bach, J. Org. Chem., 2019, 84, 1139 CrossRef CAS.
- (a) J. L. Berry, D. R. Dillin and D. G. Watson, Tetrahedron Lett., 1985, 26, 143 CrossRef CAS; (b) Y. Troin, M.-E. Sinibaldi, J.-C. Gramain, M. Rubiralta and A. Diez, Tetrahedron Lett., 1991, 32, 6129 CrossRef CAS.
- G. Grieco, J. DeAndrade, E. Dorflinger, T. Kantor, J. Saelens, A. Sunshine, R. Wang, G. Wideman and V. Zellman, Clin. Pharmacol. Ther., 1989, 42, 123 Search PubMed.
- J. C. Arnould, J. Cossy and J. P. Pete, Tetrahedron, 1980, 36, 1585 CrossRef CAS.
Footnotes |
| † In memory of Dr Lucette Duhamel. |
| ‡ Electronic supplementary information (ESI) available: Experimental accounts, spectral and analytical data, and NMR spectra. See DOI: 10.1039/d1cc05700f |
| This journal is © The Royal Society of Chemistry 2022 |