
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of tert-butyl groups on electronic communication between redox units in tetrathiafulvalene-tetraazapyrene triads†
Ping
Zhou
a,
Ulrich
Aschauer
 a,
Silvio
Decurtins
a,
Thomas
Feurer
b,
Robert
Häner
a and
Shi-Xia
Liu
*a
a,
Silvio
Decurtins
a,
Thomas
Feurer
b,
Robert
Häner
a and
Shi-Xia
Liu
*a
aDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, Bern CH-3012, Switzerland. E-mail: shi-xia.liu@unibe.ch
bInstitute of Applied Physics, University of Bern, Sidlerstrasse 5, Bern CH-3012, Switzerland
First published on 12th November 2021
Abstract
The electronic effect of tert-butyl groups on intramolecular through-bond interactions between redox units in tetrathiafulvalene-tetraazapyrene (TAP) triads is investigated. The insertion of tert-butyl groups raises the TAP-localised LUMO level by 0.21 eV, in fairly good agreement with 0.17 eV determined by DFT calculations.
Tetraazapyrene (TAP), a prototype of nitrogenated polycyclic aromatic hydrocarbons (N-PAHs), exhibits intrinsic optoelectronic and electrochemical properties as well as high thermal and chemical stability, which are important requirements for its diverse applications in the field of organic (opto)electronics.1,2 However, reports on synthetic approaches to the TAP scaffold and its derivatization are quite limited in the literature.3 In 2012, a series of 2,7-substituted TAP derivatives was prepared and tested as n-type semiconductors in organic field-effect transistors (OFETs).1 It has been demonstrated that the electronic properties are significantly affected by the nature of substituents at the core positions. Inspired by these appealing results, we have embarked on the design and synthesis of tetrathiafulvalene (TTF)-functionalized TAPs to create triads (Scheme 1) with promising material properties.
 | ||
| Scheme 1 Synthesis of a key precursor 3, triads TTF-TAP and TTF-t-Bu-TAP. | ||
Within the context of organic conductors, TTF acts as a strong π-electron donor and takes a prominent place in this class of materials.4 By virtue of its unique redox properties, it has been incorporated into a variety of (supra)molecular architectures for the development of highly efficient (opto)electronic materials.5 Among them, TTF-based electron donor–acceptor (D–A) ensembles have attracted a lot of attention with a special focus on redox modulation of intramolecular charge transfer (ICT) and chemical/optical control over multiple ICT pathways.6,7 Herein, we report a facile and efficient synthetic protocol to 2,7-di-tert-butyl-1,3,6,8-tetraazapyrene (2) as well as the further derivatization by 4-fold core-bromination for subsequent nucleophilic substitutions with a cyanoethyl-protected TTF (4), leading to the formation of two triads TTF-TAP and TTF-t-Bu-TAP with a D–A–D architecture (Scheme 1).
It is well known that bulky tert-butyl groups have been widely incorporated into large N-PAH molecules for improved solubility because of suppressed aggregation.8,9 Also, tert-butylation has been exploited to bias regioselectivity, e.g. insertion of tert-butyl groups into 2,7 positions of pyrene can avoid the reaction occurring at the electron-rich 1,3,6,8-carbons instead of the K-region (4,5,9,10-carbons).10 This work aims to reveal the effects of tert-butyl groups9,11 on the electronic characteristics of the triads. Redox and optical properties of TTF-TAP and TTF-t-Bu-TAP have been investigated in detail. The insertion of tert-butyl groups at positions 2 and 7 of the TAP core has indeed a very pronounced effect on the TAP-localised LUMO energy level.
As illustrated in Scheme 1, our synthetic approach to the 2,7-substituted TAP scaffold 2 involves a twofold cyclocondensation of naphthalene-1,4,5,8-tetraamine tin(II) salt (1)12 with an excess of pivalic anhydride, followed by oxidation of the intermediate 1,8-dihydropyrimido[4,5,6-gh]perimidine. This reaction was performed at reflux in THF in the presence of catalytic amounts of triethylamine, affording 2 as a pale-yellow powder in a yield of 60%. The subsequent core-bromination was accomplished to yield 3 (70%) in concentrated sulfuric acid in the presence of Br2 with catalytic amounts of iodine, similar to the synthetic procedure reported for 5.1 Both 4-fold core-brominated compounds 3 and 5 undergo nucleophilic substitution with 4 in the presence of CsOH/MeOH to produce the desired products TTF-t-Bu-TAP and TTF-TAP in a yield of 18% and 24%, respectively. These as-prepared compounds were unambiguously characterized by 1H and 13C NMR, cyclic voltammetry, UV-Vis-NIR spectroscopy and high-resolution mass spectrometry. All analytical data match well with their chemical structures.
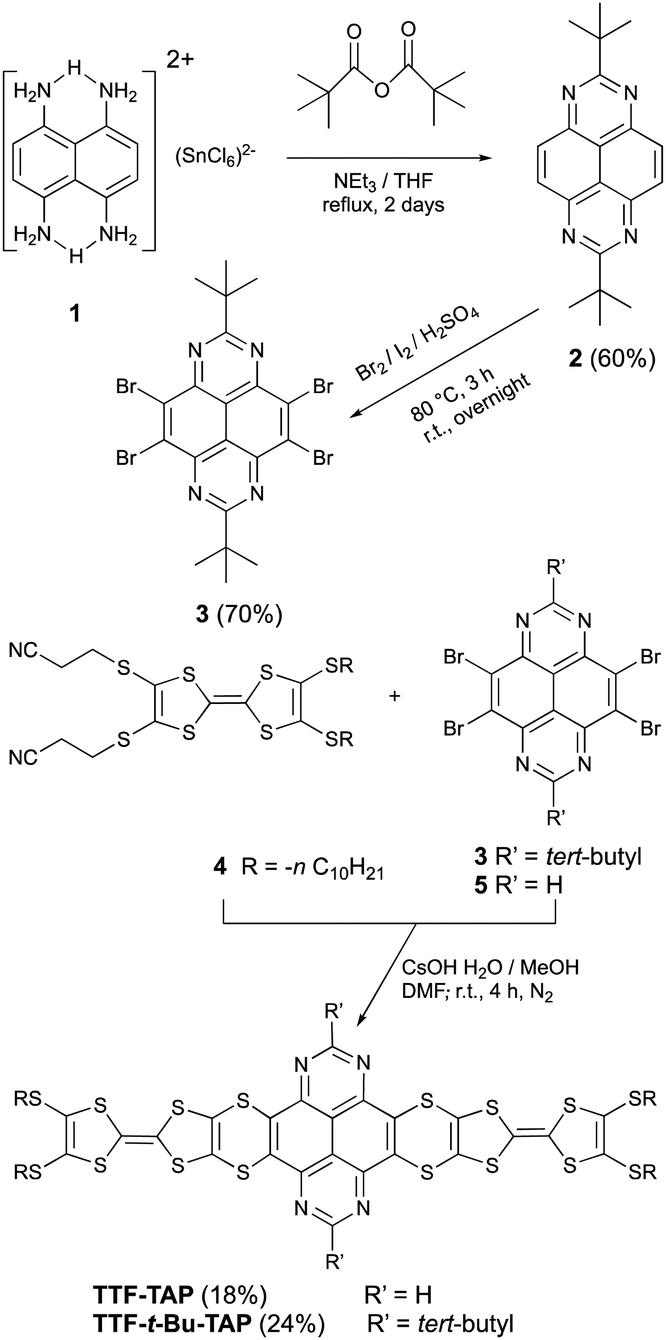
The electrochemical properties of TTF-TAP, TTF-t-Bu-TAP and the reference compounds 2 and 4 were investigated by cyclic voltammetry and differential pulse voltammetry in CH2Cl2 (Fig. 1 and Table S1, Fig. S1, ESI†). TTF-TAP undergoes three distinct reversible oxidation processes at 0.63 V, 0.77 V and at 1.06 V (vs. Ag/AgCl), indicating that two TTF units are successively oxidized to TTF radical cation species and simultaneously to TTF dication species. This phenomenon remains even in a very diluted solution (Fig. S2, ESI†), indicating that the possibility of associations between TTF-TAP molecules is ruled out, which can further be verified by concentration-dependent UV-Vis spectra (Fig. S3, ESI†). The occurrence of step-wise oxidation processes for the generation of the radical cation species points to significant intramolecular through-bond interactions between two TTF units, as previously observed in analogous molecules.13 In the negative potential window, two reversible reduction waves at −0.46 V and −0.98 V are observed, corresponding to the sequential addition of electrons to the TAP unit. Interestingly, the incorporation of tert-butyl group has an appreciable influence on the redox properties of TTF and TAP units. TTF-t-Bu-TAP shows only two reversible oxidation waves at 0.70 V and 1.03 V (Fig. S1, ESI†), suggesting that two TTF units are simultaneously oxidized to their radical cation and dication species, respectively. Similarly, it undergoes two reversible reductions at −0.67 V and −1.19 V, which, however, are negatively shifted compared to TTF-TAP. Consequently, the insertion of tert-butyl groups raises the LUMO level by 0.21 eV, very probably due to the hyperconjugation effect of the tert-butyl groups, but an electrostatic destabilization of the TAP-localized π* MO can also be invoked based on computed electrostatic potentials at nuclear positions (Table S2, ESI†). The first reduction potential values of both triads are positively shifted with respect to that of the TAP, ref. 2 indicative of the extended conjugation via four S atoms caused by the covalent linkage of two TTF units to the central TAP core. In stark contrast, their oxidation potentials are either shifted only slightly positively or remain almost unchanged compared to those of the TTF precursor 4, most likely because of the lack of an effective electronic communication between the redox units. The latter is a consequence of nonplanar conformations of the triads, as shown by the corresponding calculated structures (Fig. S4, ESI†). As a result, both triads show pronouncedly lowered LUMO energies while maintaining similar HOMO energies with respect to the constituting moieties 2 and 4, which matches well the UV-Vis spectroscopic results presented below.
 | ||
| Fig. 1 Cyclic voltammograms of TTF-TAP (blue), TTF-t-Bu-TAP (red), t-Bu-TAP 2 (black) and TTF precursor 4 (pink) were measured in dichloromethane solution (c = 3 × 10−4 M), containing 0.1 M TBAPF6 as the supporting electrolyte at room temperature, Pt working electrode, Ag/AgCl electrode as the reference electrode and the scan rate at 100 mV s−1. | ||
TTF-TAP and TTF-t-Bu-TAP strongly absorb in the UV-Visible-NIR spectral region as evidenced by their yellow-green colour. Their absorption spectra in CH2Cl2 are depicted in Fig. 2.
 | ||
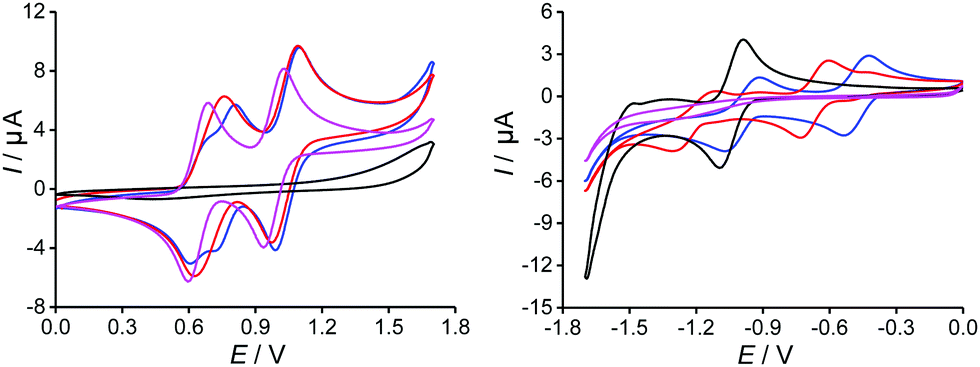
| Fig. 2 UV-Vis absorption spectra of TTF-TAP (blue), TTF-t-Bu-TAP (red), t-Bu-TAP 2 (black) and TTF precursor 4 (pink) (c = 7 × 10−6 M) in CH2Cl2 at r.t. Inset: Vis-NIR absorption spectra of TTF-TAP, TTF-t-Bu-TAP (c = 7 × 10−4 M) in CH2Cl2 at r.t. | ||
In the UV and blue part of the optical spectra, the observed strong absorption bands are attributed to π–π* transitions localized on the TTF and TAP cores, similarly to the reference compounds 2 and 4, only with bathochromic shifts owing to extended conjugation. By comparison of TTF-t-Bu-TAP (red) to TTF-TAP (blue), the insertion of the tert-butyl groups leads to noticeable bathochromic shifts of the absorption bands in a range of 360 nm to 450 nm and a slight hypsochromic shift of the absorption band around 320 nm. As these transitions correspond mainly to π–π* excitations of the TAP core, the observed shifts can be accounted for by the fact that the presence of bulky tert-butyl groups at 2,7-positions of the TAP is unfavourable for possible H- and J-aggregation and impedes effective intermolecular interactions. More importantly, both triads exhibit two weak and broad absorption bands centered at 680 nm and 1100 nm for TTF-TAP, and 630 nm and 1100 nm for TTF-t-Bu-TAP, which are not observed, however, in solutions of the acceptor 2 and donor 4 components. Consequently, these absorptions reflect ICT transitions dominated by excitations from MOs localized on the TTF units to the LUMO localized on the TAP core. These assignments are corroborated by the computational results (see below). An attempt can be made to approach with some uncertainties the onsets of the very weak and broad absorption bands of both triads in the NIR region (Fig. 2) while extrapolating the linear portions on their red sides to the abscissa. Their optical bandgaps lie approximately between 1 eV and 0.92 eV, with the higher value assigned to the more steeply falling absorption edge of the TTF-t-Bu-TAP triad, a trend compliant with the electrochemical study.
In order to shed more light on the intramolecular electronic interactions between the TTF and TAP units in TTF-TAP and TTF-t-Bu-TAP, spectroelectrochemical measurements were carried out. Chemical oxidation of TTF-TAP and TTF-t-Bu-TAP was performed by successive addition of NOSbF6 aliquots at room temperature. As shown in Fig. 3 for TTF-TAP, a progressive reduction of the absorbance of the high energy π–π* transitions is accompanied with a concomitant appearance of new absorption bands at 452 nm and 835 nm which reach their maximum values upon addition of 12 equiv. of NOSbF6. The lowest energy absorption band is continuously blue-shifted during the oxidation process. These new transitions are characteristic of the newly formed cationic TTF˙+ radical within a D–A–D triad.13,14 In contrast, the initial oxidation of TTF-t-Bu-TAP leads to a broad absorption band which emerges at 905 nm and, after addition of 1.2 equiv., suddenly moves towards higher energy (830 nm) with increasing amounts of NOSbF6 (Fig. 4). Again, it reaches its maximum value upon addition of 12 equiv. of NOSbF6. In both cases, this broad and distinctly asymmetric absorption band in the NIR region is mainly attributed to electronic transitions localized on the TTF˙+ unit with some back ICT character from the TAP unit to the radical species TTF˙+, however to a much lesser extent than previously reported in TTF-fused D–A systems.7,14,15 This result could be accounted for by their non-planar geometries (Fig. S4, ESI†). Moreover, the observed variance in the evolution of the absorption spectra in this spectral region is very probably due to different degrees of intermolecular π-dimerization of the TTF˙+ radical cation imparted by sequential or simultaneous oxidation of two TTF cores in TTF-TAP and TTF-t-Bu-TAP, as corroborated by CV measurements.
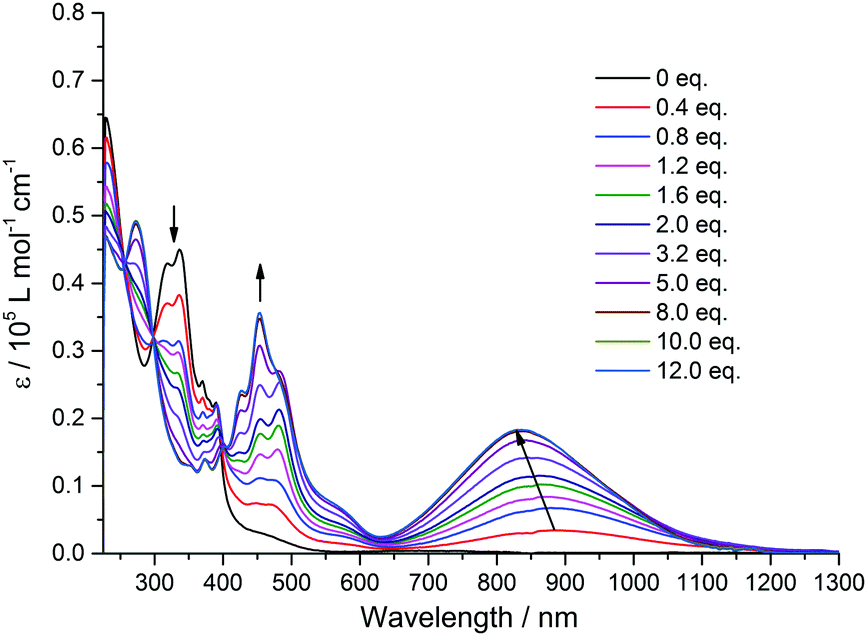 | ||
| Fig. 3 Variation of UV-Vis-NIR absorption spectra of TTF-TAP (7 × 10−6 M) in CH2Cl2 upon successive addition of aliquots of NOSbF6 at room temperature. | ||
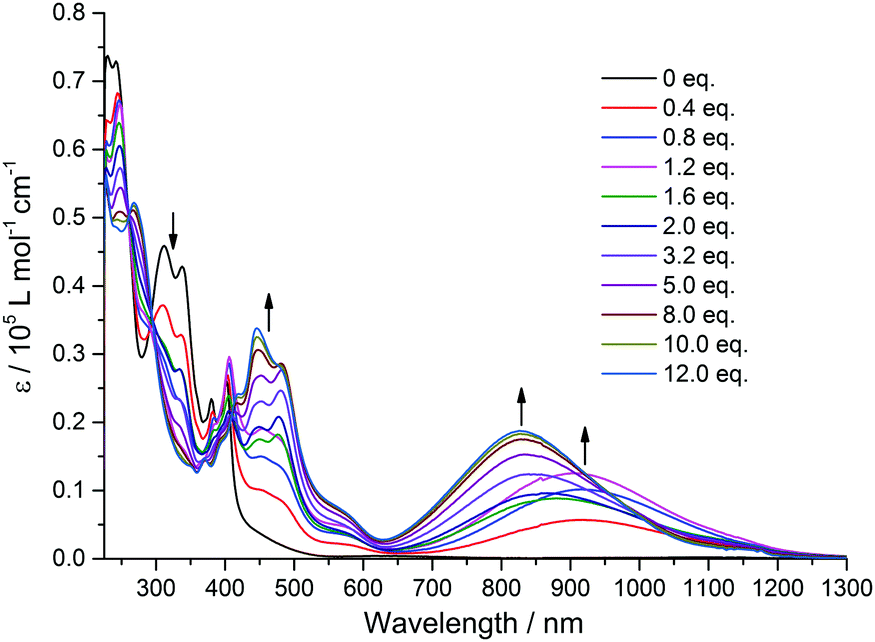 | ||
| Fig. 4 Variation of UV-Vis-NIR absorption spectra of TTF-t-Bu-TAP (7 × 10−6 M) in CH2Cl2 upon successive addition of aliquots of NOSbF6 at room temperature. | ||
To characterize and verify the various electronic transitions, TD-DFT calculations were performed using the Gaussian 16 package at the B3LYP/6-31G(d,p) level of theory.16 The predicted absorption spectra of TTF-TAP and TTF-t-Bu-TAP (Fig. S5, ESI†) are in fairly good agreement with experimental results. Fig. 5 illustrates the frontier MOs of TTF-TAP which are involved in the ICT transitions. The HOMO and HOMO−1 represent the symmetric and asymmetric combinations of the two donor TTF MOs. Analogously, HOMO−2 and HOMO−3 combine both pending units, however, the TAP core is now partly involved in their electron densities (Fig. S6, ESI†). The LUMO is localized at the acceptor TAP core with some extension to the adjacent sulfur atoms. The frontier MOs of TTF-t-Bu-TAP (Fig. S6, ESI†) show the same characteristics. The calculated HOMO–LUMO gap amounts to 1.63 eV for TTF-TAP and 1.76 eV for TTF-t-Bu-TAP.
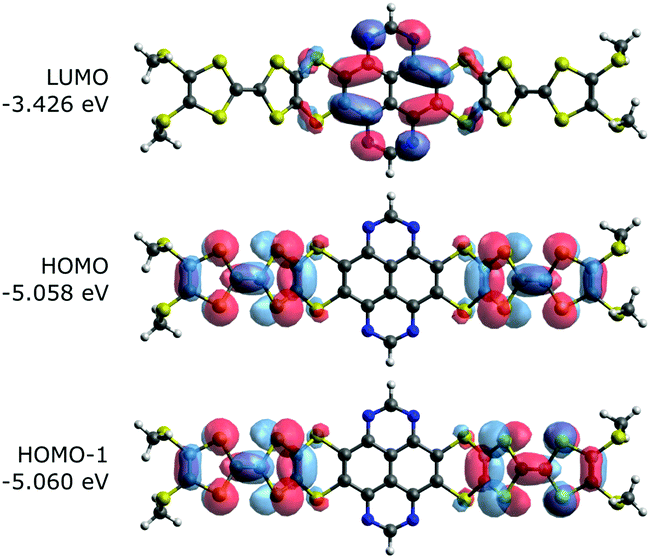 | ||
| Fig. 5 Frontier molecular orbitals of TTF-TAP that are involved in the ICT transition. | ||
The predicted absorption spectra for both triads (Fig. S5, ESI†) show a manifold of π–π* transitions with high oscillator strengths at wavelength below 450 nm, in good agreement with the measured absorption spectra. The measured low intensity absorptions around 630–680 nm are well characterized by calculated HOMO−2/HOMO−3 → LUMO excitations, fairly close to that energy range and also with low oscillator strengths (Tables S3 and S4, ESI†). The same holds for the very weak longest wavelength absorptions which are now attributed to HOMO/HOMO−1 → LUMO excitations. Clearly, the orbital overlap among these MOs is little, which leads to a small transition dipole moment, hence low intensities for their transitions. To note, there is a distinct blue-shift for these calculated excitations for the TTF-t-Bu-TAP triad compared to TTF-TAP. Fig. S7 and Tables S5–S8 (ESI†) show the calculated transitions and the involved open-shell molecular orbitals (Fig. S8–S11, ESI†) for the +1 and +2 cationic states of both triads which by and large look similar. Upon oxidation of two TTF units to their radical states, the involved molecular orbitals of the NIR transitions reveal that the majority of charge remains on the TTF units.
In conclusion, the newly synthesized TTF-TAP and TTF-t-Bu-TAP absorb in the UV-Vis-NIR spectral region due to ICT excitations from TTF units to the central TAP core. Interestingly, the two TTF units of TTF-TAP are sequentially oxidized to the TTF radical cation species while this process occurs simultaneously in TTF-t-Bu-TAP. With respect to TTF-TAP, the LUMO level of TTF-t-Bu-TAP is raised by 0.21 eV. Also it is worthy of note that a modification at the TTF side would potentially be a tuning factor in the molecule as well.17 Our findings represent a novel concept in the manipulation of the relative HOMO and LUMO energetic positions of organic donor–acceptor ensembles by chemical modification.
We greatly appreciate preliminary clarifications on these systems by Dirk Guldi. This work was financially supported by the Swiss NSF through the NCCR MUST “Molecular Ultrafast Science and Technology”, the Swiss NSF (200020_188468 and 200021_204053), and the Swiss NSF professorship grants (PP00P2_157615 and PP00P2_187185). Computations were performed on Ubelix, the HPC cluster at the University of Bern.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Geib, S. C. Martens, U. Zschieschang, F. Lombeck, H. Wadepohl, H. Klauk and L. H. Gade, J. Org. Chem., 2012, 77, 6107 CrossRef CAS PubMed.
- S. C. Martens, L. Hahn, F. Lombeck, A. Rybina, H. Wadepohl and L. H. Gade, Eur. J. Org. Chem., 2013, 5295 CrossRef CAS; M. J. Sienkowska, J. M. Farrar, F. Zhang, S. Kusuma, P. A. Heiney and P. Kaszynski, J. Mater. Chem., 2007, 17, 1399 RSC; Y.-R. Shi, H.-L. Wei and Y.-F. Liu, New J. Chem., 2019, 43, 5706 RSC.
- A. V. Aksenov, D. S. Ovcharov, N. A. Aksenov, D. A. Aksenov, O. N. Nadein and M. Rubin, RSC Adv., 2017, 7, 29927 RSC; A. V. Aksenov, A. S. Lyakhovnenko, T. S. Perlova and I. V. Aksenova, Chem. Heterocycl. Compd., 2011, 47, 916 CrossRef CAS.
- D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS PubMed; N. Martin, Chem. Commun., 2013, 49, 7025 RSC.
- A. Jana, S. Bahring, M. Ishida, S. Goeb, D. Canevet, M. Salle, J. O. Jeppesen and J. L. Sessler, Chem. Soc. Rev., 2018, 47, 5614 RSC; Y. Wu, M. Frasconi, W. G. Liu, R. M. Young, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 11835 CrossRef CAS PubMed; L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530 RSC; J. Wu, N. Dupont, S.-X. Liu, A. Neels, A. Hauser and S. Decurtins, Chem. – Asian J., 2009, 4, 392 CrossRef PubMed; K. Herve, S.-X. Liu, O. Cador, S. Golhen, Y. Le Gal, A. Bousseksou, H. Stoeckli-Evans, S. Decurtins and L. Ouahab, Eur. J. Inorg. Chem., 2006, 3498 CrossRef.
- F. Pop and N. Avarvari, Chem. Commun., 2016, 52, 7906 RSC; J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863 RSC; E. J. Rohwer, Y. Geng, M. Akbarimoosavi, L. M. L. Daku, O. Aleveque, E. Levillain, J. Hauser, A. Cannizzo, R. Häner, S. Decurtins, R. J. Stanley, T. Feurer and S. X. Liu, Chem. – Eur. J., 2021, 27, 5399 CrossRef CAS PubMed.
- P. Zhou, U. Aschauer, S. Decurtins, T. Feurer, R. Häner and S. X. Liu, Chem. Commun., 2020, 56, 13421 RSC.
- B. Gao, M. Wang, Y. Cheng, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2008, 130, 8297 CrossRef CAS PubMed; L. Zoephel, V. Enkelmann, R. Rieger and K. Muellen, Org. Lett., 2011, 13, 4506 CrossRef PubMed; N. Kulisic, S. More and A. Mateo-Alonso, Chem. Commun., 2011, 47, 514 RSC.
- Z.-H. Wu, Z.-T. Huang, R.-X. Guo, C.-L. Sun, L.-C. Chen, B. Sun, Z.-F. Shi, X. Shao, H. Li and H.-L. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13031 CrossRef CAS PubMed.
- H. Bock, D. Subervie, P. Mathey, A. Pradhan, P. Sarkar, P. Dechambenoit, E. A. Hillard and F. Durola, Org. Lett., 2014, 16, 1546 CrossRef CAS PubMed; T. H. El-Assaad, K. N. Parida, M. F. Cesario and D. V. McGrath, Green Chem., 2020, 22, 5966 RSC.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045 RSC.
- V. Vitske, C. König, O. Hübner, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2010, 115 CrossRef CAS.
- H.-P. Jia, S.-X. Liu, L. Sanguinet, E. Levillain and S. Decurtins, J. Org. Chem., 2009, 74, 5727 CrossRef CAS PubMed; C. Jia, S. Liu, C. Tanner, C. Leiggener, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. Commun., 2006, 1878 RSC.
- C. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804 CrossRef CAS PubMed.
- M. Jaggi, C. Blum, B. S. Marti, S.-X. Liu, S. Leutwyler and S. Decurtins, Org. Lett., 2010, 12, 1344 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- P. J. Skabara and M. Sallé, Beilstein J. Org. Chem., 2015, 11, 1528 CrossRef CAS PubMed; J. Sly, P. Kasák, E. Gomar-Nadal, C. Rovira, L. Górriz, P. Thordarson, D. B. Amabilino, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2005, 1255 RSC; E. Gomar-Nadal, L. Mugica, J. Vidal-Gancedo, J. Casado, J. T. L. Navarrete, J. Veciana, C. Rovira and D. B. Amabilino, Macromolecules, 2007, 40, 7521 CrossRef; A. M. Amacher, J. Puigmarti-Luis, Y. Geng, V. Lebedev, V. Laukhin, K. Kraemer, J. Hauser, D. B. Amabilino, S. Decurtins and S.-X. Liu, Chem. Commun., 2015, 51, 15063 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05671a |
| This journal is © The Royal Society of Chemistry 2021 |