
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing stress granule formation by small molecules to inhibit the cellular replication of SARS-CoV-2†
Wan Gi
Byun
a,
Jihye
Lee
b,
Seungtaek
Kim
b and
Seung Bum
Park
 *a
*a
aCRI Center for Chemical Proteomics, Department of Chemistry, Seoul National University, Seoul, 08826, Korea. E-mail: sbpark@snu.ac.kr
bZoonotic Virus Laboratory, Institut Pasteur Korea, Seongnam 13488, Korea
First published on 26th October 2021
Abstract
We identified small-molecule enhancers of cellular stress granules by observing molecular crowding of proteins and RNAs in a time-dependent manner. Hit molecules sensitized the IRF3-mediated antiviral mechanism in the presence of poly(I:C) and inhibited the replication of SARS-CoV-2 by inducing stress granule formation. Thus, modulating multimolecular crowding can be a promising strategy against SARS-CoV-2.
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused an unprecedented public health and economic crisis around the world, resulting in over 218 million reported cases of COVID-19 and 4.5 million deaths worldwide as of August 2021.1,2 Additionally, there are now more than 1000 confirmed variants of SARS-CoV-2 deposited in the viral genome sequence databases, and several variants, such as B.1.1.7. (alpha) and B.1.617.2. (delta), identified in the UK and India, respectively, have greater infectivity and severity to human beings.3,4 New variants may even escape the immunity elicited by vaccination and weaken the antiviral effects of conventional drugs.4,5 Despite this situation, there are only a few vaccines and SARS-CoV-2-targeted antiviral reagents available to date. Therefore, effective therapeutics are in high demand to end this pandemic, and a host-targeted antiviral strategy can be one of the most efficient options.
Stress granules are cytosolic membraneless organelles mainly composed of RNAs and proteins via multimolecular crowding.6 The formation of stress granules is induced by cellular stress, such as viral infection, to reduce the burden of translation, one of the most energy-consuming mechanisms in eukaryotic cells.7 Upon recognizing various types of stress, four kinds of eIF2α kinases can induce the phosphorylation of eIF2α, resulting in the disruption of initiation codon recognition by the 48S pre-initiation complex, thereby inhibiting cellular translation and minimizing cellular damage.8 Concurrently, Ras GTPase-activating protein-binding protein 1 (G3BP1) captures polysome-released mRNAs and reversibly nucleates hundreds of proteins and RNA molecules through its RNA-binding domain and intrinsically disordered regions, which mediates multiple macromolecular interactions.9,10 Under viral infection, cellular translation is arrested by the phosphorylation of protein kinase R (PKR) via its recognition of viral double-stranded RNA (dsRNA) produced in viral replication cycles (Fig. 1).11 Particularly, temporal suspension of cellular translation in the host can directly inhibit the viral replication because many viruses including coronaviruses utilize host translational machinery to proliferate themselves within the cell.12,13
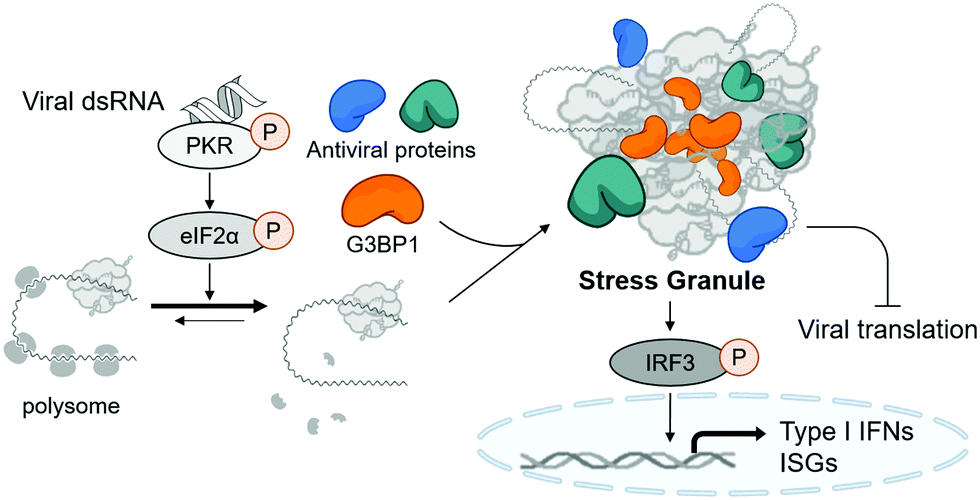 | ||
| Fig. 1 The schematic figure of stress granule formation and its antiviral function. | ||
Stress granules are also the central hub for innate antiviral immune responses. In the presence of viral stress, host antiviral immune regulators, such as RIG-I and MDA5, are recruited and assembled together with mRNAs and translation machinery proteins to form the multimolecular condensate. This antiviral stress granule can serve as a signaling platform to activate various antiviral signaling pathways that converge to phosphorylate IFN-regulatory factor 3 (IRF3), resulting in the induction of type I interferons and interferon-stimulated genes (ISGs) (Fig. 1).12,14,15 Most viruses including SARS-CoV-2 are known to alter multiple signaling pathways related to stress granule formation and subsequent type I interferon and ISG responses,16–18 indicating the importance of stress granules in antiviral responses. Considering that type I interferons have been used clinically as antiviral drugs for years and have beneficial effects on COVID-19 patients,19 enhancing the formation of stress granules using small molecules, thereby activating type I interferons, could be a novel strategy to treat viral diseases including COVID-19.
Given that stress granules are reversible and dynamic organelles, a time-dependent observation of stress granules in live cells could be informative. Thus, we utilized U2OS human osteosarcoma epithelial cells stably expressing the green fluorescent protein (GFP) tagged G3BP1, the marker protein of stress granules, for screening a drug-like small-molecule library. By simply counting cytoplasmic G3BP1 puncta without fixation in live cells, hit compounds at different time points can be discovered in a high-throughput manner. To confirm that the assay system can monitor the dynamics of stress granules in a 96-well plate format, we monitored GFP-G3BP1-expressed U2OS cells upon treatment with two well-known stress granule enhancers, sodium arsenite20 and thapsigargin,21 at various concentrations and clearly observed a dose-dependent induction of stress granules (Fig. S1, ESI†). We observed that the number of stress granules was maximized at 2 h after treatment of both enhancers and gradually reduced afterward. In sodium arsenite-treated cells, the time for maximal stress granules was postponed as its concentration decreases, which is consistent with previous reports.20
Since candidate compounds should be able to induce stress granule formation in virus-infected cells, we used polyinosinic:polycytidylic acid (poly(I:C)), a synthetic dsRNA analogue, to mimic virus-infected cells. Poly(I:C) is known to induce stress granule formation and activate the innate antiviral response upon recognition of viral dsRNA by PKR.22 Indeed, the transfection of poly(I:C) to human airway epithelial cells, Calu-3, activates the PKR-eIF2α-IRF3 pathway and arrests global translation, confirmed by immunoblot analysis (Fig. S2a, ESI†). Immunofluorescence detection of G3BP1 also revealed that poly(I:C) transfection can time-dependently increase the number of stress granules in both Calu-3- and G3BP1-GFP-expressing U2OS cells, indicating that poly(I:C) is an appropriate viral dsRNA mimic (Fig. S2b and S3, ESI†).
Subsequently, we applied the time-course monitoring system of cellular stress granules to discover small-molecule enhancers of the stress granule formation in virus-infected cells from a 464 member representative set of privileged substructure-based diversity-oriented synthesis (pDOS) library.23 Briefly, we transfected U2OS reporter cells with poly(I:C), treated them with individual compounds, and performed image-based monitoring of changes in cellular stress granules at 5 different time points (Fig. 2a). Compounds that enhanced the total count of stress granules per cell by >3-fold of standard deviations over negative controls were selected as hit compounds. Using our image-based time-course assay, we identified two classes of compounds as stress granule enhancers: benzopyranylpyrazole-cored compounds (Fig. 2b) and a tamoxifen derivative (Fig. S4, ESI†). Particularly, we observed different onset times for maximal stress granules in the case of several hit compounds, showing significant stress granule enhancement only after 3 h, which may not be detected without our time-course monitoring approach.
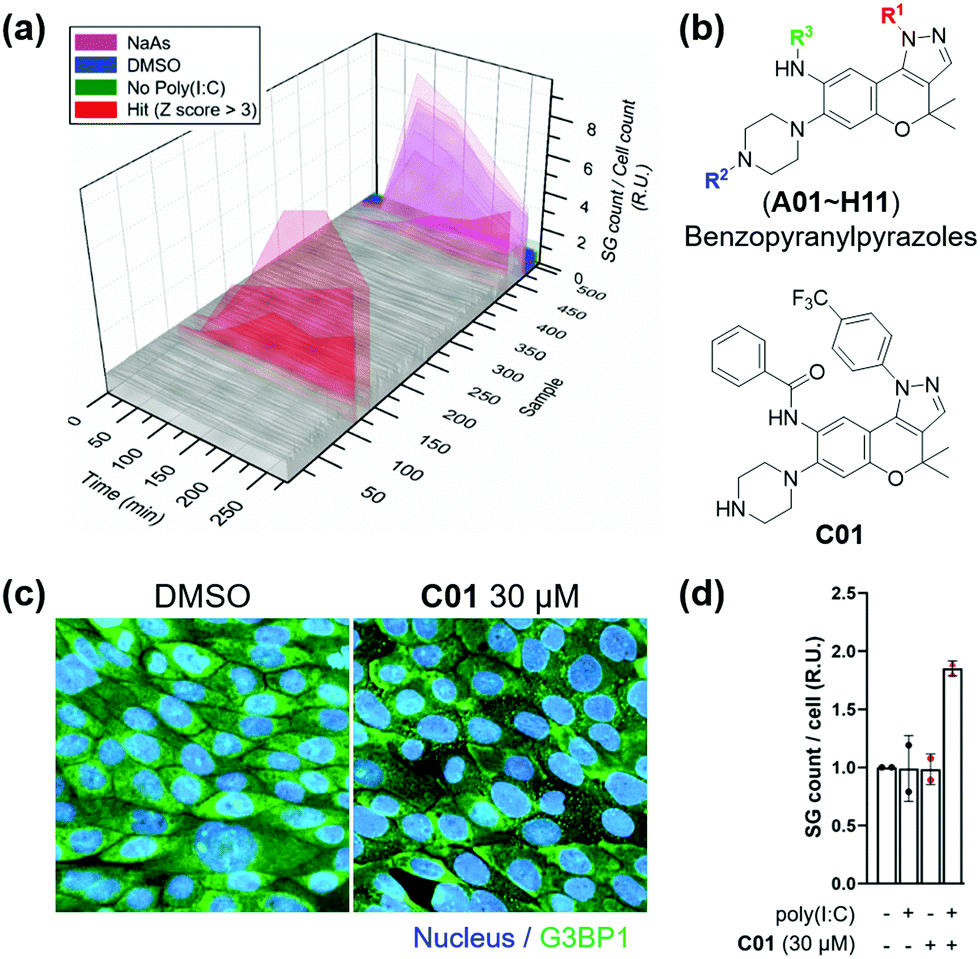 | ||
| Fig. 2 (a) Screening result of the time-course stress granule monitoring system. After transfection of G3BP1-GFP-expressed U2OS cells with 1.25 μg mL−1 poly(I:C), 10 μM compounds were treated. (b) Core structure of hit candidates and the chemical structure of optimized hit. (c) Immunofluorescence assay upon treatment of C01 in Calu-3 cells. (d) Poly(I:C)-dependent induction of stress granule formation of C01 in Calu-3 cell. 2.5 μg mL−1 poly(I:C) was treated. Data represent mean ± SD from two independent biological replicates. | ||
To improve the potency of initial hits, we performed a structure–activity relationship (SAR) study of benzopyranylpyrazole-cored compounds having novel core structures with better activities compared to tamoxifen derivatives. Through a systematic combination of substituted benzopyranyl β-keto aldehydes with various arylhydrazines, we synthesized over 100 analogues in a parallel process (Scheme S1, ESI†)24 and performed an image-based time-course evaluation of these analogues in a 96-well plate format. Several compounds consistently showed the induction of stress granule formation after 3 h, which is consistent with those from our initial screening. As shown in Fig. S5 (ESI†), the benzopyranylpyrazoles containing para-CF3-phenyl substituents were the key elements for the enhancement of cellular stress granules in the presence of poly(I:C), and analogues (C01–C11) containing various substituents with amide, urea, thiourea, and sulfonamide linkages at the R3 position generally showed good to moderate stress granule enhancement activities. Most analogues with different substituents at the R1 position did not show any activities at 5 μM concentration, and analogues containing the trifluoromethyl group at either the ortho or the meta position did not induce the stress granule formation (Fig. S6, ESI,†NI, NJ, and NK12). The modification of the secondary amine group of piperazine at the R2 position failed to enhance the stress granules and significantly decreased their solubility (Fig. 2b and Fig. S6, ESI,†NI01–NI11). Based on our SAR studies, we concluded that C01–C04 effectively enhance the cellular stress granules in the presence of poly(I:C), which led us to conduct further evaluation. To examine whether compounds induce stress granule formation in lung epithelial cells where SARS-CoV-2 causes atypical respiratory failure,2 we performed an immune-fluorescence assay for G3BP1 in Calu-3 human epithelial cells. C01, C02 and C04 increased the total number of stress granules, whereas C03 weakly induced stress granule formation (Fig. 2c and d and Fig. S7, ESI†). In a tetrazolium-based viability assay with Calu-3 cells (Fig. S8, ESI†), C04 showed a more cytotoxic effect (CC50 = 37.5 μM) compared to C01–C03 (CC50 > 60 μM). C01 and C02 also induced stress granule assembly in Vero kidney epithelial cells from the African green monkey, especially in poly(I:C)-transfected cells, commonly used for COVID-19 antiviral experiments (Fig. S9a, ESI†). In particular, C01 showed more specific induction of stress granule formation than C02 in both Calu-3 and Vero cells (Fig. S9b, ESI†). Based on these observations, we selected compound C01 for subsequent biological studies.
Having found that C01 can facilitate the formation of cellular stress granules under poly(I:C)-treated conditions, we then investigated the compound effects on the innate antiviral responses. The C01-only treatment showed a marginal impact on the PKR-eIF2α-IRF3 signaling pathway in Calu-3 cells, but C01 significantly activated this antiviral pathway in a dose-dependent manner and induced translation arrest, confirmed by the puromycin experiment, in the presence of poly(I:C), which mimics the viral infection (Fig. 3a). However, the negative analogue NI02, without enhancing the stress granule formation, did not activate antiviral responses. We then examined whether the antiviral efficacies of C01 were influenced by the quantity of treated dsRNA, and observed that C01-based antiviral responses were positively correlated with the concentration of treated poly(I:C) confirmed by western blotting of the PKR-eIF2α-IRF3 signaling pathway, indicating that C01 specifically upregulates innate antiviral responses in the presence of external dsRNA (Fig. 3b). Next, we performed a quantitative real-time PCR (qRT-PCR) to examine whether C01 treatment induced the cellular production of type I interferons and ISG56. As shown in Fig. 3c and d, C01 increased the cellular levels of IFNB1 and ISG56 in poly(I:C)-treated Calu-3 cells; however, both mRNA levels were not significantly changed in the absence of poly(I:C) or upon treatment of NI02, which is consistent with our previous image-based observations of stress granule formation. Therefore, we concluded that C01 inhibits viral replication by enhancing the formation of cellular stress granules and subsequent innate antiviral immune responses.
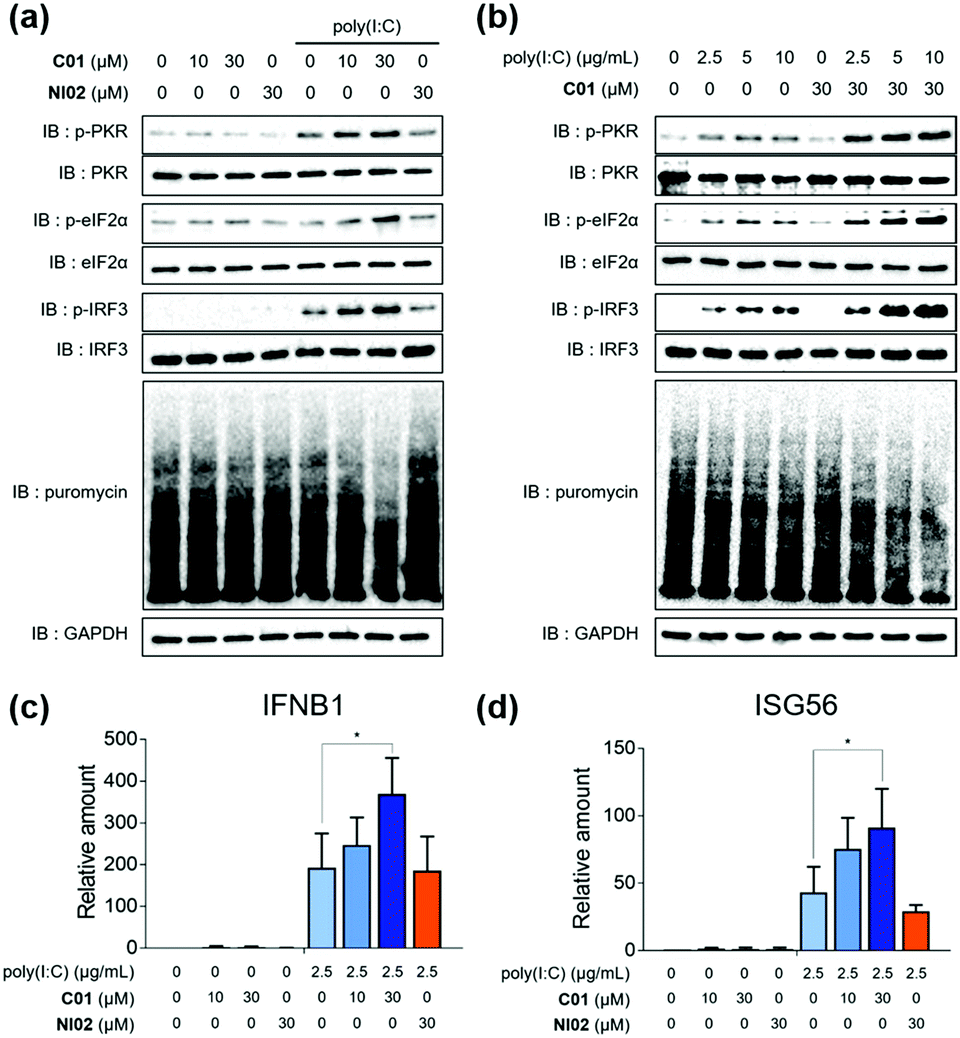 | ||
| Fig. 3 (a) Poly(I:C)-specific dose-dependent activation of the antiviral response. 2.5 μg mL−1 poly(I:C) was treated. (b) Poly(I:C)-dependent activation of the antiviral response. qPCR quantification of (c) IFNB1 and (d) ISG56. Calu-3 cells were used. Data represent mean ± SD from at least two independent biological replicates. *p <0.05 (Student's t test). | ||
Next, we investigated the antiviral effects of C01–C04 against SARS-CoV-2. Briefly, Calu-3 and Vero cells were infected with SARS-CoV-2 at multiplicity of infection (MOI) values of 0.2 and 0.0125, respectively. Then, the cells were treated with various concentrations of compounds. At 24 h post viral infection, immunofluorescence detection of the SARS-CoV-2 N protein and host cell nucleus was performed to quantitatively measure the inhibition of viral infection as well as cell viability. We observed that C01–C04 showed dose-dependent inhibition of SARS-CoV-2 replication (Fig. 4a and Fig. S10, ESI†). In particular, C01 showed the highest selective index (S.I.), the ratio of CC50 to IC50, in Calu-3 cells (Table S1, ESI†). On the other hand, the negative analogue NI02 failed to inhibit the replication of SARS-CoV-2 in both Calu-3 and Vero cells. To elucidate the inhibitory mechanism of the compounds, we performed an immunofluorescence assay to G3BP1 for SARS-CoV-2-infected cells. SARS-CoV-2 infection triggered weak stress granule formation in Vero cells, and sodium arsenite-induced stress granule formation was inhibited by SARS-CoV-2 (Fig. S11, ESI†). In contrast, the C01 and C02 treatments, but not the NI02 treatment, induce stress granule formation in the virus-infected cells, indicating that the stress granules induced by C01 and C02 activate innate immune responses against foreign viral dsRNA and block the viral translation (Fig. 4b and Fig. S12, ESI†). To specify the effective window of treatment concentration, which is limited by its cytotoxicity at high concentrations, we conducted a drug combination assay. In the presence of lopinavir (LPV), an antiviral reagent targeting viral proteases,19C01 showed significantly enhanced antiviral activity (IC50 from 7.64 to <0.78 μM) and selective index (from 2.09 to >17.91) against SARS-CoV-2 in Vero cells, whereas NI02 did not (Fig. 4c and Fig. S13, ESI†). These results suggested that the drug combination of our stress granule enhancers and other antiviral reagents with different modes of action can be a novel strategy for treating viral diseases including COVID-19.
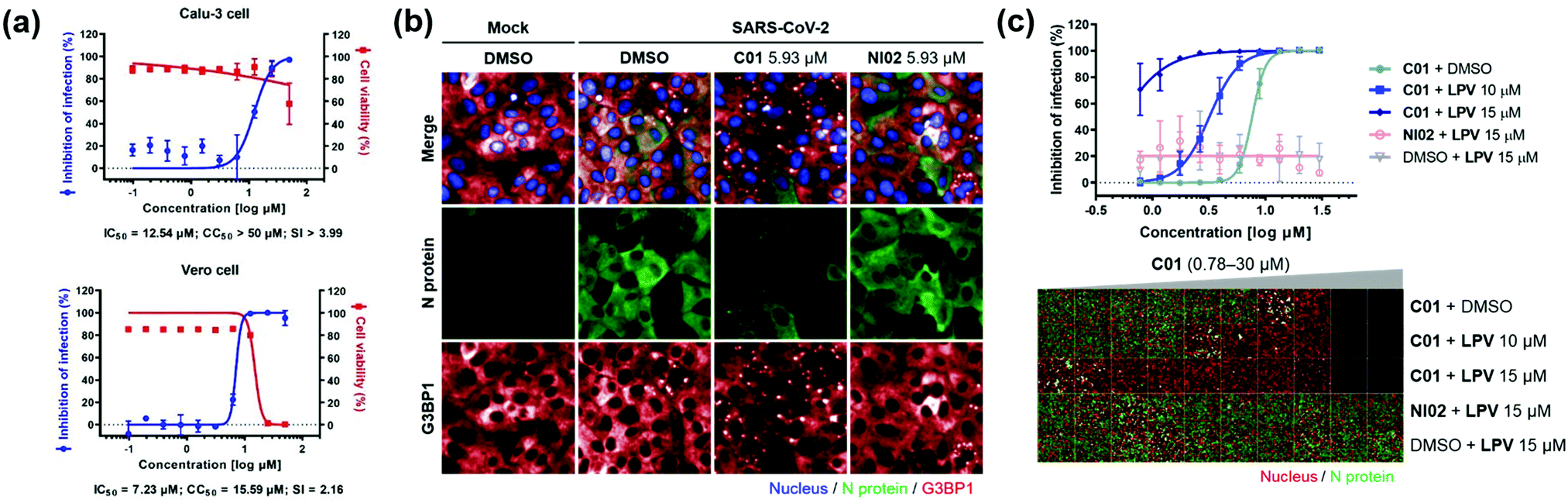 | ||
| Fig. 4 (a) Dose–response curves of C01 against SARS-CoV-2 in Calu-3 and Vero cells. (b) Co-immunofluorescence assay of C01 and NI02 (negative control) using viral N protein and G3BP1 antibodies in Vero cells. (c) Drug combination assay via co-treatment of C01 with lopinavir in Vero cells. See the ESI† for details. Data represent mean ± SD from two independent biological replicates. | ||
In summary, we discovered small-molecule stress granule enhancers by monitoring the time-course formation of cellular stress granules, cytoplasmic aggregates by multimolecular crowding of proteins and RNAs. To the best of our knowledge, this is the first dynamic screening of small molecules for stress granule regulators only in the presence of dsRNA and its potential application for antiviral disease treatment. We confirmed that our benzopyranylpyrazole-based hit compounds activated innate antiviral immune responses and increased the expression of type I interferons and ISGs in a dsRNA-dependent manner in various epithelial cells. Hit compounds also increased the quantity of stress granules in SARS-CoV-2-infected cells and inhibited the viral replication of SARS-CoV-2. A drug combination strategy with the FDA-approved drug, lopinavir, enhanced the antiviral effects of our hit compounds against SARS-CoV-2. Our host immune-targeted strategy harnessing the role of antiviral stress granules might overcome the evolution and mutation of SARS-CoV-2 variants, which render the existing vaccines and virus-targeting drugs ineffective.
This work was supported by the Creative Research Initiative Grant (2014R1A3A2030423 to S. B. P.) and the Bio & Medical Technology Development Program (2012M3A9C4048780 to S. B. P. and 2017M3A9G6068245 to S. K.) through the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science & ICT). The pathogen resource (NCCP43326) for this study was provided by the National Culture Collection for Pathogens.
Conflicts of interest
There are no conflicts to declare.Notes and references
- World Health Organization, Coronavirus (COVID-19) Dashboard, https://covid19.who.int/, accessed August 31, 2021.
- A. G. Harrison, T. Lin and P. Wang, Trends Immunol., 2020, 41, 1100 CrossRef CAS PubMed
.
- B. Giles, P. Meredith, S. Robson, G. Smith and A. Chauhan, Lancet Infect. Dis., 2021, 21, 1213 CrossRef CAS
- C. del Rio, P. N. Malani and S. B. Omer, JAMA, 2021, 326, 1001 CrossRef CAS PubMed
- W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut, S. J. Peacock and D. L. Robertson, Nat. Rev. Microbiol., 2021, 19, 409 CrossRef CAS PubMed
- P. Anderson and N. Kedersha, Nat. Rev. Mol. Cell Biol., 2009, 10, 430 CrossRef CAS PubMed
- P. Anderson and N. Kedersha, Trends Biochem. Sci., 2008, 33, 141 CrossRef CAS PubMed
- A. Marcelo, R. Koppenol, L. P. de Almeida, C. A. Matos and C. Nóbrega, Cell Death Dis., 2021, 12, 592 CrossRef CAS PubMed
- P. Yang, C. Mathieu, R. M. Kolaitis, P. Zhang, J. Messing, U. Yurtsever, Z. Yang, J. Wu, Y. Li, Q. Pan, J. Yu, E. W. Martin, T. Mittag, H. J. Kim and J. P. Taylor, Cell, 2020, 181, 325 CrossRef CAS PubMed
- T. Matheny, B. V. Treeck, T. N. Huynh and R. Parker, RNA, 2021, 27, 174 CrossRef CAS PubMed
- S. J. DeWitte-Orr and K. L. Mossman, Future Virol., 2010, 5, 325 CrossRef CAS
- C. McCormick and D. A. Khaperskyy, Nat. Rev. Immunol., 2017, 17, 647 CrossRef CAS PubMed
- P. D. Slaine, M. Kleer, N. K. Smith, D. A. Khaperskyy and C. McCormick, Viruses, 2017, 9, 388 CrossRef PubMed
- L. C. Reineke and R. E. Lloyd, J. Virol., 2015, 89, 2575 CrossRef PubMed
- N. Eiermann, K. Haneke, Z. Sun, G. Stoecklin and A. Ruggieri, Viruses, 2020, 12, 984 CrossRef CAS PubMed
- D. Campos-Melo, Z. C. E. Hawley, C. A. Droppelmann and M. J. Strong, Front. Cell Dev. Biol., 2021, 9, 621779 CrossRef PubMed
- H. H. Rabouw, M. A. Langereis, R. C. Knaap, T. J. Dalebout, J. Canton, I. Sola, L. Enjuanes, P. J. Bredenbeek, M. Kikkert, R. J. de Groot and F. J. van Kuppeveld, PLoS Pathog., 2016, 12, e1005982 CrossRef PubMed
- B. Gao, X. Gong, S. Fang, W. Weng, H. Wang, H. Chu, Y. Sun, C. Meng, L. Tan, C. Song, X. Qiu, W. Liu, M. Forlenza, C. Ding and Y. Liao, PLoS Pathog., 2021, 17, e1008690 CrossRef CAS PubMed
- S. Alhumaid, A. A. Mutair, Z. A. Alawi, N. Alhmeed, A. R. Z. Zaidi and M. Tobaiqy, Trop. Med. Infect. Dis., 2020, 5, 180 CrossRef PubMed
- A. P. Sfakianos, L. E. Mellor, Y. F. Pang, P. Kritsiligkou, H. Needs, H. Abou-Hamdan, L. Desaubry, G. B. Poulin, M. P. Ashe and A. J. Whitmarsh, Cell Death Differ., 2018, 25, 1766 CrossRef CAS PubMed
- M. G. Thomas, L. J. Martinez Tosar, M. A. Desbats, C. C. Leishman and G. L. Boccaccio, J. Cell Sci., 2009, 122, 563 CrossRef CAS PubMed
- G. Clavarino, N. Cláudio, T. Couderc, A. Dalet, D. Judith, V. Camosseto, E. K. Schmidt, T. Wenger, M. Lecuit, E. Gatti and P. Pierre, PLoS Pathog., 2012, 8, e1002708 CrossRef CAS PubMed
- J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629 CrossRef CAS PubMed
- S. O. Park, J. Kim, M. Koh and S. B. Park, J. Comb. Chem., 2009, 11, 315 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, bioassay protocols, additional figures and the characterization of all new compounds. See DOI: 10.1039/d1cc05508a |
| This journal is © The Royal Society of Chemistry 2021 |