
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective and inducible acylation of thrombin using aptamer-catalyst conjugates†
Jordi F.
Keijzer
 ,
Judith
Firet
and
Bauke
Albada
*
,
Judith
Firet
and
Bauke
Albada
*
Laboratory of Organic Chemistry, Wageningen University & Research, Stippeneng 4, 6807 WE, Wageningen, The Netherlands. E-mail: bauke.albada@wur.nl
First published on 10th November 2021
Abstract
Two acyl-transfer catalysts were conjugated to thrombin-binding DNA aptamers to acylate thrombin. Modification occurred site-selectively on Lys (≫Ser) residues proximal to the respective aptamer–thrombin interface, was selective for thrombin in the presence of other proteins, and the activity of both DNA-catalysts could be controlled by an external trigger.
The artificial chemical modification of proteins has led to new insights in protein function and novel applications of this important class of biomolecules. Examples of the latter include therapeutics, where antibody conjugates are used to carry toxins to specific tissues,1–3 and of the former include chemical biology approaches where fluorescently labelled proteins are used to track their localization and movement within cells.4,5 Currently, a remaining challenge is, however, the site-selective modification of native proteins. Although conditions have been established to derivatize specific residues in some proteins,6–8 tedious optimization is often required when applied to different proteins. Alternatively, catalytic modification of reactive residues9,10 has resulted in various degrees of selective protein modification, especially when guided by protein-binding ligands.11,12 Theoretically, this principle can also be applied using protein-binding aptamers,13–16 but this has not yet been shown. This is unfortunate as aptamers – which are nucleotide-based receptor molecules that bind a range of specific targets from small molecules to proteins17–19 – are not only conveniently synthesized20,21 and functionalized,22 but site-specifically bind to a protein of interest,18 and can easily be included in more complex systems23–25 that, for example, facilitate ON/OFF switchable characteristics.26,27 As such, aptamers offer a potentially rich platform for the site-selective modification of native proteins (Fig. 1).
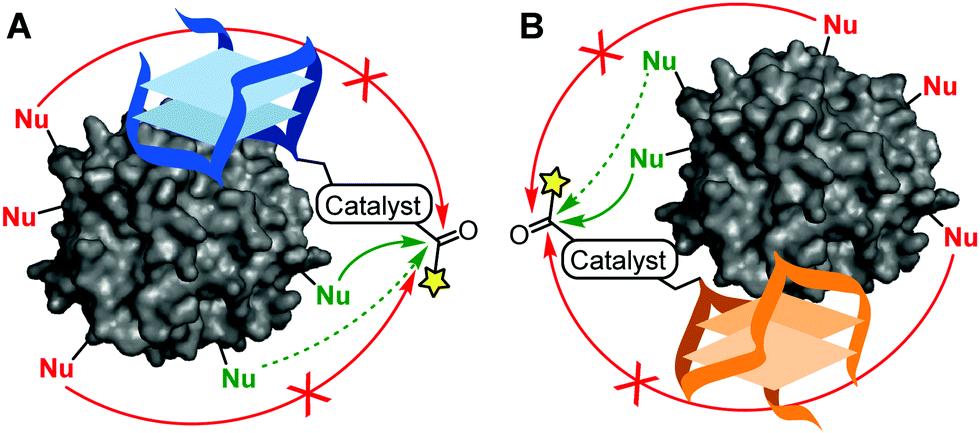 | ||
| Fig. 1 Schematic depiction of the application of DNA aptamers TBA (A) and TBA2 (B) for human α-thrombin as scaffolds for the site-selective acylation of proximal nucleophilic residues on the protein surface. Different sites of the same protein can be modified as both aptamers bind at opposite sides. | ||
In this work, we show how catalyst-functionalized aptamers can be used for the selective modification of a native protein, including its ON/OFF switchable activities. Specifically, the acyl-transfer catalysts DMAP28,29 or PyOx12 were tethered at various positions to two thrombin-binding aptamers (TBA and TBA2, 15 and 26 nucleotides, respectively; see Fig. 1A and B for a schematic of their interactions with thrombin) and we assessed the acylation of human α-thrombin, a 36 kDa serine protease.
Our first set of catalytic DNA constructs consisted of a DMAP catalyst attached to various sites on TBA. For this, a bicyclononynylmethyl-functionalized dimethylaminopyridine derivative (BCN-DMAP) was synthesized and covalently attached to an azide-functionalized thymine at one of all seven thymine residues in the TBA sequence (Fig. S1, ESI†)30,31 by means of strain-promoted azide–alkyne cycloaddition (SPAAC) chemistry. Subsequent protein modification experiments were performed by incubating human α-thrombin with one of the various TBA-DMAP constructs for 1 h at 37 °C, after which azido-thioester 1 (Fig. 2A and Scheme S1, ESI†) was added (final concentration: 150 μM). After the modification reaction and addition of an excess of BCN-PEG2000, the modification was analysed on SDS-PAGE. The various degrees of thrombin modification that we observed showed that different TBA-DMAP constructs led to different enhanced levels of single modification, with TBA3-DMAP and TBA12-DMAP as the best performing constructs with conversions of 27% and up to 49%, respectively (Table S1, ESI†). Inspection of the single crystal X-ray structure of the TBA–thrombin complex (PDB-code: 5EW1)31 confirmed that these two positions of the TBA are indeed closest to the protein surface (Fig. 3A and B). Kinetics analysis of TBA12-DMAP indicated that after 2 h the acylation reaction was mostly complete (Fig. S2, ESI†). Subsequent tryptic digestion and analysis by LC-MS/MS of azide-modified thrombin revealed that the aptamer-assisted single modification was performed in a site-selective manner on Lys residues proximal to the respective DMAP-containing thymine bases (Fig. 3A and B). Our observation that not all residues in proximity to the modified base are acylated, e.g., K77 (Fig. 3A) and K17 of the light chain (LC) (Fig. 3B), while apparently more remote residues are acylated, e.g., K154 (Fig. 3A) and K106 and K107 (Fig. 3B), confirms that the protein–aptamer interaction is quite dynamic and that subtle interactions influence the site-selectivity.16
 | ||
| Fig. 2 (A) DNA-tethered DMAP accepts thioesters 1 and 2 as substrates, whereas PyOx accepts alkylated N-acyl-N-sulfonamides 3 and 4. Both catalysts form a charged intermediate from which the acyl is transferred to a nearby nucleophile on the protein. (B and C) Details of the divalent catalyst species for (B) DMAP and (C) PyOx. | ||
 | ||
| Fig. 3 Site-selective modification of thrombin by catalyst–aptamer conjugates. The color of the residues that are modified matches the color that marks the catalyst position; the aptamer is shown in stick (blue for TBA in panels A, B, D and E, and orange for TBA2 in panel F), the base that is modified with the catalyst is shown in balls. (A and B) Thrombin residues modified with azido-thioester 1 by (A) TBA3-DMAP or (B) TBA12-DMAP in red. (C) SDS-PAGE analysis and visualization of the selectivity of TBA3-DMAP (T3D) and TBA12-DMAP (T12D) for thrombin in a selectivity assay. Proteins modified with alkyne-thioester 2 are coupled with azido-lissamine by means of CuAAC. (D) Single crystal X-ray structure of thrombin with both TBA and TBA2 as well as exposed Lys (green) and Ser (pink) residues (PDB-code: 5EW131). (E and F) Thrombin residues modified with azido-ANANS 3 by (E) TBA12-diPyOx or (F) TBA217-diPyOx and TBA223-diPyOx in yellow. Images were generated using PyMol. | ||
In order to benefit from the affinity of an aptamer for its target, we incubated a mixture of proteins (including thrombin) with TBA3-DMAP or TBA12-DMAP and exposed it to alkyne-thioester 2 (Fig. 2A). Modified proteins were visualized by click conjugation to azido-lissamine by means of the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC). As only fluorescently labelled thrombin was observed and none of the other proteins was modified (Fig. 3C), we conclude that, indeed, DMAP–aptamer conjugates specifically modify its target protein. As the high reactivity of thioesters themselves led to high background labelling (up to 20% with 300 μM 1), we synthesized divalent DMAP catalyst constructs (Fig. 2B) that could increase the acyl transfer rate by doubling the amount of activated acyl groups near the protein surface.32 Regrettably, even a divalent DMAP catalyst provided only marginally improved conversions (Table S2, ESI†). Therefore, we switched to an acyl transfer catalyst that is more nucleophilic than DMAP and can activate less reactive alkylated N-acyl-N-sulfonamide (ANANS) substrates, i.e., 4-pyridinecarbaldehyde oxime (PyOx, Fig. 2A).12 For this study, we focused on TBA positions that gave the best results in the TBA-DMAP constructs, i.e., positions 3 and 12. Thus, after synthesis of alkyne-functionalized PyOx, CuAAC with TBA3-N3 or TBA12-N3 generated constructs TBA3-PyOx and TBA12-PyOx. Azido or alkyne-functionalized ANANS derivatives 3 and 4 were also synthesized (Scheme S2, ESI†). Although exposure of thrombin to TBA3-PyOx or TBA12-PyOx and ANANS 3 under the same conditions as for TBA-DMAP revealed lower conversions, the absence of detectable background at pH 7.2 enabled us to double the substrate concentration and triple the reaction time (Fig. S3, ESI†). This resulted in 18% and 28% conversion to singly-modified thrombin for TBA3-PyOx and TBA12-PyOx (at pH 7.2), and 29% and 38%, respectively, at pH 8.0. LC-MS/MS analysis of tryptic digests of thrombin modified with azido-functionalized ANANS 3 revealed, again, site-selective acylation around the TBA-bound PyOx catalyst (Table S3, ESI†). Although overlap was observed with corresponding TBA-DMAP constructs (i.e., acylation of K21, K77, K106, K107, K154 and K18LC), modification of additional Lys residues was also observed (i.e., K83, K174 and K23LC) (Fig. 3E). To our surprise, proximal serine residues were also modified, e.g., Ser22 and Ser158 by TBA12-PyOx (Fig. 3E).
To achieve higher conversions for the modification, we also designed a divalent PyOx catalyst by mono-azidation of 1,3,5-tri(bromomethyl)benzene and subsequent attachment of PyOx to the resulting 1-(azidomethyl)-3,5-di(bromomethyl)benzene moiety, to yield azido-diPyOx (Fig. 2C and Scheme S3, ESI†). After CuAAC conjugation of azido-diPyOx to position 3 or 12 on alkyne-functionalized TBA, TBA3-diPyOx and TBA12-diPyOx were obtained (Scheme S4, ESI†). Upon testing with 300 μM of azido-ANANS 3 we found that these diPyOx constructs were much more efficient in the modification of thrombin, reaching >90% (at pH 7.2; Table S3, ESI†). That this higher percentage also entailed larger quantities of multi-modified protein (specifically: 28% single, 30% double, 22% triple and 11% quadruple modified thrombin) provides evidence that a higher concentration of activated PyOx catalyst is located at the surface of the protein. Interestingly, TBA3,12-bis(diPyOx), which contains two diPyOx catalysts at different positions, was not more active (Table S3, ESI†). Tryptic digestion analysis of thrombin modified at pH 8.0 showed that, when compared to monovalent TBA12-PyOx, divalent TBA12-diPyOx modified additional residues K196 and K23LC, while at the same time omitting modification of residues S22 and K83 (Fig. 3E). Larger differences between PyOx and diPyOx functionalized aptamers were observed at pH 7.2, with 3.9–5.2-fold enhancement when placed on TBA3 and 2.7–3.3-fold enhancement when placed on TBA12. Apparently, the diPyOx catalyst generates a higher concentration of reactive acyl-catalyst complex in proximity to the protein surface than the DMAP catalyst (Fig. 2B, see also Table S2, ESI†). As was the case for the DMAP conjugates, TBA12-diPyOx also only modified thrombin in competitive assays (Fig. S4, ESI†). LC-MS analysis of TBA12-diPyOx in the presence of ANANS 3 revealed mono- and di-acylated constructs as well as dehydrated catalyst in the absence of thrombin (Fig. S51 and S52, ESI†). Apparently, an acylated PyOx catalyst is formed that dehydrates in the absence of a protein substrate; self-acylation of the DNA was not observed.
Now that we identified a catalyst system that yields high conversions, we implemented another thrombin-binding aptamer (TBA2) in order to pursue the modification of the other side of thrombin. Indeed, the TBA2-diPyOx catalysts modified thrombin at residues positioned in close proximity to the respective positions of the catalyst (Fig. 3F), even though the conversions of 20–27% were substantially lower than for the other aptamer (Tables S4, ESI†). Nonetheless, our results show that different sides of the protein can be subjected to modification using different protein-binding aptamers. That direct conjugation of the catalyst to the protein binding aptamer is required for modification is shown by the absence of modification when the catalyst was tethered to a template strand that could hybridize with either the TBA or TBA2 aptamer that was extended with its complementary sequence (Fig. S5, ESI†). Conversions of TBA12-diPyOx and TBA217-diPyOx that were studied in the presence of native TBA and TBA2, revealed that aptamer affinity after catalyst functionalization is similar to their native counterparts (Fig. S6, ESI†). Additionally, TBA12-diPyOx conversion was unaffected by the presence of TBA2, neither was the activity of TBA217-diPyOx hampered by TBA.
In our ambition to imitate the ability of enzymes to not only site-selectively modify proteins, but also do this in a trigger-dependent manner, we incorporate an activity-control switch in our catalytic constructs. For this, we designed an ssDNA OFF-strand that could hybridize with our TBA-catalyst constructs to form a catalytically inactive dsDNA duplex as the dsDNA is unable to form the G-quadruplex structure that is essential to thrombin binding (Fig. 4A). Indeed, upon addition of this complementary ssDNA OFF-strand, the system turned to its OFF state as no protein modification was detected (Fig. 4B and Fig. S7, ESI†). After removing the OFF-strand by addition of a second DNA strand that is fully complementary to the OFF strand (including its toeholds), the original TBA is reformed and the modification of thrombin is again efficient. We could use this switch to successfully control the activity of TBA12-DMAP, TBA12-PyOx and TBA12-diPyOx, and the activity of the latter could be regulated up to at least three full cycles (Fig. 4C). The switch also worked in situ, where a 4 h reaction generated the same conversion as a system that was in OFF modus for 4 of the 8 h, even with different ON–OFF patterns, i.e., 2–4–2 h ON–OFF–ON or 2–2–2–2 h ON–OFF–ON–OFF (Fig. S8, ESI†).
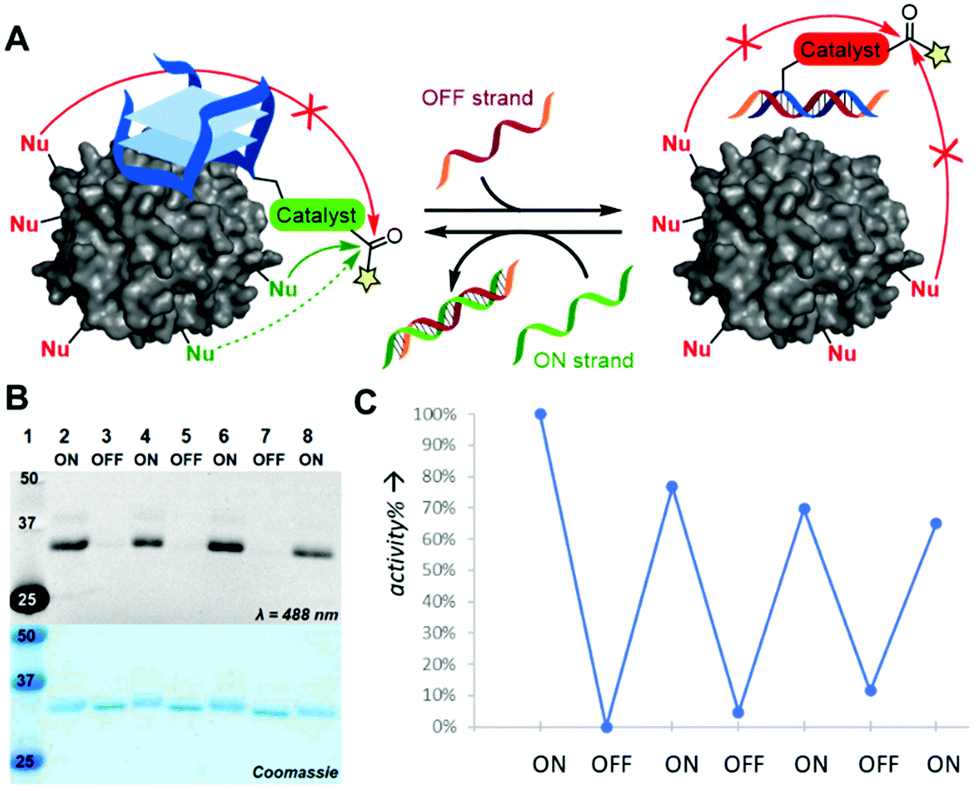 | ||
| Fig. 4 Activity control of TBA-catalyst constructs. (A) Schematic depiction of the activity switch where ssDNA OFF or ON strands suppress or regenerate, respectively, the protein-binding ability of the aptamer. In the OFF-state, the construct should not modify the protein. (B) SDS-PAGE analysis and visualization of the activity switch of TBA12-diPyOx after CuAAC modification of thrombin-bound alkyne-ANANS 4 with azido-lissamine. (C) The activity of TBA12-diPyOx could be switched OFF and ON for three cycles (quantities were determined from SDS-PAGE after modification with azido-ANANS 3 and BCN-PEG2000 using ImageJ software). | ||
In conclusion, we present the first DNA-based catalyst for the site-selective modification of a specific protein using its affinity for an aptamer. We show that both DMAP and PyOx can be used as acyl transfer catalysts, although the latter leads to significantly higher conversions and uses a less reactive substrate that supresses uncatalyzed labelling. Furthermore, we show that nucleobases 3 and 12 of the TBA are the best sites for catalyst conjugation, and that nucleobase 17 is the best for TBA2. Both TBA and TBA2 catalytic constructs modified Lys and, to a lesser extent, Ser residues that are in proximity to the catalyst. Importantly, the activity of our DNA-based catalysts could be repeatedly regulated by an external stimulus. As protein modification efficiency, substrate specificity, site-selectivity, and external control over the activity are four essential features that define enzymatic protein modification,1 our DNA-catalyst constructs are biomimetics of naturally occurring acylating enzymes. In view of the existence of aptamers for other proteins,13 we expect that our methodology will become applicable for other proteins in the future, and will be a valuable addition to the protein acylation toolbox.
J. F. K. and B. A. conceived the project, J. F. K. performed the experiments and analysis, J. F. K. and J. F. synthesized the various small molecules, J. F. K. and B. A. wrote the manuscript.
We thank Frank Claassen for his assistance on the LC-MS analysis. This work is funded by an ECHO grant from the Dutch Organization for Scientific Research (NWO) (project number 711.017.004).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. J. Bruins, D. Blanco-Ania, V. van der Doef, F. L. van Delft and B. Albada, Chem. Commun., 2018, 54, 7338–7341 RSC.
- T. Wu, M. Liu, H. Huang, Y. Sheng, H. Xiao and Y. Liu, Chem. Commun., 2020, 56, 9344–9347 RSC.
- J. J. Bruins, C. van de Wouw, K. Wagner, L. Bartels, B. Albada and F. L. van Delft, ACS Omega, 2019, 4, 11801–11807 CrossRef CAS.
- M. A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58(34), 11625–11630 CrossRef CAS PubMed.
- Y. Song, F. Xiong, J. Peng, Y. M. E. Fung, Y. Huang and X. Li, Chem. Commun., 2020, 56, 6134–6137 RSC.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- D. Hwang, K. Tsuji, H. Park, T. R. Burke and C. Rader, Bioconjugate Chem., 2019, 30, 2889–2896 CrossRef CAS.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripahti, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS.
- C. A. Pérez Rízquez, O. Abian and J. M. Palomo, Chem. Commun., 2019, 55(86), 12928–12931 RSC.
- J. F. Keijzer and B. Albada, Bioconjugate Chem., 2020, 31, 2283–2287 CrossRef CAS.
- S. Tsukiji, M. Miyagawa, T. Ogawa, Y. Koshi, I. Hamachi and E. Nakata, J. Am. Chem. Soc., 2007, 130, 245–251 Search PubMed.
- T. Tamura, Z. Song, K. Amaike, S. Lee, S. Yin, S. Kiyonaka and I. Hamachi, J. Am. Chem. Soc., 2017, 139, 14181–14191 CrossRef CAS.
- J. L. Vinkenborg, G. Mayer and M. Famulok, Angew. Chem., Int. Ed., 2012, 51, 9176–9180 CrossRef CAS PubMed.
- L. Zhang, X. Fang, X. Liu, H. Ou, H. Zhang, J. Wang, Q. Li, H. Cheng, W. Zhang and Z. Luo, Chem. Commun., 2020, 15, 1–19 Search PubMed.
- M. Kohlberger, S. Wildner, C. Regl, C. G. Huber and G. Gadermaier, PLoS One, 2017, 114, 2898–2903 Search PubMed.
- F. Rohrbach, F. Schäfer, M. A. H. Fichte, F. Pfeiffer, J. Müller, B. Pçtzsch, A. Heckel and G. Mayer, Angew. Chem., Int. Ed., 2013, 52, 11912–11915 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- S. Klussmann, The Aptamer Handbook: Functional Oligonucleotides and Their Applications, 2006 Search PubMed.
- A. A. Bastian, A. Marcozzi and A. Herrmann, Nat. Chem., 2012, 4, 789–793 CrossRef CAS PubMed.
- I. Russo Krauss, A. Merlino, A. Randazzo, E. Novellino, L. Mazzarella and F. Sica, Nucleic Acids Res., 2012, 40, 8119–8128 CrossRef CAS PubMed.
- F. X. Montserrat, A. Grandas, R. Eritja and E. Pedroso, Tetrahedron, 1994, 50, 2617–2622 CrossRef CAS.
- M. S. Kupryushkin, M. D. Nekrasov, D. A. Stetsenko and D. V. Pyshnyi, Org. Lett., 2014, 16, 2842–2845 CrossRef CAS PubMed.
- N. C. Seeman, Annu. Rev. Biophys. Biomol. Struct., 2002, 27, 225–248 CrossRef PubMed.
- S. Wintermans, J. F. Keijzer, M. Dros, H. Zuilhof and B. Albada, ChemCatChem, 2021, 13, 4618–4624 CrossRef CAS.
- T. A. Ngo, H. Dinh, T. M. Nguyen, F. F. Liew, E. Nakata and T. Morii, Chem. Commun., 2019, 55, 12428–12446 RSC.
- B. Albada, E. Golub and I. Willner, Chem. Sci., 2016, 7, 3092–3101 RSC.
- T. Li, S. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 13156–13157 CrossRef CAS PubMed.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569–583 CrossRef.
- M. S. Xie, B. Huang, N. Li, Y. Tian, X. X. Wu, Y. Deng, G. R. Qu and H. M. Guo, J. Am. Chem. Soc., 2020, 142, 19226–19238 CrossRef CAS PubMed.
- D. M. Tasset, M. F. Kubik and W. Steiner, J. Mol. Biol., 1997, 272, 688–698 CrossRef CAS PubMed.
- A. Pica, I. R. Krauss, V. Parente, H. Tateishi-Karimata, S. Nagatoishi, K. Tsumoto, N. Sugimoto and F. Sica, Nucleic Acids Res., 2017, 45, 461–469 CrossRef CAS PubMed.
- K. Shiraiwa, R. Cheng, H. Nonaka, T. Tamura and I. Hamachi, Cell Chem. Biol., 2020, 27, 970–985 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, synthesis of DNA constructs, details of the protein modification studies. See DOI: 10.1039/d1cc05446e |
| This journal is © The Royal Society of Chemistry 2021 |