
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlumination of aryl methyl ethers: switching between sp2 and sp3 C–O bond functionalisation with Pd-catalysis†
Ryan K.
Brown‡
a,
Thomas N.
Hooper‡
 a,
Feriel
Rekhroukh‡
b,
Andrew J. P.
White
a,
Paulo J.
Costa
b and
Mark R.
Crimmin
*a
a,
Feriel
Rekhroukh‡
b,
Andrew J. P.
White
a,
Paulo J.
Costa
b and
Mark R.
Crimmin
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
bBioISI – Biosystems & Integrative Sciences Institute, Faculty of Sciences, University of Lisboa, 1749-016 Lisboa, Portugal
First published on 12th October 2021
Abstract
The reaction of [{(ArNCMe)2CH}Al] (Ar = 2,6-di-iso-propylphenyl) with aryl methyl ethers proceeded with alumination of the sp3 C–O bond. The selectivity of this reaction could be switched by inclusion of a catalyst. In the presence of [Pd(PCy3)2], chemoselective sp2 C–O bond functionalisation was observed. Kinetic isotope experiments and DFT calculations support a catalytic pathway involving the ligand-assisted oxidative addition of the sp2 C–O bond to a Pd–Al intermetallic complex.
Lignin from lignocellulosic biomass is derived from a radical polymerisation of monolignols (p-coumaryl, coniferyl, and sinapyl alcohol) and its composition varies from genus to genus.1–3 The depolymerization of lignin has gained significant attention over the last 10 years, and while there is a breadth of research in this area, a clear finding of this work is that the products of depolymerization are often structurally related to monolignols themselves.4 Many of these depolymerisation products contain aryl methyl ether moieties.5–7 One example of a product made directly from lignin is vanillin, which is widely used in commercial applications (Fig. 1).8
 | ||
| Fig. 1 Monolignols (building blocks for lignin) along with vanillin. | ||
Despite the importance of aryl methyl ethers as sustainable chemical building blocks, reactions that break the strong C–O bonds in these substrates are rare,9 those which form reactive fragments amenable for synthetic diversification are rarer still. For example, nickel catalysed silylation reactions of sp2 C–O bonds have been pioneered by Martin and coworkers with initial reactions focussing on silylation of aryl and benzyl pivalates with a silyl-borane reagent.10,11 This was later expanded to include aryl, benzyl and vinyl ethers.12 The nickel catalysed stannylation of C–O bonds of aryl esters with a silyl stannane has also been reported using a similar protocol.13 Related reactions with boron reagents are known. Chatani and coworkers have reported the Rh catalysed borylation of pyridyl ethers,14 while Martin and co-workers have used diboranes to borylate aryl and vinyl ethers in the presence of a nickel catalyst.15 Very recently, Nakao and coworkers reported the borylation of methyl aryl ethers using a heterometallic Rh–Al complex.16 Despite these important examples, most studies to date have focused on derivatives of phenol, in the case of aryl methyl ethers the substrate of choice is anisole. Application of this type of methodology to electron-rich and ortho-substituted motifs, such as 1,2-dimethoxybenzene, remains uncommon.
Monolignol derivatives are structurally complex and potentially contain multiple adjacent sp2 C–O and sp3 C–O bonds in the same molecule. A particular challenge in this area of research is controlling chemoselectivity. The bond dissociation energies of the sp2 C–O bond of phenyl methyl ether is 101 ± 1 kcal mol−1 while the sp3 C–O bond of dimethyl ether is 83.2 ± 0.9 kcal mol−1.17 In this paper, we report a method for the transformation of C–O bonds of aryl methyl ethers into C–Al bonds. The work builds on an established program from our lab focusing on main group reagents for C–O bond activation.18–20 We show that the chemoselectivity of the reaction can be precisely controlled through inclusion or exclusion of a homogeneous palladium catalyst, allowing a tuneable approach to either sp3 C–O bond activation or sp2 C–O bond activation of aryl methyl ethers.
Anisole and derivatives thereof can be functionalised by reaction with the aluminium reagent 1 (Scheme 1a).21,22 The non-catalysed reaction of an excess of anisole (2a, 100 equiv. or neat) with 1 resulted in cleavage of the sp3 C–O bond and formation of new Al–CH3 and Al–OPh functional groups. The reaction is entirely chemoselective and a large excess of anisole is required.23 Reaction of 1 with 1,2-dimethoxybenzene (2b) also resulted in the chemoselective alumination of the sp3 C–O bond. In contrast, the reaction of 1 with the methyl protected vanillyl alcohol 2c (derived from vanillin) resulted in the selective reaction of a dialkyl ether moiety to form 3c, despite the presence of two distinct aryl methyl ether groups. 3a and 3b show peaks in the 1H NMR spectrum at δ = −0.68 (s, 3H) and −0.65 ppm (s, 3H) respectively. These high-field resonances are characteristic of Al–CH3 groups. In contrast, the benzylic CH2 group of 3c is considerably more deshielded and resonates at δ = 1.46 ppm in the 1H NMR spectrum. 3a and 3c have also been characterised by single crystal X-ray diffraction (Fig. 2).
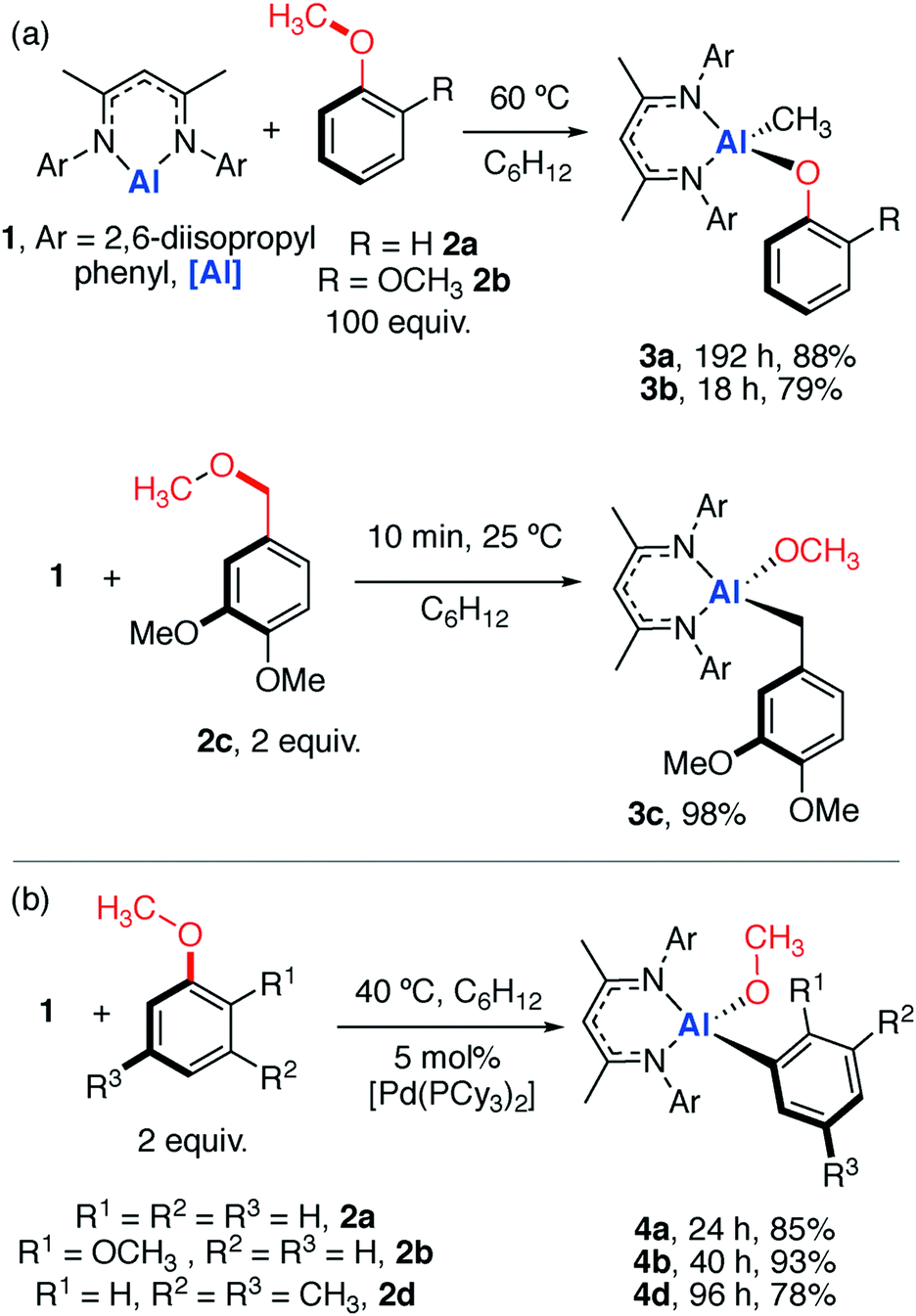 | ||
| Scheme 1 (a) Non-catalysed and (b) catalysed reactions of 1 with aryl methyl ethers. | ||
 | ||
| Fig. 2 Structures of 3a, 3c, 4a, and 4b determined by single crystal X-ray diffraction studies. Hydrogen atoms and components of disorder omitted for clarity. 50% probability ellipsoids. Bond lengths reported in Å. | ||
The non-catalysed reactions of 1 with aryl methyl ethers are consistent with established reactivity patterns of these functional groups. Direct attack of nucleophiles on the CH3 moiety can occur due to the ability of OPh− to act as a leaving group. Typically, an SN2 pathway is invoked to explain this behaviour. Recently a number of aluminyl anions (aluminium based nucleophiles) related to 1 have been shown to effect sp3 C–O bond alumination of aryl methyl ethers.24,25
Notable changes in selectivity and reaction times were observed when the reactions were conducted in the presence of a catalyst. Addition of [Pd(PCy3)2] (5 mol%) to a mixture of 1 and 2 equiv. of anisole (2a) resulted in sp2 C–O bond functionalisation to form 4a (Scheme 1b).§ Similarly, 1,2-dimethoxybenzene and 1-methoxy-3,5-dimethylbenzene resulted in clean catalytic reactions with 1 to form 4b and 4d respectively. The catalytic protocol occurs under milder conditions than the non-catalysed (40 vs. 60 °C) and proceeds with high chemoselectivity for the sp2 C–O bond of the substrate. 4a–b and 4d were characterised by distinct resonances at δ = 3.38 to 3.75 ppm in the 1H NMR spectrum assigned to the –OCH3 group derived from C–O bond functionalisation. To unambiguously determine their structures 4a–b and 4d were also characterised by single crystal X-ray diffraction (Fig. 2). The ortho–OCH3 group in 4b shows a close contact with aluminium centre; the Al–O2 bond length is ∼3.0 Å and within the sum of the van der Waals radii (3.36 Å).26
We have previously shown the combination of 1 with catalytic [Pd(PCy3)2] is able to achieve the C–H, C–O and C–F alumination of unactivated arenes, furans, and fluoroarenes under mild conditions. In certain cases, remarkable changes in selectivity have been observed between catalytic and non-catalytic protocols.19,20,27–29 Others have shown that modification of reagents or conditions can result in switches in selectivity between the functionalisation of sp2 and sp3 C–O bonds with Ni-based catalysts.15,30
To shed further light on the mechanisms of C–O bond activation and the switch in selectivity, further experiments and calculations were undertaken. The non-catalysed reaction of 1 with 2a was presumed to occur by an SN2 mechanism in which 1 acts as an aluminium-centered nucleophile.31,32 Under pseudo-first order conditions (excess anisole), kinetics data could be fitted to 1st order in [1] across a series of initial concentrations. Competition experiments between anisole (2a), 1,2-dimethoxybenzene (2b) and 1 revealed that the latter substrate reacts fastest. This finding is consistent with the established Lewis-acidity of 1 and indicates the ortho-methoxy moiety can act as a directing group. DFT calculations suggest that the SN2 pathway is not only plausible, but also potentially facilitated by general base catalysis and/or Lewis-acid catalysis (see ESI† for further details).
Given the switch in selectivity under catalytic conditions, it is highly likely an alternative mechanism is in operation in the presence of [Pd(PCy3)2]. A series of plausible pathways for the reaction of [Pd(1)2]28,29 were investigated by DFT calculations. The lowest energy calculated pathway involves the ligand-assisted oxidative addition of the sp2 C–O bond of anisole to [Pd(1)2] and leads directly to a Pd-coordinated analogue of 4a (Fig. 3). Approach of anisole to [Pd(1)2] forms Int-1 which involves a weak interaction between Pd and the π-system of anisole through the Cipso–O bond. The potential formation of transition metal π-complexes as unstable intermediates in the C–O bond functionalisation of aryl esters, aryl carbamates, aryl sulfamates, and aryl ethers has been highlighted previously.33–35Int-1 evolves to Int-2 by TS-1 and this step leads to the coordination of the oxygen lone pair to the 3p orbital of one of the aluminylene ligands of the [Pd(1)2] fragment with synchronous interaction of the electron-rich Pd centre with the sp2 C atom. Int-2 is the precursor for the ligand assisted oxidative addition step. Activation and breaking of the sp2 C–O bond occurs by TS-2 and this is the highest barrier on the pathway ΔG‡ = 15.5 kcal mol−1. The coordinated reaction product Int-3 is generated directly from TS-2 with migration of both the CH3 and OPh fragments to Al occurring in this step. Across the sequence Int-1–TS-1–Int-2–TS-2–Int-3 the sp2 C–O bond of anisole stretches from 1.36–1.43–1.47–1.64–3.05 Å, the Wiberg Bond index of the C–O bond decreases from 1.01–0.85–0.79–0.57–0.02, and the NPA charge of the key Al atom increases from 0.99–1.09–0.99–1.39–2.16.
 | ||
| Fig. 3 DFT calculated pathway for the Pd-catalysed reaction of 1 with anisole (2a). Gibbs energies in kcal mol−1. | ||
QTAIM calculations return data for a four-membered ring constructed from Pd, C, O and Al atoms for Int-2, which is again consistent with a ligand-assisted oxidative addition pathway. Bond paths are found between each atom and a ring critical point located in the centre of the four-membered ring. In addition to the short Al–O distance of 2.15 Å detected in the optimized geometry of Int-2, the presence of a primarily ionic interaction between Al and O atoms was evidenced by the attributes of the BCP (ρ = 0.033 e Bohr−3, ∇2ρ = 0.12 e Bohr−5). Further analysis of the QTAIM data is consistent with a strengthening of the Al–O interaction and weakening of the C–O interaction along the reaction coordinate (see ESI†).
Alternative pathways involving either the direct oxidative addition of the sp2 C–O bond (ΔG‡ = 44.8 kcal mol−1) or the ortho sp2 C–H bond (ΔG‡ = 26.5 kcal mol−1) of anisole to [Pd(1)2] were calculated to occur with higher energy transition states and would not be expected to be competitive. To further exclude the possibility that the reaction occurs through a mechanism involving C–H bond activation on or before the turnover-limiting step, the kinetic isotope effect (KIE) was determined experimentally. The KIE for the reaction of 2a and d8-2a was measured by both independent rates (1.3 ± 0.1) and an intermolecular competition experiment (1.2 ± 0.1). The lack of a significant KIE is consistent with a turnover-limiting step that involves the direct cleavage of the C–O bond.
Ligand-assisted oxidative addition has been invoked to explain the reaction of aryl halides with complexes containing M–PR3,36 M–SiR3,37 and M–BR2 bonds.38 Such a pathway has even been considered to explain the assistive role of magnesium reagents in the cross-coupling of fluoroarenes.39 We proposed that ligand-assisted oxidative addition may be a key step during the reaction of 1 with fluorobenzene with the same catalyst system reported herein.27 Ligand-assisted oxidative addition involves a four-membered transition state (rather than a classical three-membered oxidative addition transition state) and is reliant on the ligand acting as an acceptor. There is a clear analogy to Lewis-acid assisted processes and it is notable that a number of reactions involving C–O bond activation of aryl methyl ethers with transition metals have been shown to be promoted by Al3+ compounds.40,41
In summary, we report the reactions of a low-valent aluminium(I) reagent with a series of aryl methyl ethers to form aluminium building blocks containing polar Al–C and Al–O bonds. In the absence of a catalyst, selective functionalisation of sp3 C–O bonds occurs in preference to the sp2 C–O bond. In the presence of catalytic [Pd(PCy3)2] there is a complete switch in selectivity for functionalisation of sp2 C–O bonds. DFT calculations in combination with measurements of KIEs support a pathway involving the ligand-assisted oxidative addition of the sp2 C–O bond of the substrate to a known Pd–Al intermetallic intermediate. Catalyst switching of chemoselectivity provides access to isomeric organoaluminium building blocks from a single starting material. The new methodology was applied to upgrading a derivative of vanillin.
We are grateful to the European Research Council for and ERC StG (FluoroFix:677367) and Marie Curie Fellowship to F. R. Johnson Matthey are thanked for generous donation of PdCl2. P. J. C. thanks Fundação para a Ciência e a Tecnologia (FCT), Portugal, for projects UIDB/04046/2020, UIDP/04046/2020 (BioISI). P. J. C. also acknowledges Programa Operacional Regional de Lisboa (Lisboa 2020), Portugal 2020, FEDER/FN, and the European Union for project number 28455 (LISBOA-01-0145-FEDER-028455, PTDC/QUI-QFI/28455/2017).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. J. Ragauskas, Science, 2006, 311, 484–489 CrossRef CAS PubMed
.
- Y. Pu, D. Zhang, P. M. Singh and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2008, 2, 58–73 CrossRef CAS
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS
- Y. Xin, Y. Jing, L. Dong, X. Liu, Y. Guo and Y. Wang, Chem. Commun., 2019, 55, 9391–9394 RSC
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang, G. Yang and B. Han, Chem. Sci., 2020, 11, 1347–1352 RSC
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2015, 4, 35–46 CrossRef
- E. Wenkert, E. L. Michelotti and S. C. Swindell, J. Am. Chem. Soc., 1979, 101, 2246–2247 CrossRef CAS
- C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed
- R. J. Somerville, L. V. A. Hale, E. Gómez-Bengoa, J. Bures and R. Martin, J. Am. Chem. Soc., 2018, 140, 8771–8780 CrossRef CAS PubMed
- C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed
- Y. Gu and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 3187–3190 CrossRef CAS PubMed
- H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593–1600 CrossRef CAS PubMed
- C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6757 CrossRef CAS PubMed
- R. Seki, N. Hara, T. Saito and Y. Nakao, J. Am. Chem. Soc., 2021, 143, 6388–6394 CrossRef CAS PubMed
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed
- S. Yow, A. E. Nako, L. Neveu, A. J. P. White and M. R. Crimmin, Organometallics, 2013, 32, 5260–5262 CrossRef CAS
- M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 RSC
- T. N. Hooper, R. K. Brown, F. Rekhroukh, M. Garçon, A. J. P. White, P. J. Costa and M. R. Crimmin, Chem. Sci., 2020, 11, 7850–7857 RSC
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC
- T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363–5365 CrossRef CAS
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed
- S. Kurumada, K. Sugita, R. Nakano and M. Yamashita, Angew. Chem., Int. Ed., 2020, 59, 20381–20384 CrossRef CAS PubMed
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS
- F. Rekhroukh, W. Chen, R. K. Brown, A. J. P. White and M. R. Crimmin, Chem. Sci., 2020, 11, 7842–7849 RSC
- T. N. Hooper, M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC
- W. Chen, T. N. Hooper, J. Ng, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2017, 56, 12687–12691 CrossRef CAS PubMed
- B.-T. Guan, S.-K. Xiang, B.-Q. Wang, Z.-P. Sun, Y. Wang, K.-Q. Zhao and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 3268–3269 CrossRef CAS PubMed
- N. A. Jasim, A. C. Whitwood, A. Lledós, R. N. Perutz and M. A. Ortuño, Dalton Trans., 2016, 45, 18842–18850 RSC
- M. E. van der Boom, S.-Y. Liou, Y. Ben-David, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 1998, 120, 6531–6541 CrossRef CAS
- Z. Li, S.-L. Zhang, Y. Fu, Q.-X. Guo and L. Liu, J. Am. Chem. Soc., 2009, 131, 8815–8823 CrossRef CAS PubMed
- K. W. Quasdorf, A. Antoft-Finch, P. Liu, A. L. Silberstein, A. Komaromi, T. Blackburn, S. D. Ramgren, K. N. Houk, V. Snieckus and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 6352–6363 CrossRef CAS PubMed
- M. C. Schwarzer, R. Konno, T. Hojo, A. Ohtsuki, K. Nakamura, A. Yasutome, H. Takahashi, T. Shimasaki, M. Tobisu, N. Chatani and S. Mori, J. Am. Chem. Soc., 2017, 139, 10347–10358 CrossRef CAS PubMed
- S. A. Macgregor, D. C. Roe, W. J. Marshall, K. M. Bloch, V. I. Bakhmutov and V. V. Grushin, J. Am. Chem. Soc., 2005, 127, 15304–15321 CrossRef CAS PubMed
- A. L. Raza, J. A. Panetier, M. Teltewskoi, S. A. Macgregor and T. Braun, Organometallics, 2013, 32, 3795–3807 CrossRef CAS
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed
- C. Wu, S. P. McCollom, Z. Zheng, J. Zhang, C.-C. Sha, M. Li, P. J. Walsh and N. C. Tomson, ACS Catal., 2020, 10, 7934–7944 CrossRef CAS PubMed
- P. Kelley, G. A. Edouard, S. Lin and T. Agapie, Chem. – Eur. J., 2016, 22, 17173–17176 CrossRef CAS PubMed
- X. Liu, C.-C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093–6098 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of calculations and characterization data (PDF). Coordinates for DFT calculations (xyz). Crystallographic data for 3a, 3c, 4a, 4b, and 4d (cif). CCDC 1973118–1973121. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05408b |
| ‡ These authors contributed equally and are listed in alphabetical order. |
| § A small impurity was detected during the reaction of anisole which was removed in the recrystallisation process. This by-product was identified as the result of C–H alumination of the meta position of anisole and estimated to form in <5% yield. This minor component is enriched on recrystallisation of 4a. |
| This journal is © The Royal Society of Chemistry 2021 |