
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented superstructure in the type I family of clathrates†
Philip
Yox
ab,
Oleg I.
Lebedev
c,
Davide
Donadio
 d and
Kirill
Kovnir
*ab
d and
Kirill
Kovnir
*ab
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: kovnir@iastate.edu
bAmes Laboratory, U.S. Department of Energy, Ames, IA 50011, USA
cLaboratoire Crismat, Ensicaen, CNRS UMT 6508, 6 Boulevard du Marechal Juin, F-14050 Caen, France
dUniversity of California, Davis, Davis, CA 95616, USA
First published on 17th November 2021
Abstract
The first arsenic-based clathrate exhibiting superstructural ordering due to optimization of Au–As, As–As, and Ba–Au bonding is reported. Ba8Au16As30 crystallizes in a unique P21/c monoclinic clathrate structure. The synthesis, crystal and electronic structure, and transport properties are discussed.
Clathrates have “guest atoms” that reside in cages and “framework” atoms that compose the cages. Type I clathrates are an expansive group of compounds typically crystallizing in the Pm
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n space group with a framework composed of tetrel elements, Si, Ge, or Sn.1,2 As the type I clathrate has expanded into the regime of late transition metals and pnictogens forming the framework, there has been evidence of different superstructural orderings in selected compounds.3 Most notably, Ba8Cu16P30 crystallizes in Pbcn with a primitive cell containing 4 formula units.4,5 Ba8Au16P30 was shown to be twinned down to the nano domain using transmission electron microscopy and was determined to crystallize with the same superstructure as the Cu analogue.6 Several ternary clathrates with late transition metals and arsenic are reported: Ba8Cu16As30, Cs8Zn18As28, Rb8Zn18As28, and Cs8Cd18As28.7,8 All the arsenide-based clathrates have been reported to crystallize in the archetype clathrate I structure (cubic Pmn space group) with mixed M/As sites in the framework. Moving down the pnictogen group, a new story has emerged for antimony-based clathrates. Originally investigated in 2009, single crystal X-ray diffraction (SXRD) suggested that Cs8Zn18Sb28 and Cs8Cd18Sb28 crystallize in Pmn.9 Recent reinvestigation has shown that the antimony-based clathrates Cs8Zn18Sb28 and Cs8Cd18Sb28 exhibit ordered superstructures with segregation of M and Sb over different framework sites as evidenced using transmission electron microscopy and high-resolution synchrotron X-ray powder diffraction (HR-PXRD).10 A similar ordering was found for In-Sb and Ga-Sb clathrates.11
n space group with a framework composed of tetrel elements, Si, Ge, or Sn.1,2 As the type I clathrate has expanded into the regime of late transition metals and pnictogens forming the framework, there has been evidence of different superstructural orderings in selected compounds.3 Most notably, Ba8Cu16P30 crystallizes in Pbcn with a primitive cell containing 4 formula units.4,5 Ba8Au16P30 was shown to be twinned down to the nano domain using transmission electron microscopy and was determined to crystallize with the same superstructure as the Cu analogue.6 Several ternary clathrates with late transition metals and arsenic are reported: Ba8Cu16As30, Cs8Zn18As28, Rb8Zn18As28, and Cs8Cd18As28.7,8 All the arsenide-based clathrates have been reported to crystallize in the archetype clathrate I structure (cubic Pmn space group) with mixed M/As sites in the framework. Moving down the pnictogen group, a new story has emerged for antimony-based clathrates. Originally investigated in 2009, single crystal X-ray diffraction (SXRD) suggested that Cs8Zn18Sb28 and Cs8Cd18Sb28 crystallize in Pmn.9 Recent reinvestigation has shown that the antimony-based clathrates Cs8Zn18Sb28 and Cs8Cd18Sb28 exhibit ordered superstructures with segregation of M and Sb over different framework sites as evidenced using transmission electron microscopy and high-resolution synchrotron X-ray powder diffraction (HR-PXRD).10 A similar ordering was found for In-Sb and Ga-Sb clathrates.11
Contrary to the observation of superstructural ordering among the phosphorous- and antimony-based clathrates, no superstructural ordering has been reported for arsenide clathrates. The superstructural ordering can be due to the energy gain attained by avoiding metal–metal bonds. In Pmn, the framework filling of the 6c, 16i, and 24k sites cannot simultaneously fulfill the stoichiometry while avoiding metal–metal bonds and the structure is forced to reduce the overall symmetry and enlarge the unit cell volume. For phosphides, the known type I clathrates have crystallized in either Pbcn or P42/ncm, while the antimonides exhibited complex ordering in the Ia, Iad, and P2 space groups (the latter was proposed using microscopy studies). Herein, we report the first arsenide clathrate Ba8Au16As30 with unique superstructural ordering in the monoclinic space group P21/c.
The direct synthesis from elements at 600 °C resulted in a polycrystalline sample of Ba8Au16As30. Elemental energy dispersive analysis confirms presence of only Ba, Au, and As in the sample. A separate reaction utilizing a CsBr/CsI flux and an excess of As resulted in crystals suitable for single crystal X-ray diffraction (SCXRD). Ba8Au16As30 crystallizes in the P21/c space group as determined using SCXRD with a = 15.147(1) Å, b = 15.218(1) Å, c = 10.954(1) Å, and β = 90.502(2)° (Table S1, ESI†). Refinement of the crystal structure shows that all atomic sites are fully occupied (Fig. 1A). Like many other pnictide clathrates with superstructures, no metal–metal (Au–Au) bonds are formed in this framework. There are five Ba positions inside the Au–As cages: Ba1@Au6As14, Ba2@Au8As12, Ba3@Au9As15, Ba4@Au8As16, and Ba5@Au8As16. Ba1 and Ba2 occupy the 512 pentagonal dodecahedra (Fig. 1A, red cage), while Ba3, Ba4, and Ba5 occupy the 51262 tetrakaidecahedra (Fig. 1A, blue cage). Although the data suggested a monoclinic space group, the lattice parameters and beta angle were suspiciously close to tetragonal symmetry. The supergroups Pmn, P42/mmc, and P42/ncm were considered as possible solutions but failed to give a good refinement. To confirm the small but robust beta angle deviation from 90° and the correct symmetry, a sample synthesized from elements was prepared and studied with HR-PRXD.
 | ||
| Fig. 1 (A) Crystal structure of Ba8Au16As30, Ba: blue/red; Au: gold; As: grey. (B) Experimental (black) HR-PXRD pattern and theoretical patterns for P21/c (blue) and hypothetical Pbcn derived from Ba8Au16P30 (red); (C and D) zoomed-in regions of (B). | ||
Aside from a few minor peaks attributed to admixtures, the calculated diffraction pattern from the single crystal model fit extremely well once the room temperature unit cell parameters (a = 15.197 Å, b = 15.261 Å, c = 10.979 Å, and β = 90.444°) were refined (Fig. 1). HR-PXRD ruled out the aristotype cubic and orthorhombic (Pbcn) structural models. In Fig. 1C a zoomed-in region from 1.9 to 2.3 Q explicitly demonstrates that the Pbcn model fails to generate enough peaks to even begin fitting the experimental diffraction pattern. In Fig. 1D a similar conclusion can be made while noticing an agreeable peak splitting from the P21/c model and the experimentally collected data. The formation energy and stability of Ba8Au16As30 was calculated and compared with other ternary Ba–Au–As phases using density functional theory (Fig. S1, ESI†). The atomization energy is negative (−0.429 eV per atom) and Ba8Au16As30 is a thermodynamically stable phase when considering all experimentally reported binary and ternary compounds in the Ba–Au–As system. However, inclusion of the hypothetical BaAu2As4 (isostructural to BaAu2P4)12,13 shows that Ba8Au16As30 could decompose to BaAu2As4, BaAuAs, and Au (Fig. S1, ESI†). Our experimental efforts to synthesize BaAu2As4 have instead resulted in the formation of Ba8Au16As30.
The reduction of symmetry from Pmn to P21/c stems from the ordering of the framework elements (Au and As). The Bärnighausen tree (Fig. S2, ESI†) shows the relationship between the two space groups as well as the Wyckoff site splitting. In the first reduction of symmetry to the space group P42/mmc almost all the framework sites remain mixed occupied except for the 2e site. The second transformation to P42/ncm is accompanied by a doubling of the unit cell volume (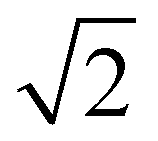 ×
×  × 1). In this space group a hypothetical distribution of Au and As among the Wyckoff sites could result in a completely ordered structure that fulfills the stoichiometry of the framework (16Au:30As) in analogy with the reported ordered structure for La1.6Ba6.4Cu16P30.14 Further reduction of symmetry may be caused by optimization of the As–As and Ba–Au interactions (Table S2, ESI†). The next transformation lowers the symmetry to the orthorhombic space group Pccn. In the final transformation to P21/c all the framework sites are identified as general position 4e sites and are fully occupied by either Au or As. All the Au atoms are surrounded by four As atoms and no Au–Au distance shorter than 3.91 Å is present.
× 1). In this space group a hypothetical distribution of Au and As among the Wyckoff sites could result in a completely ordered structure that fulfills the stoichiometry of the framework (16Au:30As) in analogy with the reported ordered structure for La1.6Ba6.4Cu16P30.14 Further reduction of symmetry may be caused by optimization of the As–As and Ba–Au interactions (Table S2, ESI†). The next transformation lowers the symmetry to the orthorhombic space group Pccn. In the final transformation to P21/c all the framework sites are identified as general position 4e sites and are fully occupied by either Au or As. All the Au atoms are surrounded by four As atoms and no Au–Au distance shorter than 3.91 Å is present.
The Bärnighausen tree demonstrates that single atom type site occupation is possible in P21/c but the same ordering can be accomplished in Pbcn, as was found for Ba8Au16P30 and Ba8Cu16P30. What is the driving force for this reduction of symmetry as opposed to Pbcn found in Ba8Au16P30? In the structure of Ba8Au16Pn30 (Pn = P, As) pnictogen atoms account for almost 2/3 of the framework atoms. The replacement of all smaller P atoms with As leads to an overall 13.7% increase of the unit cell volume per formula unit. Assuming an isotropic structure expansion in the Pbcn space group, this would lead to the increase of the Ba-framework distances and the volume of the polyhedral cages, which may become oversized for the Ba guest cations. Reducing the crystal symmetry of Ba8Au16As30 down to P21/c allows optimization of the Ba-framework interactions. The volumes of the Ba polyhedra in Ba8Au16As30 are 14% larger than those of the respective polyhedra in Ba8Au16P30 (Table S2, ESI†). However, the average and shortest Ba-framework distances in both polyhedra increase only by 4% as compared to the distances in the phosphide analogue. The simplest model of Pbcn Ba8Au16As30 was produced using the atomic coordinates of Ba8Au16P30 and isotropically expanding the volume per formula unit to match that of Ba8Au16As30. The analysis of the consequent atomic distances shows that for cages of the same volume, P21/c allows for optimized Ba–Au distances (Table S2.2, ESI†). Relaxing of the produced hypothetical Pbcn Ba8Au16As30 using DFT still results in the structure with ∼0.1 Å longer Ba–Au distances, e.g. the shortest Ba–Au distance in relaxed Pbcn is 3.49 Å while in P21/c the shortest Ba–Au distance is 3.39 Å. Another important difference between Ba8Au16As30 and Ba8Au16P30 is the pnictogen–pnictogen bonding. While there are no Au–Au bonds in either structure, there are homoatomic pnictogen–pnictogen bonds. The sum of covalent radii for P is 2.20 Å while the sum of covalent radii for As is 2.40 Å. In the case of Ba8Au16P30, very long P–P bonds are observed with distances up to 2.72 Å.6 This is not the case for Ba8Au16As30 where the longest As–As distance is 2.53 Å. The Zintl phases Ba3As1415 and Ba2As7I16 feature As7 clusters where the longest As–As bond is 2.52–2.53 Å, while BaAs217 features one-dimensional As chains with a longest distance of 2.50 Å, which suggests that the As–As distances in Ba8Au16As30 are not unusual. The substantial differences between Ba8Au16As30 and Ba8Au16P30 are in the polypnictide anions. While there are two fragments in Ba8Au16P30, P12 and P18, only one type of As fragment, As15, exists in Ba8Au16As30 (Fig. S3, ESI†). We hypothesize that the differences in the superstructural ordering for Ba8Au16As30 and Ba8Au16P30 are due to optimization of pnictogen–pnictogen bonding and Ba–Au interactions.
To confirm the superstructural ordering determined using HR-PXRD and SCXRD, transmission electron microscopy was employed. The selected area electron diffraction (ED) patterns collected from the main crystallographic zone axes show agreement with the P21/c space group determined using the other methods (Fig. 2). In the [001] zone axis, the absence of 4-fold symmetry excludes tetragonal or cubic symmetry and the absence of the (010) diffraction peak is in line with the systematic absence condition for 0k0, k = 2n, which is a general condition for P21/c as well as Pccn. The presence of the (100) reflection shows that Pccn cannot be used to index the ED patterns, because of the violation of the h00, h = 2n condition. The [011] zone axis, which corresponds to [111] in the cubic aristotype structure, confirms an absence of the 3-fold rotation axis and shows only the presence of two-fold symmetry. This zone confirms the presence of the (100) reflection seen in the [001].
 | ||
| Fig. 2 Top: ED of the [001], [011], and [112] zones of Ba8Au16As30 indexed in the P21/c model; middle: low magnification HAADF-STEM image of the single crystallite and the corresponding EDX elemental mapping of Au M (red), Ba L (green), As K (blue), and the overplayed image; bottom: high resolution [011] and [112] HAADF-STEM images. The simulated images are given as insets. | ||
Elemental mapping and high resolution HAADF-STEM imaging show the homogeneous distribution of all chemical elements within a composition close to nominal (As – 54.6 at%, Ba – 15.0 at%, and Au – 31.8 at%) and good crystallinity (free of twinning and defects). The simulated high resolution [001] and [011] HAADF-STEM images are in agreement with the experimental ones (inset in Fig. 2, bottom panel).
Low thermal conductivity is ubiquitous among clathrates.2 The large number of atoms in the unit cell and the rattling guest atoms give rise to localized modes, which reduces the lattice contribution to thermal conductivity (κlattice). Additionally, the presence of heavy atoms in the framework reduces κlattice by lowering the optical phonon frequency range. Au is the third heaviest atom that has been incorporated into the framework of any clathrate, behind Bi and Hg.2,18–20 The incorporation of Au and other heavy framework atoms has been shown to lower the thermal conductivity significantly. For the Ba8Ga16−3xAuxGe30+2x system, substitution of Au (x = 4) lowered the lattice thermal conductivity from 1.6 W K−1 m−1 in Ba8Ga16Ge30 to 1.0 W K−1 m−1.21 Ba8Au16P30 has an exceptionally low lattice thermal conductivity even for clathrates.6 Considering the substitution of As for P it is reasonable to expect an even further reduction of the κlattice for Ba8Au16As30. However, that is not the case. As seen in Fig. 3, the lattice thermal conductivity for Ba8Au16As30 is ∼0.85 W K−1 m−1 at room temperature. The thermal conductivity is comparable to that for Ba8Au6Ge40 considering that Ge and As are comparable in atomic mass and Ba8Au16As30 has substantially more Au allowing the lattice thermal conductivity to be lowered from ∼1.0 W K−1 m−1 for Ba8Au6Ge4021 to ∼0.85 W K−1 m−1 for Ba8Au16As30. The thermal conductivity of Ba8Au16As30 is lower than most type I clathrates including Ba8Cu16P30 however it is still ∼50% higher than Ba8Au16P30. It may be that Ba8Au16P30 is an outlier due to an extrinsic lowering of the thermal conductivity caused by the grain boundaries and nanoscale twinning detected using HAADF-STEM.6 The extent of the twinning in Ba8Au16As30 is currently unknown but the presence of twinning to nano-domains seems unlikely as a structural solution from a single crystal was easily obtained and no twinning was detected during HAADF-STEM studies.
 | ||
| Fig. 3 Left: projected density of states in the GGA+U approximation. Right: temperature dependence of resistivity (blue), Seebeck coefficient (black), and thermal conductivity (red). The total thermal conductivity trend is shown with a black line to guide the eye. | ||
Ba8Au16As30 exhibits a metal-like temperature behavior of electrical resistivity with values similar to that for Ba8Au16P30. This is expected for compositions that are not electron balanced. Ba8Au16As30 has 182 electrons per 46 framework atoms, 8 × 2(Ba) + 16 × 1(Au) + 30 × 5(As) = 182, while an electron balanced composition requires 46 × 4 = 184 electrons. Electronic structure calculations confirm the expected metallic nature of Ba8Au16As30. The Fermi level is about 0.25 eV below the maximum of the valence band, which makes this material p-type. The Seebeck coefficient of Ba8Au16As30 is positive and increases with temperature. The room temperature absolute value (13 μV K−1) is close to those for other metallic pnictide clathrates, such as Ba8Au16P30 (15 μV K−1)6 and Cs8Cd18As28 (11 μV K−1).8 The major charge carriers are holes, which is expected for an electron deficient clathrate. The absolute values for the Seebeck coefficient below 75 K were lower than 1 μV K−1 and were omitted due to a high level of noise. While the Seebeck coefficient is low; zT ∼ 2.2 × 10−3, the low thermal conductivity and electron deficiency means that Ba8Au16As30 is well positioned for substitution and alloying to optimize the electronic transport properties, as was shown for clathrates developed from Ba8Cu16P30.14,22–24 To probe potential of Ba8Au16As30 for thermoelectric applications, the transport properties were calculated as a function of the energy of the Fermi level (Fig. S4, ESI†). Shifting the Fermi level via substitutional doping to the top of the valence band increases the Seebeck coefficient by an order of magnitude. However, the resistivity also increased by a similar amount. Due to the counteracting effects of resistivity and Seebeck coefficient, the overall zT of the material did not surpass 0.07. Even if the material was doped to n-type, zT will not exceed 0.1 (Fig. S4, ESI†). These results indicate that only modifying the carrier concentration will not provide a feasible route to a “state-of-the-art” thermoelectric material. Adjusting the hole-mobility may be an alternative route to further increase the thermoelectric performance of Ba8Au16As30, as was demonstrated for LaxBa8–xCu16P30.14
In conclusion, Ba8Au16As30 is a new type I clathrate crystallizing in the unprecedented superstructural space group P21/c that features a singular repeating As fragment and the absence of Au–Au bonds. We hypothesize that factors such as cage volume, and optimization of the Au–As, As–As, and Ba-framework interactions are the reasons for Ba8Au16As30 crystallizing in a different superstructure compared to Ba8Au16P30. The transport properties of Ba8Au16As30 are expected for a metallic clathrate but point to Ba8Au16P30 as an outlier in terms of its thermal conductivity. The theoretical transport properties indicate that modification of carrier mobility is required to substantially increase the zT of Ba8Au16As30-based thermoelectric materials.
The authors would like to thank Dr Saul Lapidus (APS) for help collecting synchrotron the HR-PXRD data and Prof. J. Zaikina (ISU) for access to the SPS. This research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering, grant DE-SC0022288. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility, operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Conflicts of interest
There are no conflicts to declare.References
- G. Nolas, The Physics and Chemistry of Inorganic Clathrates, Springer, Netherlands, 1 edn, 2014 Search PubMed.
- J.-A. Dolyniuk, B. Owens-Baird, J. Wang, J. V. Zaikina and K. Kovnir, Mater. Sci. Eng., 2016, 108, 1–46 CrossRef.
- J. Wang, J.-A. Dolyniuk and K. Kovnir, Acc. Chem. Res., 2018, 51, 31–39 CrossRef CAS PubMed.
- J. Dünner and A. Mewis, Z. Anorg. Allg. Chem., 1995, 621, 191–196 CrossRef.
- D. Huo, T. Sasakawa, Y. Muro and T. Takabatake, Appl. Phys. Lett., 2003, 82, 2640–2642 CrossRef CAS.
- J. Fulmer, O. I. Lebedev, V. V. Roddatis, D. C. Kaseman, S. Sen, J.-A. Dolyniuk, K. Lee, A. V. Olenev and K. Kovnir, J. Am. Chem. Soc., 2013, 135, 12313–12323 CrossRef CAS PubMed.
- K. Wei, A. R. Khabibullin, D. Hobbis, W. Wong-Ng, T. Chang, S. G. Wang, I. Levin, Y.-S. Chen, L. M. Woods and G. S. Nolas, Inorg. Chem., 2018, 57, 9327–9334 CrossRef CAS PubMed.
- H. He, A. Zevalkink, Z. M. Gibbs, G. J. Snyder and S. Bobev, Chem. Mater., 2012, 24, 3596–3603 CrossRef CAS.
- Y. Liu, L.-M. Wu, L.-H. Li, S.-W. Du, J. D. Corbett and L. Chen, Angew. Chem., 2009, 121, 5409–5412 CrossRef.
- B. Owens-Baird, P. Yox, S. Lee, X. B. Carroll, S. Grass Wang, Y.-S. Chen, O. I. Lebedev and K. Kovnir, Chem. Sci., 2020, 11, 10255–10264 RSC.
- B. Owens-Baird, J. Wang, S. G. Wang, Y.-S. Chen, S. Lee, D. Donadio and K. Kovnir, J. Am. Chem. Soc., 2020, 142, 2031–2041 CrossRef CAS PubMed.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, JOM, 2013, 65, 1501–1509 CrossRef CAS.
- J. Fulmer, D. Kaseman, J. Dolyniuk, K. Lee, K. S. Sen and K. Kovnir, Inorg. Chem., 2013, 52, 7061–7067 CrossRef CAS PubMed.
- J. Wang, Y. He, N. E. Mordvinova, O. I. Lebedev and K. Kovnir, Chem, 2018, 4, 1465–1475 CAS.
- W. Schmettow and H. G. Von Schnering, Angew. Chem., Int. Ed. Engl., 1977, 16, 857 CrossRef.
- J.-A. Dolyniuk, S. Lee, N. Tran, J. Wang, L.-L. Wang and K. Kovnir, J. Solid State Chem., 2018, 263, 195–202 CrossRef CAS.
- F. Emmerling, D. Petri and C. Röhr, Z. Anorg. Allg. Chem., 2004, 630, 2490–2501 CrossRef CAS.
- T. F. Fassler and C. Kronseder, Z. Anorg. Allg. Chem., 1998, 624, 561–568 CrossRef.
- A. Kaltzoglou, S. Ponou and T. F. Fässler, Eur. J. Inorg. Chem., 2008, 4507–4510 CrossRef CAS.
- A. Kaltzoglou, S. Ponou and T. F. Fässler, Z. Anorg. Allg. Chem., 2008, 538–542 CAS.
- Z. Ye, J. Y. Cho, M. M. Tessema, J. R. Salvador, R. A. Waldo, J. Yang, H. Wang, W. Cai, M. J. Kirkham, J. Yang and W. Zhang, J. Alloys Compd., 2014, 587, 747–754 CrossRef CAS.
- J. Wang, O. I. Lebedev, K. Lee, J.-A. Dolyniuk, P. Klavins, S. Bux and K. Kovnir, Chem. Sci., 2017, 8, 8030–8038 RSC.
- J. Dolyniuk, P. S. Whitfield, K. Lee, O. I. Lebedev and K. Kovnir, Chem. Sci., 2017, 8, 3650–3659 RSC.
- J. Dolyniuk, J. Wang, M. Marple, S. Sen, Y. Cheng, A. J. Ramirez-Cuesta and K. Kovnir, Chem. Mater., 2018, 30, 3419–3428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2109533. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05167a |
| This journal is © The Royal Society of Chemistry 2021 |