
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInexpensive gram scale synthesis of porous Ti4O7 for high performance polymer electrolyte fuel cell electrodes†
Mitsuharu
Chisaka
 *a,
Waka
Nagano
*b,
Byambasuren
Delgertsetseg
*b and
Tatsuya
Takeguchi
*b
*a,
Waka
Nagano
*b,
Byambasuren
Delgertsetseg
*b and
Tatsuya
Takeguchi
*b
aDepartment of Sustainable Energy, Hirosaki University, 3 Bunkyo-cho, Hirosaki, Aomori 036-8561, Japan. E-mail: chisaka@hirosaki-u.ac.jp
bFaculty of Science and Engineering, Iwate University, 4-3-5 Ueda, Morioka, Iwate 020-8551, Japan. E-mail: wnagano@iwate-u.ac.jp; delgerts@iwate-u.ac.jp; takeguch@iwate-u.ac.jp
First published on 10th November 2021
Abstract
As a fuel cell catalyst support, more than 2 g of Magnéli phase Ti4O7 fine-particles were synthesized in a single reaction via an inexpensive route. The single-cell performance reached that of commercial carbon-supported platinum, with an excellent load cycle durability, one of the highest ever reported for oxide-supported platinum catalysts.
Following the commercialization of polymer electrolyte fuel cell (PEFC)-powered passenger vehicles in the 2010s, they remain a niche market despite the longer travel distances (>500 km) and higher power density compared with battery-driven electric vehicles.1 The high cost of platinum-based catalysts has been assumed to present the highest barrier to widespread use.2 Currently, platinum-based nanoparticle catalysts are supported on conductive carbon black to maximize the “active” platinum surface area where the three reactants, i.e. gases, protons, and electrons, can access the platinum surface simultaneously. Carbon black is an excellent support material, delivering: (1) a high Brunauer–Emmett–Teller surface area, SBET, of above 100 m2 g−1; and (2) high electrical conductivity, σ, of above 1 S cm−1 at low compression pressures, typically below 10 MPa. The low cost is another advantage, although additional costs are required to protect the material from oxidation.3 Several oxide materials, including titanium dioxides,4,5 titanium suboxides,6,7 and tin dioxides,8,9 have been developed as support materials because they are stable in PEFC cathodes, where four times higher platinum loading is currently needed compared with the anodes.10 However, both the SBET and σ values of the oxides are generally insufficient; the values are often one or even two orders of magnitude lower than those of carbon black, reducing the cell performance. Recently, He et al. successfully prepared antimony-doped tin dioxide with an exceptionally high σ of 6.2 S cm−1 without disclosing the compression pressure, while the SBET was below 50 m2 g−1.9 The preparation of oxide supports exceeding both thresholds (1) and (2) is a major challenge. Magnéli phase titanium suboxides, with a composition that can be expressed as TinO2n−1, are well known as conductive oxides with a maximum σ at n = 4.11 The highest SBET of a Ti4O7 support for PEFCs remains low, at 26 m2 g−1.7 In this study, fine Ti4O7 particles were synthesized as supports in an attempt to meet these challenging targets. The particles were synthesized from titanium oxysulfate (TiOSO4) and polyethylene glycol with an average molecular weight of 360–440 (PEG400) via a carbothermal reduction reaction. This route was originally reported by the Nazar group, and uses conductive Ti4O7 as a lithium sulfur battery cathode, with the Ti4O7 synthesized from titanium ethoxide and PEG400.12 We selected TiOSO4 as it is stable in air and easy to handle, making it more suited to the mass production of Ti4O7 than titanium ethoxide or other titanium sources that react readily with moisture in the air. The details of the catalyst synthesis and characterization are described in S1 and S2, ESI.†
After optimizing the precursor composition and annealing conditions (S3, ESI†), fine Ti4O7 particles were synthesized as shown in Fig. 1(a). Submicron particles formed aggregates with a broad pore size distribution (S4, ESI†). Magnéli phase Ti4O7 is generally synthesized at higher temperatures, typically 1323 K or above, and for much longer durations.13 The PEG400-derived carbon species produced a reductive atmosphere and thus the annealing temperature and duration were successfully suppressed to 1240 K and 3 h, respectively, yielding a high SBET of 172 m2 g−1. When commercial TiO2 powders were annealed under the flow of an identical gas mixture, 10% v/v H2/Ar, a higher temperature of 1323 K and longer duration of 6 h was needed to obtain much larger particle sizes of Ti4O7 powders, Ti4O7-L, as shown in Fig. 1(b). The composition can be easily controlled to yield the further reduced Ti3O5 and Ti2O3 phases by controlling the annealing temperature and starting material of the present carbothermal reduction route, respectively (S3, ESI†). The batch size, i.e., the mass of Ti4O7 per annealing, was also easily increased to more than 2 g by simply increasing the mass of the precursor mixture (S3, ESI†).
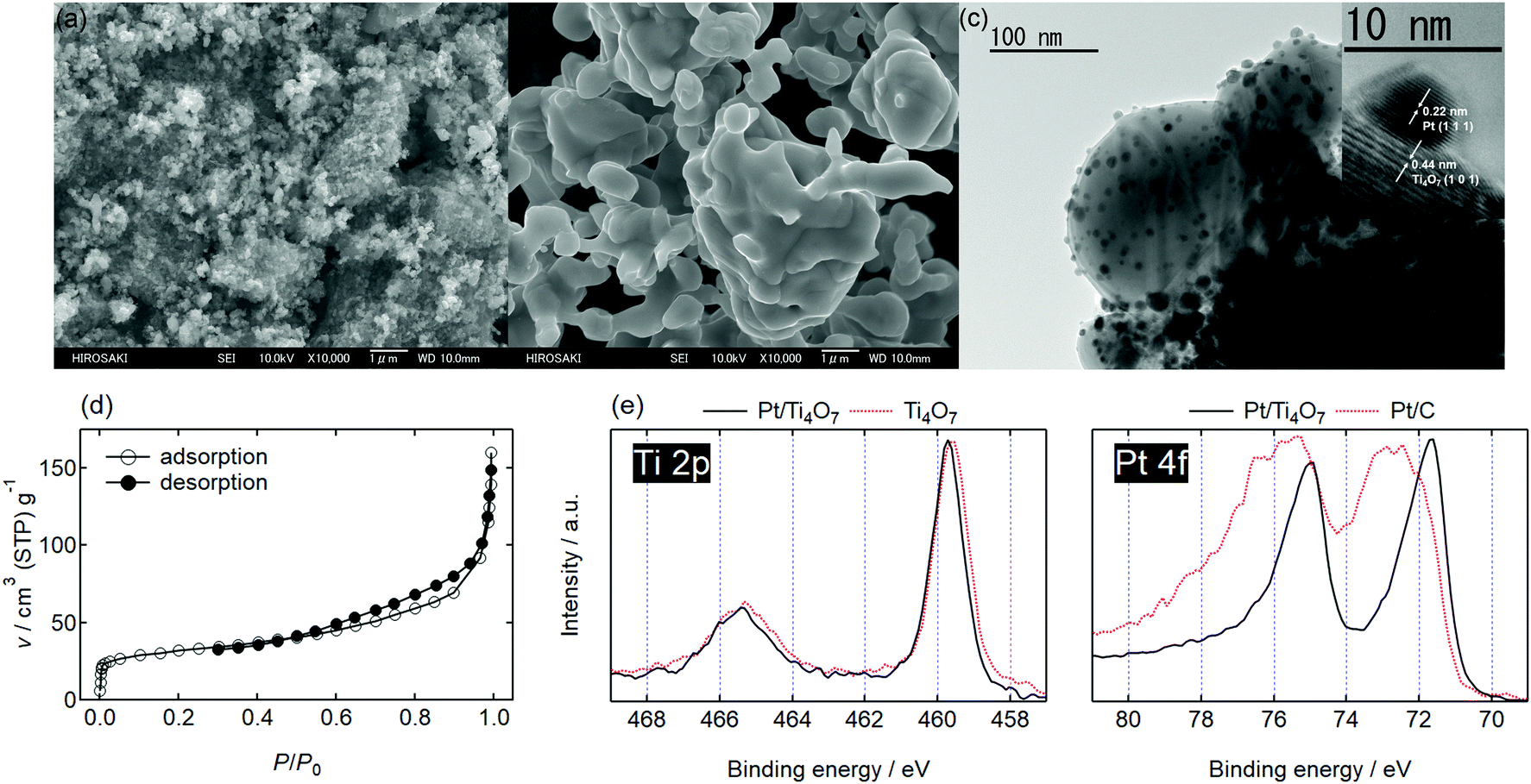 | ||
| Fig. 1 Field emission-scanning electron microscopy (FE-SEM) images of (a) Ti4O7 particles synthesized from titanium oxysulfate/polyethylene glycol mixture and (b) Ti4O7 powders synthesized from commercial TiO2, Ti4O7-L. (c) A transmission electron microscopy (TEM) image, (d) an N2 adsorption/desorption isotherm, and (e) Ti 2p and Pt 4f X-ray photoelectron (XP) spectra of 20% w/w Pt/Ti4O7 catalyst. The inset in (c) shows the high-resolution TEM (HR-TEM) image. For reference, a Ti 2p spectrum of Ti4O7 and a Pt 4f spectrum of commercial Pt/C are shown by the dashed curves in (e). | ||
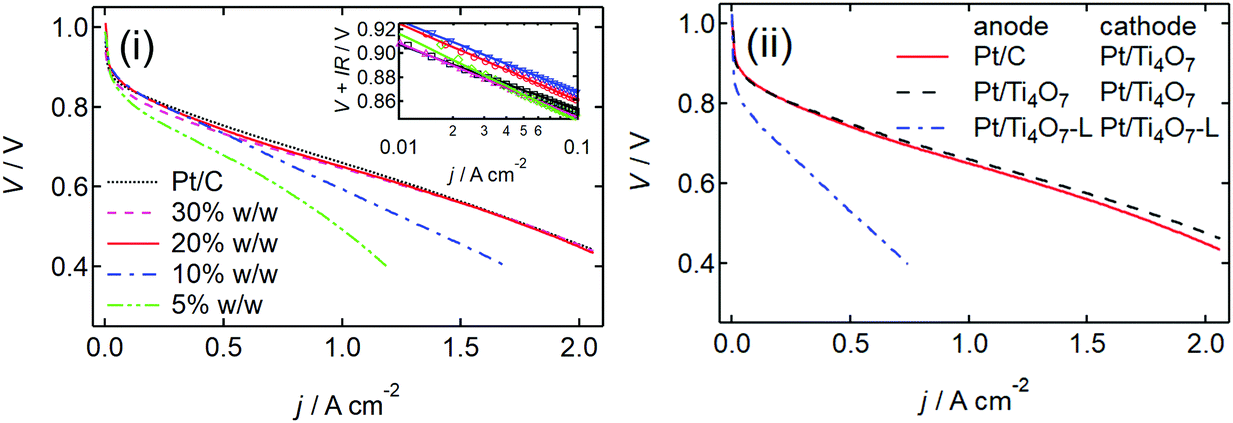 | ||
| Fig. 2 (i) j – V curves of five membrane electrode assemblies (MEAs) fabricated using 46% w/w Pt/C and Pt/Ti4O7 with four different platinum fractions, wPt, of 5%, 10%, 20%, and 30% w/w in the cathode. The Pt/C was used in the anode of all MEAs. (ii) j–V curves of three MEAs fabricated using Pt/C in the anode and 20% w/w Pt/Ti4O7 in the cathode, 20% w/w Pt/Ti4O7 in both the anode and the cathode, and 20% w/w platinum on Ti4O7 powder as shown in Fig. 1(b), Pt/Ti4O7-L in both the anode and cathode. The platinum loading at the anode and cathode was at 0.2 and 0.5 mgPt cm−2, respectively. | ||
Platinum nanoparticles were supported on the Ti4O7 particles using a recently reported ethanol reduction method.14 The transmission electron microscopy (TEM) images of 20% w/w Pt/Ti4O7 are shown in Fig. 1(c). Platinum particles of less than 10 nm in diameter were highly dispersed on the Ti4O7 support. The HR-TEM image shown in the inset indicates that platinum nanoparticles were in direct contact with the Ti4O7 particles. The N2 adsorption–desorption isotherm is displayed in Fig. 1(d). A type IIb isotherm with a H3 hysteresis loop was observed according to the classification by Rouquerol et al.15 and no plateau appears at a high relative pressure, P/P0. It is a typical isotherm for porous materials in which pores originate from the space between aggregates;15,16 in this case, the aggregates were composed of the fine Pt/Ti4O7 particles.
The surface chemical states of the Pt/Ti4O7 catalysts were investigated using X-ray photoelectron spectroscopy (XPS) and their Ti 2p and Pt 4f spectra are displayed in Fig. 1(e). The Ti 2p and Pt 4f core levels split into Ti 2p3/2/Ti 2p1/2 and Pt 4f7/2/Pt 4f5/2, as shown by doublets in the spectra. The Ti4O7 supports displayed a Ti 2p3/2 peak at approximately 459 eV in Fig. 1(e), which was assigned to Ti4+ in TiO2.17 No clear peaks from Ti3+ were observed in the Ti 2p spectrum of Ti4O7, indicating that the surface of Ti4O7 was oxidized to form a thin layer of TiO2−x.6 The corresponding Raman spectrum showed rutile TiO2 with oxygen vacancies (S3, ESI†), and agreed well with these XPS analyses. The Hwang group also reported that the Ti4O7 surface was oxidized to form a rutile TiO2 layer.18 The Ti 2p3/2 peak shifted to higher binding energy regions after platinum nanoparticles were supported, as shown in Fig. 1(e), indicating that the electron density of titanium decreased. The Pt 4f spectrum of Pt/Ti4O7 showed lower Pt 4f7/2 peak binding energy compared with that of commercial Pt/C, indicating that the electron density of platinum nanoparticles was higher in Pt/Ti4O7 than Pt/C. The Ti 2p and Pt 4f spectra presented in Fig. 1(e) indicate that the electrons from the titanium atoms in Ti4O7 supports were transferred to the platinum atoms. These results were in good agreement with previous oxide-supported platinum catalysts, in which strong metal–support interactions (SMSIs) were reported to enhance oxygen reduction reaction (ORR) activity.19 The SMSI was enhanced by increasing platinum fraction, wPt (S5, ESI†).
Both the platinum particle size, dPt, and the chemically active surface area of platinum nanoparticles in Pt/Ti4O7, SCSA, were evaluated using a CO pulse method and the electrochemically active platinum surface area, SECSA, was evaluated using CO stripping voltammetry. The results are summarized in Table 1. As wPt decreased, dPt decreased and SCSA increased, indicating that the platinum nanoparticles were highly dispersed on Ti4O7. Indeed, the corresponding XRD peaks were too broad to calculate the crystallite size when wPt < 20% w/w (S6, ESI†). The SECSA displayed a similar trend to wPt when compared with SCSA, indicating that the protons and electrons could access the surface of small platinum nanoparticles. At wPt = 5% w/w, SECSA was larger than SCSA suggesting the presence of small platinum particles which were insensitive to the CO pulse method. These Pt/Ti4O7 catalysts exhibited excellent ORR activity in a single cell cathode, as shown in Fig. 2(i). Membrane electrode assemblies (MEAs) were fabricated using commercial Pt/C at the anode and Pt/Ti4O7 or Pt/C at the cathode, and then the current density versus voltage (j–V) curves were obtained at 353 K. These curves were separately interpreted at V ≥ 0.8 V and V < 0.8 V. At V ≥ 0.8 V, the curves were analyzed using eqn (1) to reveal the ORR kinetics in a single cell:20
| V = V0 − blog(j) − jRA | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, indicating that neither HOR nor ORR performance changed over the 10000 cycles. Commercial SnO2-supported platinum displayed much lower durability. In Table S1 (S8, ESI†), the load cycle durability of platinum or platinum–cobalt supported on oxide catalysts reported to date is summarized. The changes in V at j = 1.0 A cm−2 during the cycles, which was previously utilized by Ramani group,5 were used as a measure of durability as high loads are needed in vehicles. The load cycle durability of the present Pt/Ti4O7 is among the highest of the state-of-the-art Pt/oxide catalysts,4,5,8,9 indicating that Ti4O7 stabilized the platinum nanoparticles well. Although the mechanism is still under investigation, one of the reasons may be the SMSI, as platinum nanoparticles are in contact with Ti4O7 with a large surface area; a SBET close to normal carbon black and among the highest in oxide supports reported to date, 172 m2 g−1.
000 cycles, indicating that neither HOR nor ORR performance changed over the 10000 cycles. Commercial SnO2-supported platinum displayed much lower durability. In Table S1 (S8, ESI†), the load cycle durability of platinum or platinum–cobalt supported on oxide catalysts reported to date is summarized. The changes in V at j = 1.0 A cm−2 during the cycles, which was previously utilized by Ramani group,5 were used as a measure of durability as high loads are needed in vehicles. The load cycle durability of the present Pt/Ti4O7 is among the highest of the state-of-the-art Pt/oxide catalysts,4,5,8,9 indicating that Ti4O7 stabilized the platinum nanoparticles well. Although the mechanism is still under investigation, one of the reasons may be the SMSI, as platinum nanoparticles are in contact with Ti4O7 with a large surface area; a SBET close to normal carbon black and among the highest in oxide supports reported to date, 172 m2 g−1.
| Catalyst | d Pt (nm) | S CSA (m2 gPt−1) | S ECSA (m2 gPt−1) | V 0 (V) | b (V dec−1) | RA (Ω cm2) | i m (A gPt−1) |
|---|---|---|---|---|---|---|---|
| 30% w/w Pt/Ti4O7 | 6.8 | 40.8 | 32.7 | 0.970 ± 0.000 | 0.061 ± 0.000 | 0.146 ± 0.001 | 185 |
| 20% w/w Pt/Ti4O7 | 4.2 | 66.0 | 58.2 | 0.983 ± 0.001 | 0.059 ± 0.001 | 0.157 ± 0.002 | 351 |
| 10% w/w Pt/Ti4O7 | 3.4 | 82.0 | 116.0 | 0.986 ± 0.001 | 0.059 ± 0.000 | 0.172 ± 0.001 | 404 |
| 5% w/w Pt/Ti4O7 | 2.1 | 131.6 | 210.9 | 0.985 ± 0.009 | 0.074 ± 0.005 | 0.222 ± 0.023 | 166 |
| 46% w/w Pt/C | N/A | N/A | 88.3 | 0.961 ± 0.001 | 0.053 ± 0.001 | 0.094 ± 0.003 | 245 |
| 20% w/w Pt/Ti4O7-L | N/A | N/A | N/A | 0.926 ± 0.005 | 0.065 ± 0.004 | 0.347 ± 0.066 | 28 |
In summary, Magnéli phase Ti4O7 particles were synthesized via carbothermal reduction to yield high σ and SBET values of 1.2 S cm−1 at low compression pressure (6 MPa) and 172 m2 g−1, respectively. The precursors are low-cost, stable in air, and easy to handle and to increase the mass of Ti4O7; more than 2 g were synthesized at once. The resulting Ti4O7 particles supported the platinum nanoparticles to display SMSI between the platinum and Ti4O7. The optimized 20% w/w Pt/Ti4O7 exhibited excellent HOR/ORR performances in a single cell, almost the same as those of commercial Pt/C. The load cycle durability was the highest among the state-of-the-art platinum/oxide catalysts, with no change in the cell performance after 10000 voltage cycles.
Mitsuharu Chisaka worked on conceptualization, support synthesis, physical and chemical characterizations, data curation, formal analysis, funding acquisition, investigation, writing (original draft), and writing (review & editing). Waka Nagano worked on catalyst synthesis, elemental analyses, preparation of MEAs, and single cell tests. Byambasuren Delgertsetseg worked on the CO pulse experiments and CO stripping voltammetry. Tatsuya Takeguchi worked on methodology, funding acquisition, investigation, and writing (review & editing).
We thank Mr Yusei Tsushima at Hirosaki University and Ms Kyoko Mitsunari at Horiba Techno Service Co., Ltd for performing FE-SEM/TEM imaging and particle size analyses, respectively. This work was partially supported by a grant from the New Energy and Industrial Technology Development Organization, Japan. Part of the work was conducted at the Advanced Characterization Nanotechnology Platform of the University of Tokyo, supported by the “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology in Japan, grant number JPMXP09A21UT0016.
Conflicts of interest
There are no conflicts to declare.Notes and references
- O. Gröger, H. A. Gasteiger and J. P. Suchsland, J. Electrochem. Soc., 2015, 162, A2605 CrossRef.
- M. M. Whiston, I. L. Azevedo, S. Litster, K. S. Whitefoot, C. Samaras and J. F. Whitacre, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4899 CrossRef CAS PubMed.
- U. Eberle and R. von Helmolt, Energy Environ. Sci., 2010, 3, 689 RSC.
- (a) S. Y. Huang, P. Ganesan and B. N. Popov, Appl. Catal., B, 2011, 102, 71 CrossRef CAS; (b) A. Pătrua, A. Rabis, S. E. Temmel, R. Kotz and T. J. Schmidt, Catal. Today, 2016, 262, 161 CrossRef; (c) P. Dhanasekaran, S. V. Selvaganesh and S. D. Bhat, New J. Chem., 2017, 41, 13012 RSC.
- (a) A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516 CrossRef CAS; (b) J. Parrondo, T. Han, E. Niangar, C. Wang, N. Dale, K. Adjemian and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 45 CrossRef CAS PubMed.
- T. Ioroi, H. Kageyama, T. Akita and K. Yasuda, Phys. Chem. Chem. Phys., 2010, 12, 7529 RSC.
- C. Yao, F. Li, X. Li and D. Xia, J. Mater. Chem., 2012, 22, 16560 RSC.
- C. Takei, R. Kobayashi, Y. Mizushita, Y. Hiramitsu, K. Kakinuma and M. Uchida, J. Electrochem. Soc., 2018, 165, F1300 CrossRef CAS.
- C. He, X. Wang, S. Sankarasubramanian, A. Yadav, K. Bhattacharyya, X. Liang and V. Ramani, ACS Appl. Energy Mater., 2020, 3, 5774 CrossRef CAS.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127 CrossRef CAS PubMed.
- J. R. Smith, F. C. Walsh and R. L. Clarke, J. Appl. Electrochem., 1998, 28, 1021 CrossRef CAS.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342 CrossRef CAS.
- M. M. Rahman, K. Inaba, G. Batnyagt, M. Saikawa, Y. Kato, R. Awata, B. Delgertsetsega, Y. Kaneta, K. Higashide, T. Uruga, Y. Iwasawa, K. Ui and T. Takeguchi, RSC Adv., 2021, 11, 20601 RSC.
- F. Rouquerol, J. Rouquerol, K. S. W. Sing, P. Llewellyn and G. Maurin, Adsorption by Powders and Porous Solids. Principles, Methodology and Applications, Academic Press, London, 2014, 2nd edn, p. 290 Search PubMed.
- (a) S. Abelló and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2006, 96, 102 CrossRef; (b) G. S. Shao, F. Y. Wang, T. Z. Ren, Y. Liu and Z. Y. Yuan, Appl. Catal., B, 2009, 92, 61 CrossRef CAS.
- (a) K. S. Robinson and P. M. A. Sherwood, Surf. Interface Anal., 1984, 6, 261 CrossRef CAS; (b) N. C. Saha and H. G. Tompkins, J. Appl. Phys., 1992, 72, 3072 CrossRef CAS.
- A. D. Duma, Y. C. Wu, W. N. Su, C. J. Pan, M. C. Tsai, H. M. Chen, J. F. Lee, H. S. Sheu, V. T. T. Ho and B. J. Hwang, ChemCatChem, 2018, 10, 1155 CrossRef CAS.
- I. Jiménez-Morales, F. Haidar, S. Cavaliere, D. Jones and J. Rozière, ACS Catal., 2020, 10(18), 10399 CrossRef.
- M. Chisaka, E. Matsuoka and H. Daiguji, J. Electrochem. Soc., 2010, 157, B1218 CrossRef CAS.
- A. Parthasarathy, S. Srinivasan, A. J. Appleby and C. R. Martin, J. Electrochem. Soc., 1992, 139, 2530 CrossRef CAS.
- (a) Y. W. Rho, O. A. Velev, S. Srinivasan and Y. T. Kho, J. Electrochem. Soc., 1994, 141, 2084 CrossRef CAS; (b) G. Li and P. G. Pickup, J. Electrochem. Soc., 2003, 150, C745 CrossRef CAS.
- M. Chisaka and H. Daiguji, J. Electrochem. Soc., 2009, 156, B22 CrossRef CAS.
- M. Zhang, Y. Wang, Y. Zhang, J. Song, Y. Si, J. Yan, C. Ma, Y. T. Liu, J. Yu and B. Ding, Angew. Chem., Int. Ed., 2020, 59, 23252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, elemental analyses, pore size distributions, XRD patterns, FE-SEM images, XP spectra and RDE voltammograms. See DOI: 10.1039/d1cc05144j |
| This journal is © The Royal Society of Chemistry 2021 |