
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation–π interactions enabling hard/soft Ti/Ag heterobimetallic cooperativity in lactide ring-opening polymerisation†‡
Chloe A.
Baker
 ,
Charles
Romain
* and
Nicholas J.
Long
*
,
Charles
Romain
* and
Nicholas J.
Long
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: c.romain@imperial.ac.uk; n.long@imperial.ac.uk
First published on 27th October 2021
Abstract
The combination of a Ti–salen complex with AgBArF reveals unique hard/soft heterobimetallic cooperativity in lactide ring-opening polymerisation (ROP), enabling significant activity at room temperature. Reactivity, mechanistic and computational studies highlight the role of cation–π interactions in the formation of heterobimetallic species and provide key insights into the role of both metals in ROP.
Taking inspiration from Nature, numerous bi- and multi-metallic catalytic species have been developed to enhance chemical transformations, for example to improve catalytic performance (e.g. activity, selectivity) or to enable in situ cascade reactions (e.g. tandem catalysis).1 The use of at least two different metals is of interest to enable the complementary reactivities offered by each metal to be exploited, either in tandem or synergistically.2 Thus, heterobimetallic catalysts have been successfully applied to organic catalytic1a reactions as well as to polymerisation reactions.3 As recently reviewed, heterobimetallic catalytic species can outperform their homobimetallic counterparts, such as in olefin polymerisation,3b–d cyclic ester ring-opening polymerisation (ROP),3a,4 as well as epoxide/CO2 and epoxide/anhydride ring-opening copolymerisation (ROCOP).3a ROP and ROCOP are particularly timely as they provide access to well-defined bio-based and/or non-durable polyesters and polycarbonates. Polylactic acid (PLA), a flagship biopolymer, being both bio-derived and industrially compostable, is currently industrially obtained by the Sn-catalysed ROP of lactide, the dimer of lactic acid.
Several successful combinations of hard and intermediate Lewis acidic metals (as per Pearson's classification)5 have been investigated as hetero bi- and-multi-metallic catalysts in lactide ROP,3a including Li/Mg,6 Li/Zn,7 Li/Ge,8 Li/Y,9 Li/Sn,8 Na/Zn,6a Mg/Ti,10 Al/Zn,11 Al/Y,12 Al/Sm,12 K/Zn2,13 and Ti/Zn.10,14 As proposed by Garden and co-workers,3a,11,13 the nature of the two metals is highly important, with large differences in electronegativity and at least one metal with a large ionic radius being particularly desirable. Interestingly, to the best of our knowledge, no examples of heterobimetallic catalysts including soft metals [e.g. Ag(I), Cu(I)] have been reported for lactide ROP so far.
The synthesis and characterisation of well-defined, discrete heterobimetallic catalysts remains challenging, and the formation of some of the aforementioned examples may be attributed to residual salt traces from the complex synthesis.4,15 Dinucleating ligands providing at least two different pockets tailored towards two different metals have been successfully designed to overcome this, but their synthesis and modulations can still be challenging. In line with previous work by some of us16 and others17 exploiting non-covalent interactions (NCI) in ROP, we decided to investigate how cation–π interactions could favour the in situ formation of aggregated heterobimetallic species, and provide a new use of NCI as a strategy to decipher metal cooperativities. Metal–π interactions have been found to be of importance in biology, materials and chemistry,18 with Ag+–π interactions being one of the most widely investigated and used in catalysis.19
Thus, with the aim of positioning a soft metal cation near a hard oxophilic metal via cation–π interactions, we considered the combination of a Ti–salen ROP catalyst with silver tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (AgBArF). Titanium alkoxide complexes bearing Schiff base salen ligands have a strong track record in lactide ROP,20 and provide both a π system via the phenolate moieties of the salen ligand and an “hard” oxophilic metal. AgBArF is a common reagent in organometallic and inorganic chemistry with a weakly coordinating anion,21 and thus provides an accessible source of “naked” Ag+. It is noteworthy that few successful heterobimetallic catalysts containing Ti have been reported for cyclic ester ROP. Using a reduced Robson-type macrocycle, Williams and co-workers14 reported a Ti(IV)/Zn(II) catalyst for LA ROP where activity at room temperature is observed upon addition of a second metal (i.e. Zn). Lin and co-workers reported Ti(IV)/Mg(II) and Ti(IV)/Zn(II) bis(phenolate) complexes showing enhanced catalytic activities towards LA ROP (91% in 30 min and 89% in 3.5 hours respectively), compared to their monometallic Ti(IV) counterparts (76% in 94 hours).10 Sarazin et al. reported a Ti(III)/Na complex showing enhanced activity in ε-caprolactone ROP compared to a monometallic Ti(IV) analogue.22
Here, we report the unique and successful hard/soft combination of a well-defined Ti–salen complex with a silver salt leading to activity enhancement in lactide ROP and activity at room temperature. Reactivity, mechanistic and computational studies highlight the nature of the observed heterobimetallic cooperativity.
Following a modified literature procedure,23 the bis(iso-propoxide) titanium salen complex 1 was prepared in a good yield (88%) from the reaction of one equivalent of salen pre-cursor H2L with one equivalent of Ti(OiPr)4. The 1H NMR spectrum (CDCl3, 298 K) suggests that the complex adopts a C2-symmetric structure in solution with only two signals for the tBu groups (δ1H = 1.32 ppm and 1.49 ppm), two signals in the aromatic region (δ1H = 7.48 ppm and 7.16 ppm), and one imine signal (δ1H = 8.27 ppm), in line with a meridional coordination of the tetradentate salen ligand as previously observed for other bis(alkoxide) titanium salen complexes.24
Complex 1 was tested for rac-lactide (rac-LA) ROP in standard literature conditions (1 mol% catalyst, [rac-LA]0 = 1 mol L−1, 90 °C, toluene) and was able to convert 73% of rac-LA in 24 hours (entry 1, Table 1) to afford atactic PLA with well-controlled polymerisation characteristics (i.e. controlled molar masses, low dispersities <1.3, Fig. S12, ESI‡). In contrast, complex 1 gave no activity within 4 hours for rac-LA ROP in dichloromethane (DCM) at room temperature (entry 2, Table 1). However, when 1 equivalent of AgBArF was initially added, the resulting catalytic system of an equimolar amount of 1 + AgBArF afforded well-controlled polymerisation of rac-LA at room temperature, leading to 76% conversion to PLA after 1 hour (entry 4, Table 1 and Fig. S13, ESI‡). It is noteworthy that AgBArF alone does not exhibit any activity towards LA ROP (entry 3, Table 1); only the combination of both metal reagents in situ leads to an active system, highlighting the cooperativity between these two entities (Scheme 1).
| Entry | Cat./Co-cat. | Solvent | T (°C) | t (h) | Conv.a (%) | M n(exp) (Đ)b (kg mol−1) | M n(calc) (kg mol−1) |
|---|---|---|---|---|---|---|---|
| Reaction conditions cat./co-cat./rac-LA:1/1/100, [rac-LA]0 = 1 mol L−1.a Determined by 1H NMR spectroscopy by relative integration of the signals at 5.06 ppm (rac-LA) vs. 5.20 ppm (PLA).b Determined by SEC calibrated with polystyrene standards and corrected by factor of 0.58 for PLA.c Calculated using formular Mn(calc) = conv. (%) × 144.13 × 10−3 (kg mol−1), end groups omitted. | |||||||
| 1 | 1/— | Toluene | 90 | 24 | 73 | 6.6 (1.29) | 10.5 |
| 2 | 1/— | DCM | 25 | 4 | 0 | — | — |
| 3 | —/AgBArF | DCM | 25 | 4 | 0 | — | — |
| 4 | 1/AgBArF | DCM | 25 | 1 | 76 | 9.5 (1.22) | 10.9 |
| 5 | 1/AgBArF | Toluene | 90 | 0.25 | 49 | 7.2 (1.21) | 7.1 |
The polymers feature low dispersity (Đ ∼ 1.2), and molar masses in close agreement with the calculated Mn assuming only one polymer chain per Ti centre (i.e. only one OiPr group initiating the polymerisation, as observed for 1 initiating rac-LA ROP at 90 °C). A plot of ln([rac-LA]0/[rac-LA]t) versus time (Fig. S15, ESI‡) shows a linear trend, as commonly reported for LA ROP initiated by well-defined metal catalysts. Polymer analysis by MALDI-ToF mass spectrometry (Fig. S17, ESI‡) shows no evidence of transesterification (i.e. peaks are spaced by 144 a.u.) or cyclic polymers, and suggests the formation of linear polymers (OiPr or OH end-capped), in line with a polymerisation occurring via a coordination–insertion mechanism.
The catalytic system 1 + AgBArF was also tested at 90 °C (in toluene), and showed high activity under these conditions, converting 49% of rac-LA in 15 minutes, compared to 12% in 1 hour without AgBArF in similar conditions.
To further highlight the need of both Ti and Ag metals not only to initiate but also to propagate the reaction, further investigations were carried out. Thus, whereas 1 alone (1 equiv.) is unable to initiate rac-LA ROP in DCM at room temperature (0% in 30 minutes), subsequent addition of AgBArF (1 equiv.) immediately triggers the polymerisation and leads to almost full conversion of rac-LA in 4 hours (Fig. S19, ESI‡). Conversely, once the polymerisation is initiated (i.e. using an equimolar amount of 1 and AgBArF), addition of a silver chelating agent such as bipyridine (bipy) immediately stops the polymerisation, presumably via formation of a Ag(bipy)2+ complex and thus inhibiting any beneficial Ag–Ti cooperativity for rac-LA ROP.
 | ||
| Scheme 1 rac-LA ROP initiated by 1 + AgBArF (DCM, 25 °C, [rac-LA]0 = 1 mol L−1). | ||
In order to gain a better understanding of the observed cooperativity, a stoichiometric reaction between 1 and AgBArF was monitored by 1H NMR (400 MHz, C6D6) and UV-vis. spectroscopies. Addition of 1 equivalent of AgBArF to 1 equivalent of 1 in C6D6 at room temperature leads to immediate and significant shifts in the 1H NMR spectrum to all signals previously associated to 1, suggesting interactions of 1 with AgBArF and formation of a new species, referred to as 1-AgBArF (Scheme 2). Interestingly, the resulting 1H NMR spectrum of 1-AgBArF shows two distinct signals attributed to two distinct OiPr groups (δ1H = 3.91 ppm, and d1H = 3.56 ppm), suggesting potential interactions of Ag+ with one OiPr group resulting in a loss of symmetry in solution compared to 1 (δ1H(OiPr) = 4.13 ppm). Imine and phenolate signals are also shifted compared to 1 (see Fig. S20–S23, ESI‡) suggesting potential interactions with the phenolate moieties, presumably via cation–π interactions as depicted in Scheme 2. It should be noted that despite moderate solubility in C6D6, AgBArF completely dissolves in the presence of 1 supporting the presence of attractive interactions between both species leading to 1-AgBArF (variable temperature 1H NMR analysis in Fig. S9–S11, ESI‡). In addition, the hypothesised 1-AgBArF was found be stable in C6D6 for up to 6 hours as determined by 1H NMR spectroscopy, which is well within the timescale for the observed polymerisation reaction to reach completion. Finally, as determined by 1H NMR spectroscopy (Fig. S22, ESI‡), addition of 2 equivalents of bipy to a solution of 1-AgBArF in C6D6 leads to the re-formation of 1 along with the formation of a new species presumed to be Ag(bipy)2·BArF. Overall, this confirms the existence of weak attractive interactions between 1 and Ag+ to form a heterobimetallic species 1-Ag+, presumably via interactions involving one OiPr group and the phenolate moieties of the salen ligand.
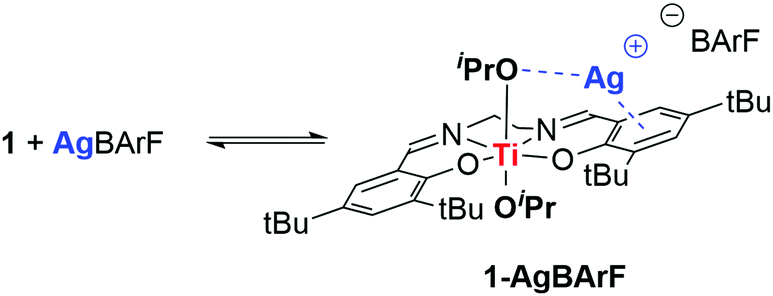 | ||
| Scheme 2 Proposed interactions of 1 with AgBArF leading to 1-AgBArF. | ||
Upon addition of 1 equiv. of AgBArF to 1 equiv. of 1 in C6D6, DOSY analysis (Fig. S23–S25, ESI‡) confirms the formation of a single species with a diffusion coefficient in line with an aggregated species comprising the characteristic signals attributed to the BArF counter-anion and the salen ligand. Upon the addition of 2 equiv. of bipy, multiple species with distinct diffusion coefficients are observed in line with the formation of 1 and a hypothesised cationic Ag(bipy)2+ complex with a dissociated BArF−counter anion.
UV-vis absorption spectroscopy analysis was also conducted to gather further structural information on 1-AgBArF (Fig. S26, ESI‡). Both 1 and 1-AgBArF (in situ generated) show absorption bands at λ ∼ 350–450 nm assigned to ligand-centred transitions.25 Thus, upon addition of 1 equivalent of AgBArF to a solution of 1 in DCM, the absorption maximum wavelength is blue-shifted from λmax = 380 nm to λmax = 368 nm (Δλ = 12 nm), in line with Ag+ interactions affecting the conjugated π system of the salen ligand.
Finally, in order to gain better evidence of the molecular structure of 1-AgBArF and insights into the role of both metals during the polymerisation reaction, DFT calculations were carried out (see details in ESI‡). The coordination of Ag(I) salts to metal salen complexes has previously been reported.26 Ag(I) ions have been found to coordinate via two oxygen atoms of the phenolate moieties, as well as via π interactions with the phenolate rings of Cu salen complexes.26b Attempts to coordinate an Ag+ atom to the oxygen atoms of the phenolate moieties (i.e. k2 fashion coordination) as previously observed for unsubstituted Cu salen complexes were unsuccessful due to the bulkiness of the tBu groups.26 Instead, the Ag+ ion was found to coordinate with one of the phenolate moieties via cation–π interactions and with one oxygen atom of the alkoxide groups (i.e.I-Ag+ in Scheme 3). This is supported by non-covalent interaction (NCI) surfaces (Fig. S35, ESI‡) and the molecular electrostatic potential surface of I indicating regions of high electron density above the phenolate moieties defining a suitable ‘pocket’ for Ag+ cation docking (Fig. S33, ESI‡). Silver cations have been known to act as bifunctional Lewis acids via π-binding to carbon–carbon multiple bonds, as well as σ-binding to heteroatoms.27 Such a coordination of Ag+ leads to two non-equivalent Ti–OMe bonds (Fig. S29, ESI‡), in line with the NMR spectrum showing two distinct signals for both OiPr groups. The Ti–OMe bond with the OMe group binding to Ag+ is lengthened to around 2.02 Å (vs. 1.85 Å in I), leading to a reduced π donation from the O atom to Ti, which was previously proposed to enhance catalytic activity in Ti-initiated ROP (Fig. S42, ESI‡).10
 | ||
| Scheme 3 Energy profile for l-LA ring-opening by 1-Ag+ (ωB97xD/def2-SVP cpcm = benzene), data available at DOI: http://10.14469/hpc/8730. | ||
Considering the reaction of one molecule of L-lactide with I-Ag+, the presence of Ag+ was found to offer ‘electrophilic assistance’ which renders one of the methoxy ligands more labile and therefore facilitates the nucleophilic attack onto the carbonyl of the lactide, also coordinated to Ag+ (III-Ag+-TS, in Scheme 3 and Fig. S38, ESI‡). To some extent, Ag+ not only ‘activates’ the monomer (i.e. enhanced electron deficiency on the C atom of the carbonyl) but also contributes to activating the nucleophilic actor (i.e. elongated Ti–OMe bond). Contrary to a monometallic coordination-insertion mechanism where both monomer activation and nucleophilic attack occur at the same metal, the presence of a second metal (i.e. Ag+) near the Ti catalytic site allows the distribution of both mechanistic elements across both metals, hence providing cooperativity.11 Thus, whereas the Ti atom offers restricted coordination sites, the Ag cation does not feature an initiating group but combining both allows Ag+ to activate the monomer (i.e. as an electrophilic partner) and the Ti alkoxide moiety to act as the nucleophilic partner. Similarly, in the ring-opening step (VII′-Ag+-TS in Scheme 3 and Fig. S38, ESI‡), the presence of an Ag+ atom facilitates the ring-opening by reducing geometrical constraints in the transition state. The overall energy barrier was found to be ∼24 kcal mol−1 (in line with a reaction occurring at room temperature) and supports a cooperative Ti/Ag mechanism where both hard and soft metals act cooperatively. These results are in line with previous experimental observations by Garden and co-workers suggesting that less electronegative metals with large radii preferably activate monomers whereas more Lewis acidic metals are preferably involved in the nucleophilic attack.3a,13 In addition, the “malleable” cation–π interactions allow Ag+ to adjust its position above the phenolate ring and to adapt to the different geometries of key intermediates lowering overall energy barriers.
The combination of a Ti–salen complex with AgBArF reveals unprecedented hard/soft metal cooperativity in lactide ROP. Whereas both entities separately do not exhibit any activities, the combination of both leads to an active catalytic system for lactide ROP at room temperature. To the best of our knowledge, this is the first evidence of metal cooperativity involving silver in cyclic ester ROP.
Mechanistic, reactivity and computational studies suggest that cation–π interactions between the phenolate moieties of the salen ligand and the Ag+ cation enables the formation of a Ti/Ag heterobimetallic species responsible for the cooperativity. These findings open up new strategies exploiting NCI to reveal heterobimetallic cooperativity with existing metal phenolate complexes and inspire new ligand designs.
Finally, preliminary DFT calculations on a ROP mechanism initiated by a Ti/Ag heterobimetallic species provides some insights into the role of both metals during the polymerisation. The silver cation not only contributes towards activating the monomer but also renders the nucleophilic attack more facile by weakening the M–alkoxide bond. In addition, the flexible cation–π interaction between Ag+ and the phenolate ring allows the silver cation to easily adapt to the geometries of key intermediates during the polymerisation lowering overall energy barriers. This can be further exploited towards the polymerisation of “challenging” monomers which require efficient catalysts at low temperature to overcome thermodynamic constraints.28
Conflicts of interest
There are no conflicts to declare.References
- (a) R. Maity, B. S. Birenheide, F. Breher and B. Sarkar, ChemCatChem, 2021, 13, 2337–2370 CrossRef CAS; (b) C. Robert and C. M. Thomas, Chem. Soc. Rev., 2013, 42, 9392–9402 RSC; (c) T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed.
- (a) P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed; (b) D. Das, S. S. Mohapatra and S. Roy, Chem. Soc. Rev., 2015, 44, 3666–3690 RSC; (c) R. M. Haak, S. J. Wezenberg and A. W. Kleij, Chem. Commun., 2010, 46, 2713–2723 RSC.
- (a) W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed; (b) Z. Cai, D. Xiao and L. H. Do, Comments Inorg. Chem., 2019, 39, 27–50 CrossRef CAS; (c) J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed; (d) M. Khoshsefat, Y. Ma and W.-H. Sun, Coord. Chem. Rev., 2021, 434, 213788 CrossRef CAS.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) Y. Sun, L. Wang, D. Yu, N. Tang and J. Wu, J. Mol. Catal. A: Chem., 2014, 393, 175–181 CrossRef CAS; (b) M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS.
- J. Char, E. Brulé, P. C. Gros, M.-N. Rager, V. Guérineau and C. M. Thomas, J. Organomet. Chem., 2015, 796, 47–52 CrossRef CAS.
- L. Wang, S.-C. Roşca, V. Poirier, S. Sinbandhit, V. Dorcet, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2014, 43, 4268–4286 RSC.
- N. Maudoux, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5740–5748 CrossRef CAS.
- H.-Y. Chen, M.-Y. Liu, A. K. Sutar and C.-C. Lin, Inorg. Chem., 2009, 49, 665–674 CrossRef PubMed.
- A. J. Gaston, Z. Greindl, C. A. Morrison and J. A. Garden, Inorg. Chem., 2021, 60, 2294–2303 CrossRef CAS PubMed.
- J. Hao, J. Li, C. Cui and H. W. Roesky, Inorg. Chem., 2011, 50, 7453–7459 CrossRef CAS PubMed.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- (a) S. Gesslbauer, G. Hutchinson, A. J. P. White, J. Burés and C. Romain, ACS Catal., 2021, 11, 4084–4093 CrossRef CAS; (b) S. Gesslbauer, R. Salvela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 9, 7912–7920 CrossRef CAS; (c) S. Gesslbauer, H. Cheek, A. J. P. White and C. Romain, Dalton Trans., 2018, 47, 10410–10414 RSC.
- R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Chem. Commun., 2018, 54, 8024–8031 RSC.
- (a) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed; (b) K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- J. M. Maier, P. Li, J. Hwang, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2015, 137, 8014–8017 CrossRef CAS PubMed.
- S. Dagorne and C. Romain, PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd., 2016, pp. 1–44 Search PubMed.
- D. J. Liston, Y. J. Lee, W. R. Scheidt and C. A. Reed, J. Am. Chem. Soc., 1989, 111, 6643–6648 CrossRef CAS.
- Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC.
- C. K. Gregson, I. J. Blackmore, V. C. Gibson, N. J. Long, E. L. Marshall and A. J. White, Dalton Trans., 2006, 3134–3140 RSC.
- (a) H. Chen, P. S. White and M. R. Gagné, Organometallics, 1998, 17, 5358–5366 CrossRef CAS; (b) R. K. Gurung, C. D. McMillen, W. L. Jarrett and A. A. Holder, Inorg. Chim. Acta, 2020, 505, 119496 CrossRef CAS.
- (a) J. Cheng, K. Wei, X. Ma, X. Zhou and H. Xiang, J. Phys. Chem. C, 2013, 117, 16552–16563 CrossRef CAS; (b) K. Y. Hwang, H. Kim, Y. S. Lee, M. H. Lee and Y. Do, Chem. – Eur. J., 2009, 15, 6478–6487 CrossRef CAS PubMed.
- (a) M. Nayak, S. Sarkar, S. Hazra, H. A. Sparkes, J. A. K. Howard and S. Mohanta, CrystEngComm, 2011, 13, 124–132 RSC; (b) A. Biswas, S. Mondal, L. Mandal, A. Jana, P. Chakraborty and S. Mohanta, Inorg. Chim. Acta, 2014, 414, 199–209 CrossRef CAS; (c) N. Şenyüz, B. Yılmaz, H. Batı, E. Bozkurt and O. Büyükgüngör, Spectrochim. Acta, Part A, 2013, 101, 167–171 CrossRef PubMed.
- (a) Y. Yamamoto, J. Org. Chem., 2008, 73, 5210 CrossRef CAS; (b) K. Maeda, T. Takahashi, R. Tomifuji, N. Hirao, T. Kurahashi and S. Matsubara, Chem. Lett., 2018, 47, 532–535 CrossRef CAS.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05083d |
| ‡ Data are available at DOI: 10.14469/hpc/8729 |
| This journal is © The Royal Society of Chemistry 2021 |