
Construction of an isoquinolinone framework from carboxylic-ester-directed umpolung ring opening of methylenecyclopropanes†
Hao-Zhao
Wei
a,
Yin
Wei
*b and
Min
Shi
 *ab
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: mshi@mail.sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling-Ling Lu, Shanghai, 200032, China. E-mail: weiyin@sioc.ac.cn
First published on 27th September 2021
Abstract
An interesting type of reaction involving functionalized methylenecyclopropanes (MCPs) has been revealed. Here, a nucleophilic attack of an anionic species onto a partially polarity-reversed MCP was realized by treating a neighbouring carboxylic ester tethered to the MCP and amine with KHMDS to realize an umpolung ring opening of the MCP. This work established an operationally convenient protocol for the rapid construction of isoquinolinone frameworks.
Isoquinolin-1(2H)-one derivatives have attracted extensive interest for being a class of essential alkaloids and were documented early on to exhibit excellent biological and clinical activities, such as various antitumor and antidiabetic activities (Scheme 1).1 Moreover, the skeletons of some naturally and biologically valuable compounds such as ruprechstyril, pancratistatin and actinoplanone C consist of the significant isoquinolinone units (Scheme 1). To date, the efforts at constructing this key structural motif have been reported to mainly depend on using transition-metal-catalyzed transformations under harsh conditions, limiting their practical applications, especially in drug discovery.2
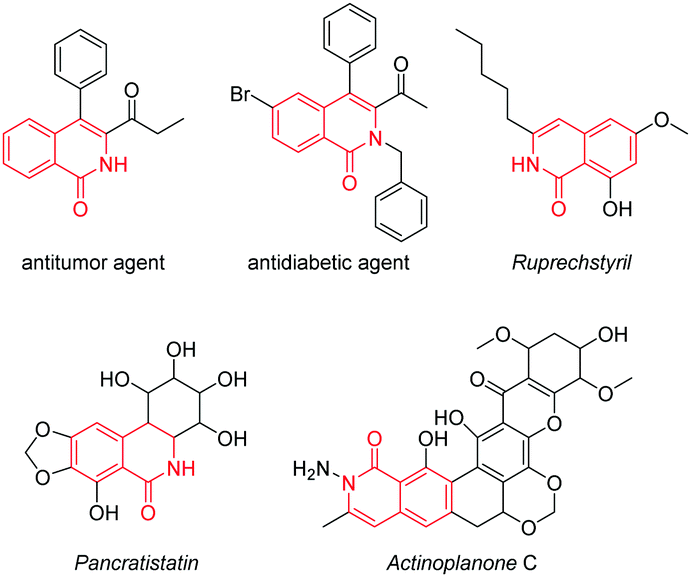 | ||
| Scheme 1 Bioactive and natural compounds each containing an isoquinolin-1(2H)-one framework. | ||
Methylenecyclopropanes (MCPs) are highly strained molecules that provide a sufficient thermodynamic driving force in organic reactions for generating carbocycles.3 Traditional types of reactions involving MCPs include (A) thermally induced or thermally promoted cyclizations,4 (B) reactions catalyzed by Lewis or Brønsted acids,5 (C) transition-metal-catalyzed reactions,6 (D) radical-promoted reactions,7 and other reactions similar to those involving alkenes (Scheme 2). However, to the best of our knowledge, the type of reaction in which an MCP is the direct target of a nucleophilic attack made by an electron-rich anionic species has not yet been reported, presumably due to the MCP unit itself being an electron-rich moiety. Hence, we envisaged that an electron-withdrawing carboxylic ester group introduced next to the MCP unit as a directing group8 might attract a nearby nucleophilic species, leading to an interesting umpolung ring opening of the cyclopropane (Scheme 2, this work).
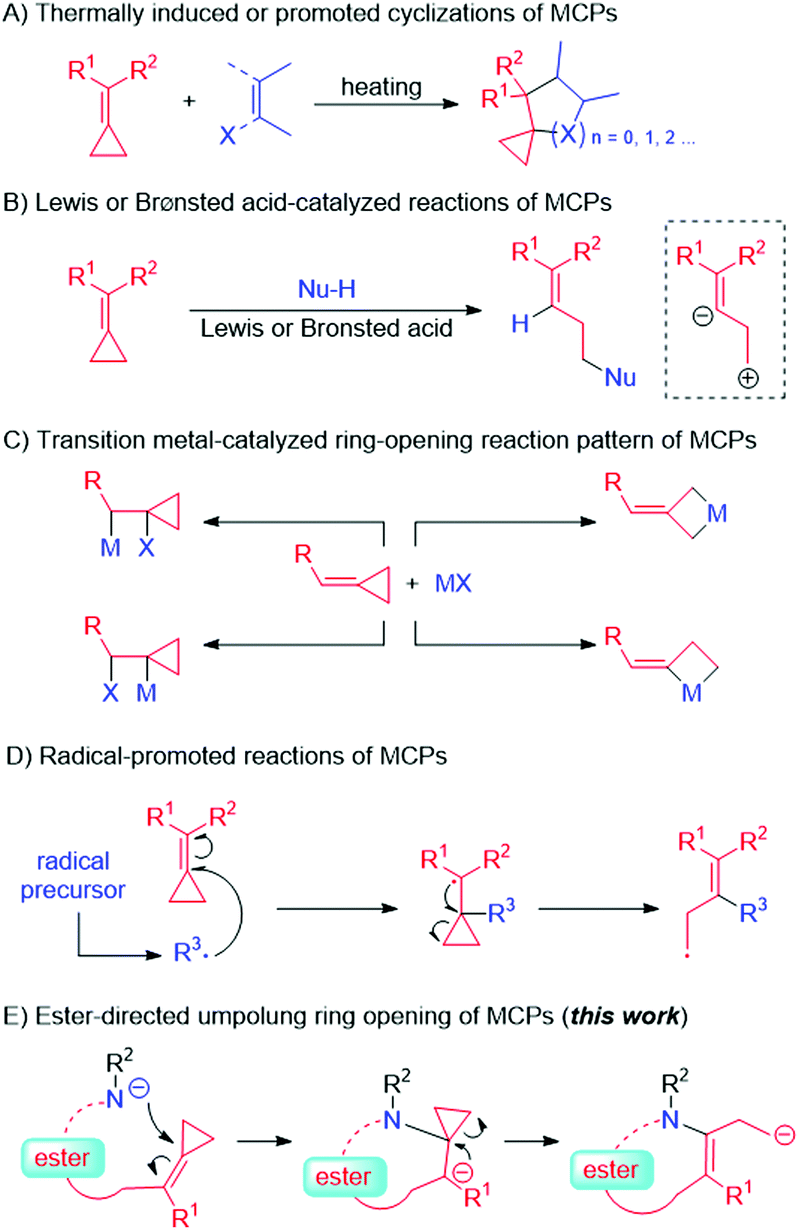 | ||
| Scheme 2 Reactions of MCPs. | ||
Our working hypothesis is shown in Scheme 3. Amides could be converted to amide anions upon being treated with a strong base having electron-withdrawing carbonyl groups and stabilized nucleophilic species. Therefore, considering that aromatic acid esters have been shown to react with anilines to provide amides in the presence of strong bases,9 we assumed that methyl-ester-tethered MCP 1a could be first transformed to amide III in the presence of p-methoxyaniline 2a and the base through a nucleophilic addition-elimination mechanism, and amide III could be next transformed to anion IV. Subsequently, the nucleophilic attack of nitrogen anion onto the partially polarity-reversed MCP unit owing to the neighbouring carbonyl group would provide the cyclized intermediate V, which would undergo aromatization and concerted protonation upon release of ring strain as the driving force to afford the ring-opened product 3a bearing an isoquinolinone motif.
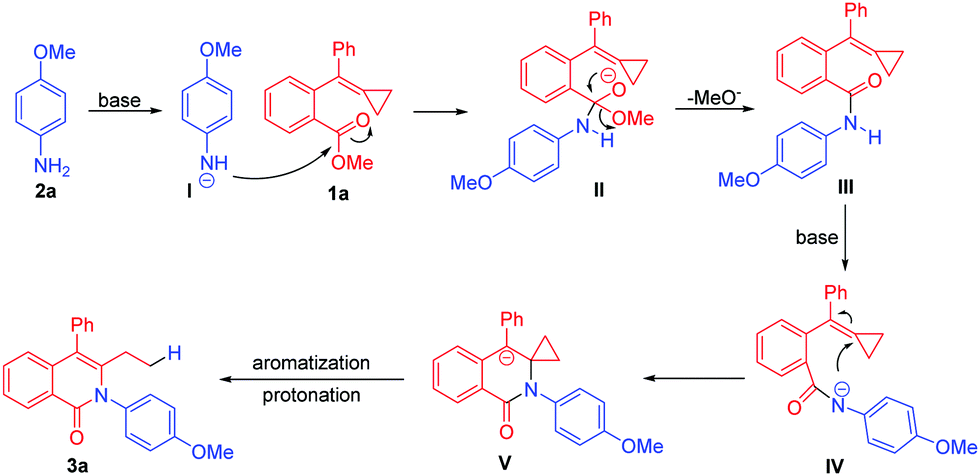 | ||
| Scheme 3 Proposed process achieving an umpolung ring opening of an MCP. | ||
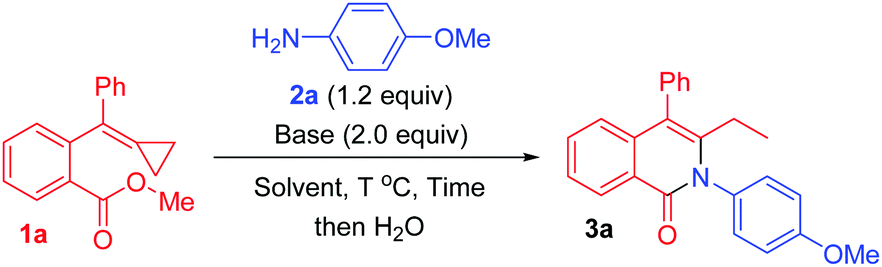
We initially used 1a as the model substrate to verify the reaction outcome in the presence of p-methoxyaniline 2a and LiHMDS (2.0 equiv.) in toluene at room temperature. To our delight, the desired cyclized product 3a was obtained in 54% yield after 24 h (Table 1, entry 1). When LiHMDS was added into the reaction mixture at 0 °C, 3a was obtained in 98% yield after 10 h (Table 1, entry 2). Considering that the nucleophilic attack of an anionic species onto the MCP unit is unusual and may be sensitive to the reaction temperature, we next carried out the reaction at 50 °C after adding LiHMDS into the reaction mixture at 0 °C. As we expected, the required reaction duration was reduced to 1.5 h, affording 3a in 86% yield (Table 1, entry 3). Subsequently, several other bases such as KHMDS, t-BuOK, DBU, and K2CO3 were examined, and we found KHMDS to be the best choice, furnishing 3a in 96% yield after 1.5 h (Table 1, entries 4–7). Some other solvents were also tested for the reaction, but no improved results were obtained (Table 1, entries 8–10), and hence toluene was considered to be the solvent of choice in this transformation, and gave 3a in 95% isolated yield (Table 1, entry 11). It should be noted that LiHMDS and KHMDS, but not DBU and K2CO3, were dissolved in THF, and thus the solvents in these cases were actually mixed solvents (for more details, see Table S1 in the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solventa | T [°C] | Time (h) | Yield/3ab [%] |
| Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.24 mmol, 1.2 equiv.), base (0.4 mmol, 2.0 equiv.), solvent, T °C, time, quenched by water.a Except DBU and K2CO3, bases were dissolved in THF, thus solvents are mixed solvents in these entries in fact.b 1H NMR yield using 1,3,5-trimethoxybenzene as an internal standard.c Isolated yield. | |||||
| 1 | LiHMDS | Toluene | r.t. | 24 | 54 |
| 2 | LiHMDS | Toluene | 0-r.t. | 10 | 98 |
| 3 | LiHMDS | Toluene | 0–50 | 1.5 | 86 |
| 4 | KHMDS | Toluene | 0–50 | 1.5 | 96 |
| 5 | t BuOK | Toluene | 0–50 | 1.5 | 0 |
| 6 | DBU | Toluene | 0–50 | 1.5 | 0 |
| 7 | K2CO3 | Toluene | 0–50 | 1.5 | 0 |
| 8 | KHMDS | MeCN | 0–50 | 1.5 | 0 |
| 9 | KHMDS | DCE | 0–50 | 1.5 | 0 |
| 10 | KHMDS | THF | 0–50 | 1.5 | 94 |
| 11 | KHMDS | Toluene | 0–50 | 1.5 | 95c |
With the optimal reaction conditions in hand, we next evaluated various aromatic acid esters each tethered to the MCP unit for this reaction (Scheme 4). A methyl group, tertiary butyl group, methoxy group and halogen atoms were each tested as the R1 group at the para-position of the benzene ring and provided the corresponding products 3b–3g in 65%–98% yields. However, when MCP 1h bearing a trifluoromethyl group was applied to the reaction, only a trace amount of 3h was obtained, and the reaction system became messy even at low temperature, presumably due to the high reactivity of the substituted trifluoromethyl group and ester moiety tethered to the MCP in this base-promoted transformation. When methyl or methoxy as the R1 group was introduced at the other positions, the desired products 3i–3k were also attained in high yields. Then, we examined a methyl group or halogen atom as the R2 position substituent, and found that the cyclization reactions proceeded smoothly, affording 3l and 3m in 87% and 98% yields, respectively. It should be pointed out that for substrate 1l, a higher temperature (80 °C) was required for the reaction to proceed to this yield, perhaps due to the induction effect of the carbonyl group having been impaired by the methyl substituent. Interestingly, using 1n as a substrate, 3n′ was obtained in 95% yield, and was derived from the substitution of in situ-generated methoxyl anion at the fluorine atom position.10
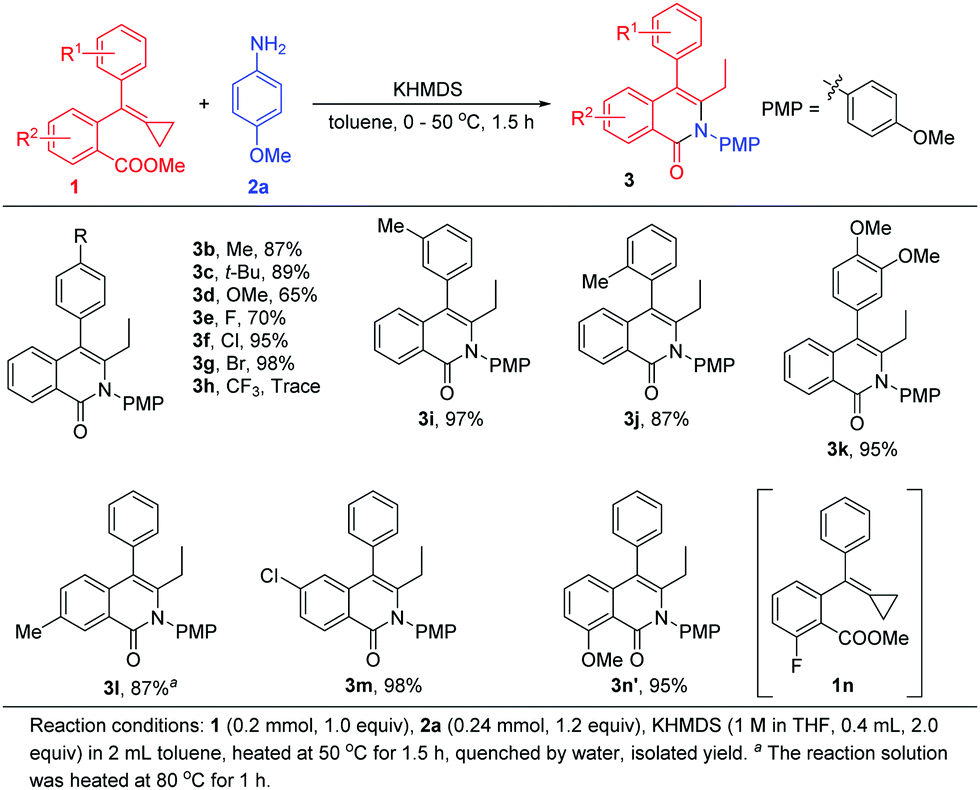 | ||
| Scheme 4 Scope of aromatic acid esters tethered to the MCP group for the cyclization. | ||
Various aromatic amines were also examined (Scheme 5). An H atom, an isopropyl group, a tertiary butyl group, halogen atoms, an N,N-dimethyl group, and an acetenyl group were each tested as an R group at the para-position, and resulted in the formation of 3o–3t and 3v in moderate to excellent yields ranging from 42% to 99%. The structure of 3o was unambiguously determined in the current work using X-ray diffraction and its ORTEP drawing is shown in Scheme 5. (For details of 3o's X-ray crystallographic data, see the ESI†). Nevertheless, for the 4-trifluoromethylaniline 2u, only a trace amount of product was obtained, perhaps owing to its weak nucleophilicity. The use of other anilines, in particular 3-methoxyaniline and 3-chloro-4-methoxyaniline, delivered the corresponding products 3w and 3x in good yields as well. When using sterically hindered anilines in this reaction, the reaction also proceeded smoothly, providing 3y and 3z in 68% and 66% yields, respectively. Heterocyclic aromatic amine 2aa was also compatible with this reaction, giving 3aa in 51% yield.
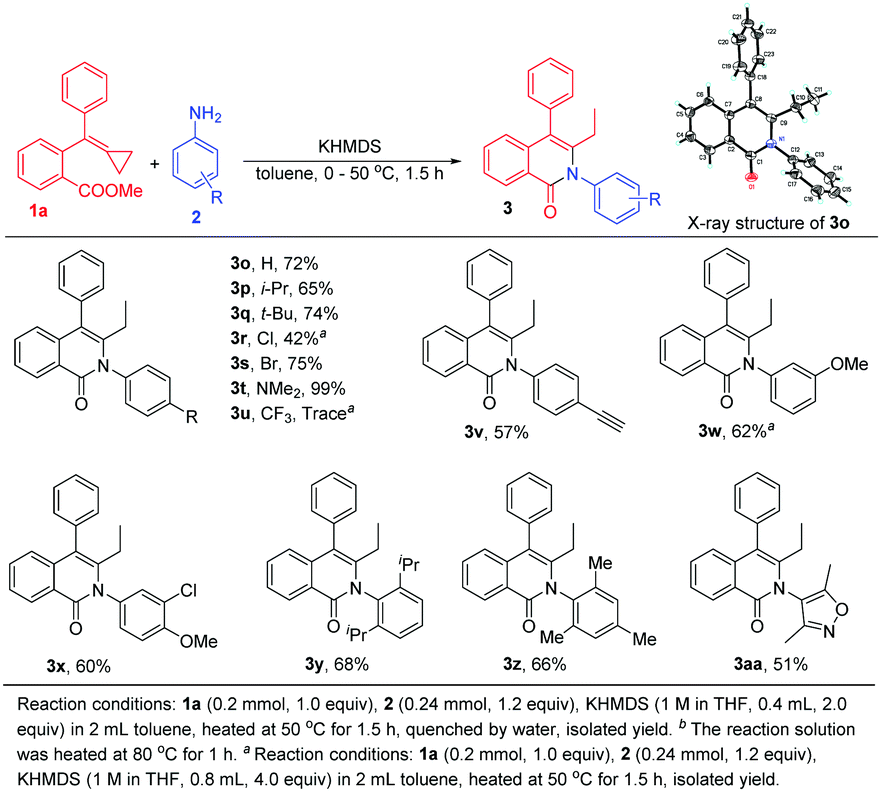 | ||
| Scheme 5 Scope of aromatic amines for the cyclization. | ||
When 2-aminopyridine 2ab was applied to this cyclization, we found an interesting migration of the pyridine ring, affording 4a in 85% yield (Scheme 6). Its structure was determined using X-ray diffraction and the corresponding products 4b and 4c were obtained in moderate yields when R was a halogen atom.
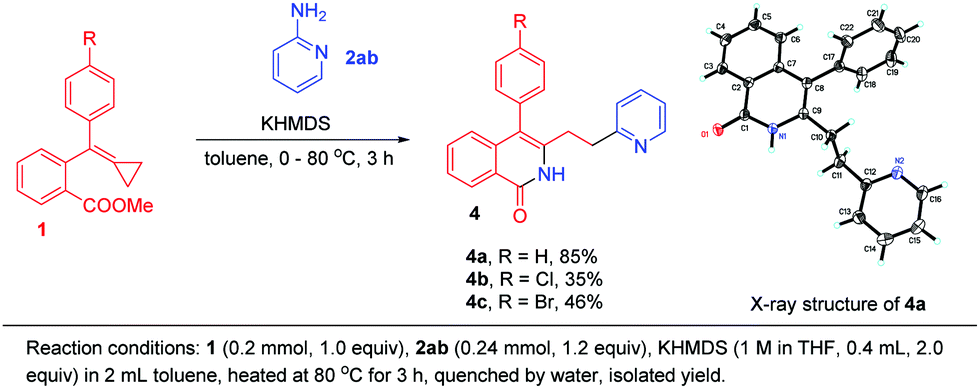 | ||
| Scheme 6 Migrations of the pyridine rings. | ||
To clarify the reaction mechanism, we carried out the reaction of 1a with 2a at room temperature and quenched the reaction with saturated aqueous NH4Cl in 10 minutes. We found that the amide 5a was isolated in 80% yield along with 5% of 3a, and that 5a was subsequently transformed to 3a in 56% yield upon adding KHMDS (for more details, see section S4 in ESI†), supporting the mechanism proposed in Scheme 3 (Scheme 7a). Therefore, the pyridine ring migration may have proceeded through a Meisenheimer complex as shown in Scheme 7b.
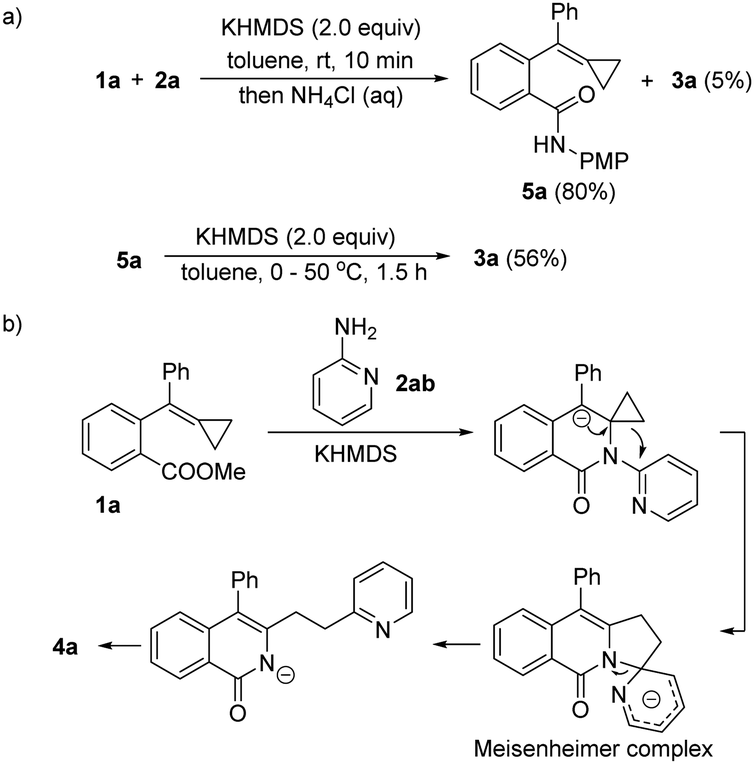 | ||
| Scheme 7 Proposed mechanism for the pyridine ring migration. | ||
To further evaluate the practicability of this novel synthetic methodology, the scale of the reaction of 1a with 2a was increased, specifically to a gram scale, and afforded 3a in 95% yield, showing the synthesis to be potentially practical for constructing heterocyclic materials and bioactive molecules (Scheme 8).
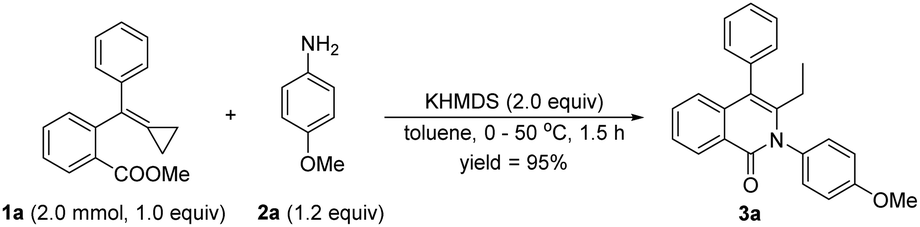 | ||
| Scheme 8 Cyclization reaction on a gram scale. | ||
When alkyl amines were used instead of aromatic amines for this cyclization reaction with 1a, none of the cyclized products formed, and 1a was not consumed at all, indicating that the amide intermediates were not generated. Therefore, we attempted to synthesize the alkyl-substituted amides from condensations between carboxylic-acid-tethered MCPs and alkyl amines to directly obtain 6a and 6b, which were applied for the cyclization in the presence of KHMDS (1.2 equiv.). To our delight, the desired products 7a and 7b were obtained in, respectively, 70% and 84% yields at 0 °C within 30 minutes, demonstrating a broad scope for this reaction (Scheme 9).
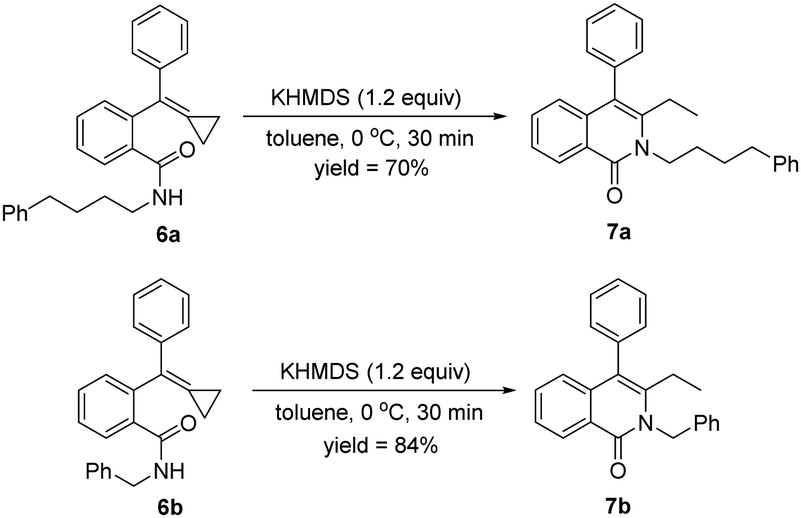 | ||
| Scheme 9 Use of alkyl amines instead of aromatic amines for the cyclization. | ||
In addition, it should be emphasized here that when the MCP unit of 1a was replaced by a methylene group, no cyclization took place, indicating the essential nature of the MCP unit for this protocol (for more details, see section S4 in ESI†).
In conclusion, we have developed an interesting type of reaction involving carrying out a nucleophilic attack of an anionic species onto a partially polarity-reversed MCP unit upon treating a neighbouring carboxylic ester tethered to the MCP and an amine with KHMDS for the rapid construction of an isoquinolinone framework through an umpolung ring opening of the MCP under mild conditions. The ester moiety served as a directing group in this transformation. This newly developed protocol represents a straightforward and environmentally friendly manipulation without using transition metal catalyst, and has provided a valuable tool for the applications of functionalized MCPs in organic synthesis.
We are grateful for the financial support from the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078) and the Fundamental Research Funds for the Central Universities 222201717003.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Q. Long, K. Zou, W. Dong, D. Xie and D. An, Synth. Commun., 2021, 1–12, DOI:10.1080/00397911.2021.1952433; (b) K. Hemalatha and G. Madhumitha, Appl. Microbiol. Biotechnol., 2016, 100, 7799–7814 CrossRef CAS PubMed; (c) S. F. Hussain, S. Nakkady, L. Khan and M. Shamma, Phytochemistry, 1983, 22, 319–320 CrossRef CAS; (d) V. U. Ahmad, R. Atta ur, T. Rasheed, R. Habib ur and A. Q. Khan, Tetrahedron, 1987, 43, 5865–5872 CrossRef CAS; (e) Y. Aly, A. Galal, L. K. Wong, E. W. Fu, F.-T. Lin, F. K. Duah and P. L. Schiff, Phytochemistry, 1989, 28, 1967–1971 CrossRef CAS; (f) B. D. Krane and M. Shamma, J. Nat. Prod., 1982, 45, 377–384 CrossRef CAS.
- (a) S. Allu and K. C. K. Swamy, J. Org. Chem., 2014, 79, 3963–3972 CrossRef CAS PubMed; (b) K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 5353–5357 CrossRef CAS PubMed; (c) T. Li, C. Zhang, Y. Tan, W. Pan and Y. Rao, Org. Chem. Front., 2017, 4, 204–209 RSC; (d) D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650–9654 CrossRef CAS PubMed; (e) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed; (f) N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361–12365 CrossRef CAS PubMed.
- (a) J. Liu, R. Liu, Y. Wei and M. Shi, Trends Chem., 2019, 1, 779–793 CrossRef CAS; (b) H. Cao, F. Chen, C. Su and L. Yu, Adv. Synth. Catal., 2019, 362, 438–461 CrossRef; (c) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379–8392 RSC; (d) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 CrossRef CAS PubMed; (e) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
- (a) L.-Z. Yu, X.-B. Hu, Q. Xu and M. Shi, Chem. Commun., 2016, 52, 2701–2704 RSC; (b) H.-Z. Wei, Q.-Z. Li, Y. Wei and M. Shi, Org. Biomol. Chem., 2020, 18, 7396–7400 RSC; (c) H.-Z. Wei, L.-Z. Yu and M. Shi, Org. Biomol. Chem., 2020, 18, 135–139 RSC.
- (a) L.-P. Liu and M. Shi, J. Org. Chem., 2004, 69, 2805–2808 CrossRef CAS PubMed; (b) L. Ackermann, S. Kozhushkov, D. Yufit and I. Marek, Synlett, 2011, 1515–1518 CrossRef CAS; (c) M. Shi, M. Jiang and L.-P. Liu, Org. Biomol. Chem., 2007, 5, 438–440 RSC; (d) B. Xu and M. Shi, Org. Lett., 2003, 5, 1415–1418 CrossRef CAS PubMed.
- (a) D. Li, W. Zang, M. J. Bird, C. J. T. Hyland and M. Shi, Chem. Rev., 2020, 121, 8685–8755 CrossRef PubMed; (b) N. Kimura, S. Katta, Y. Kitazawa, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2021, 143, 4543–4549 CrossRef CAS PubMed; (c) J. Yao, Z. Chen, L. Yu, L. Lv, D. Cao and C.-J. Li, Chem. Sci., 2020, 11, 10759–10763 RSC; (d) Y. Liu, A. A. Ogunlana and X. Bao, Dalton Trans., 2018, 47, 5660–5669 RSC; (e) Y. Liu, S. Feng and X. Bao, ChemCatChem, 2018, 10, 2817–2825 CrossRef CAS.
- (a) H.-Z. Wei, Y. Wei and M. Shi, Org. Chem. Front., 2021, 8, 4527–4532 RSC; (b) Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011–3014 RSC; (c) M. Chen, Y. Wei and M. Shi, Org. Chem. Front., 2020, 7, 374–379 RSC; (d) Z.-Z. Zhu, K. Chen, L.-Z. Yu, X.-Y. Tang and M. Shi, Org. Lett., 2015, 17, 5994–5997 CrossRef CAS PubMed.
- (a) X. He, Y. Tang, Y. Wang, J. Chen, S. Xu, J. Dou and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 10698–10702 CrossRef CAS PubMed; (b) J. Wu, Y. Tang, W. Wei, Y. Wu, Y. Li, J. Zhang, Y. Zheng and S. Xu, Angew. Chem., Int. Ed., 2018, 57, 6284–6288 CrossRef CAS PubMed.
- G. Li, C.-L. Ji, X. Hong and M. Szostak, J. Am. Chem. Soc., 2019, 141, 11161–11172 CrossRef CAS PubMed.
- J. Su, Q. Chen, L. Lu, Y. Ma, G. H. L. Auyoung and R. Hua, Tetrahedron, 2018, 74, 303–307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data. CCDC 2074397 and 2091583. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04826k |
| This journal is © The Royal Society of Chemistry 2021 |