
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H Electrophilic (phenylsulfonyl)difluoromethylation of (hetero)arenes with a newly designed reagent†
Enzo
Nobile
 ,
Thomas
Castanheiro
and
Tatiana
Besset
*
,
Thomas
Castanheiro
and
Tatiana
Besset
*
Normandie Univ, INSA Rouen, UNIROUEN, CNRS, COBRA (UMR 6014), 76000 Rouen, France. E-mail: tatiana.besset@insa-rouen.fr
First published on 8th November 2021
Abstract
The synthesis of an original electrophilic difluoromethylating reagent was successfully achieved upon a straightforward protocol (3 steps). Like a Swiss army knife, this bench-stable reagent allowed the functionalization of various classes of compounds under mild and transition metal-free conditions. Hence, an efficient and operationally simple tool for the construction of C(sp2)-, C(sp3)- and S-CF2SO2Ph bonds was provided, expanding the chemical space of PhSO2CF2-containing molecules. Late-stage functionalization of bioactive molecules and the synthesis of PhSO2CF2- and HCF2-analogs of Lidocaine were also successfully achieved.
Organofluorine chemistry is a research area of paramount importance as fluorinated molecules are prevalent derivatives in materials science, and the agrochemical and pharmaceutical industries.1 More than 40% of pharmaceuticals approved by the FDA in 2019 contain at least one fluorine atom,2 which can be explained by the fact that the incorporation of a fluorine atom or a fluorinated moiety drastically impacts the properties of the corresponding molecules.3 Therefore, over the years, the field of organofluorine chemistry has been revolutionized by impressive advances including original and efficient transformations along with the discovery and design of potent fluorinated groups, which have widened the chemical space of fluorinated compounds.4 Whilst special attention was paid to the HCF2 group,5 design of the FGCF2 moiety has emerged as an interesting lead to be followed. Among them, the PhSO2CF2 residue proved to be of high importance and has aroused the interest of the scientific community.6 Found in the analog of Fluconazole with antifungal activities,7 this fluorinated group is a real synthetic handle. Indeed, it is easily converted into other valuable difluorinated moieties such as the difluoromethyl (HCF2), the difluoromethylene (–CF2–), and the difluoromethylidene (= CF2) groups. These were found in drugs such as Eflornithine8 and Seletracetam,9 for instance. The main synthetic pathways to build up a C–CF2SO2Ph bond relied on the use of nucleophilic sources of the (phenylsulfonyl)difluoromethyl residue or proceeded via a radical process.6 In sharp contrast, methodologies involving electrophilic reagents are restricted to two major contributions from key players in the field, namely the groups of Hu10 and Shibata.11 They designed two electrophilic sources based on a hypervalent iodine and a sulfonium salt, respectively (Scheme 1). While the group of Hu showcased that its source was very efficient for the functionalization of thiols as well as for the copper-catalyzed allylic and vinylic (phenylsulfonyl)difluoro-methylation reaction, Shibata and co-workers successfully applied their reagent to the functionalization of C(sp3) centers with β-ketoesters, β-diketones and dicyanoalkylidenes. Although those reagents served as a proof-of-concept, we wondered whether it would be possible to synthesize a new electrophilic reagent, which will (1) be easily accessible on a large scale, (2) not require ozone depleting reagents for its synthesis and (3) react with a large panel of nucleophiles. Such a reagent will definitely facilitate access to difluoromethylated compounds.
 | ||
| Scheme 1 State of the art and this work. | ||
By analogy with Umemoto's reagent and its analogs,12 we anticipated that the backbone of the reagent will be important in reaching a good reactivity. Therefore, we envisaged the synthesis of an S-([phenylsulfonyl]difluoromethyl)dibenzothiophenium salt. Herein, we disclose a simple and straightforward synthesis of an electrophilic source of a PhSO2CF2 group and its broad application in various transformations.
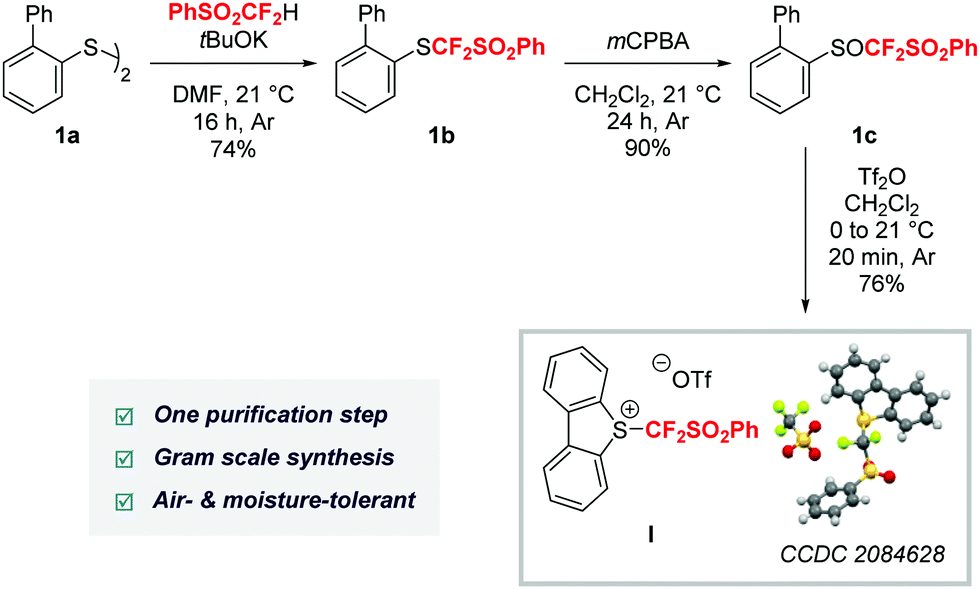
With these considerations in mind, we embarked on the preparation of reagent I. This newly designed reagent was synthesized in a three step/one purification sequence in 51% overall yield starting from the commercially available or easy-to-synthesize biaryl disulfide 1a13 and (difluoromethyl)sulfonylbenzene.14,15 First, the (phenylsulfonyl)difluoromethylation of the disulfide 1a under basic conditions provided 1b in 74% yield. Selective oxidation of the sulfane with mCPBA furnished 1c in 90% yield, without any purification. Finally, upon treatment with triflic anhydride, the sulfoxide 1c was converted into the corresponding dibenzothiophenium salt I in 76% yield. The structure of I was further ascertained using X-ray analysis.15 The reagent turned out to be air- and moisture-tolerant and bench-stable and was kept for more than three months at room temperature under air with no alteration of its reactivity. Remarkably, its transition metal-free synthesis did not require any ozone depleting fluorinated reagents (Scheme 2) and was conveniently scalable as reagent I was obtained in 46% overall yield on a 48 mmol scale.
 | ||
| Scheme 2 Synthesis of reagent I. Reaction performed on a 5 mmol scale. See the ESI† for more details. | ||
Having this reagent in hand, we first explored its reactivity towards the (phenylsulfonyl)difluoromethylation of aniline derivatives with complete selectivity towards the formation of a C(sp2)–CF2SO2Ph bond (Scheme 3). When aniline 2a was reacted with reagent I, compound 3a was obtained in a high yield as an easy-to-separate mixture of ortho and para regioisomers, the ortho isomer being the major one.15,16 As these fluorinated molecules might be of high interest for medicinal chemistry and drug discovery, access to both regioisomers in one transformation might constitute a substantial advantage. Then, a panel of original and diversely para-substituted (phenylsulfonyl)difluoromethylated anilines was synthesized. Anilines substituted with electron-donating groups (2b–2d), halogens (2e and 2f) and electron-withdrawing groups (2g and 2h) were all suitable substrates, the transformation being more efficient with electron-rich compounds. The reaction was tolerant towards various functional groups such as ketones (3g) and esters (3h). When the 3-chloroaniline 2i reacted with reagent I, the functionalization occurred on both ortho positions, and the major isomer, resulting from the (phenylsulfonyl)difluoromethylation at the less sterically hindered position, was preferentially obtained as ascertained using NMR experiments and X-ray analysis (CCDC 2084629).15 In the case of 2-methylaniline, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of both the ortho- and para- substituted products (3j) was obtained in 83% yield as confirmed using 2D-NMR experiments. Interestingly, the 2,6-xylidine 2k, a key scaffold found in several anesthetics such as Lidocaine, Bupicaine, Mepivacaine and Etidocaine, was smoothly functionalized in 84% yield. It is worth mentioning that heteroarylamines (2l and 2m) were also functionalized in good yields.15 The methodology was not restricted to the functionalization of primary anilines as N-methylaniline reacted and 3n was isolated in 48% yield for the ortho isomer.
:1 mixture of both the ortho- and para- substituted products (3j) was obtained in 83% yield as confirmed using 2D-NMR experiments. Interestingly, the 2,6-xylidine 2k, a key scaffold found in several anesthetics such as Lidocaine, Bupicaine, Mepivacaine and Etidocaine, was smoothly functionalized in 84% yield. It is worth mentioning that heteroarylamines (2l and 2m) were also functionalized in good yields.15 The methodology was not restricted to the functionalization of primary anilines as N-methylaniline reacted and 3n was isolated in 48% yield for the ortho isomer.
 | ||
| Scheme 3 (Phenylsulfonyl)difluoromethylation of anilines 2. Reaction conditions: 2 (3 equiv.), I (0.3 mmol, 1 equiv.), CH2Cl2 (0.15 M), 21 °C, 24 h, under argon. Isolated yields are given. In the case of the regioisomers, combined isolated yields were provided and the ratio of regioisomers, in parentheses, was determined using 19F NMR of the crude reaction mixture using α,α,α-trifluoro-acetophenone as an internal standard. aThe minor isomer was isolated with an inseparable impurity. b8 days. c10 days. d12 days. e4 days. fThe product was isolated with an inseparable impurity. gOnly this isomer was isolated in 48% yield. | ||
Encouraged by these results, we turned our attention to the electrophilic (phenylsulfonyl)difluoromethylation of electron-rich (hetero)arenes (Scheme 4). First indoles (4a–4e), a key scaffold for medicinal chemistry, were studied. Non-protected and N-protected 3-substituted indoles (4a–4c) were smoothly functionalized at the C2 position. When the N-methylindole 4d was used as the substrate, the expected product 5d was obtained as a mixture of regioisomers. Interestingly, the C2-functionalization of pyrroles was efficiently achieved, except in the case of 5h, which was isolated in a modest 27% yield. The (phenylsulfonyl)difluoromethylation of phenol and anisole derivatives (4i–4l) provided the corresponding products in decent to good yields. The late-stage functionalization of bioactive molecules such as Melatonin (4e), “the sleep hormone”, and Propofol (4j), a potent anesthetic, went smoothly. The expected products (5e and 5j) were obtained with complete regioselectivity in 74% and 70% yields, respectively. To further demonstrate the potential of reagent I, the functionalization of C(sp3)-centered nucleophiles was studied (Scheme 5). Pleasingly, in the presence of DBU, β-ketoesters 6 and dicyanoalkylidene 8 were efficiently functionalized at low temperature (−78 °C or −42 °C) and the corresponding (phenylsulfonyl)difluoromethylated products 7a, 7b and 9 were isolated in good to excellent yields with complete selectivity for the C-functionalization: a valuable alternative to the pioneering work from Shibata.11 Due to the recent interest in the (phenylsulfonyl)difluoromethyl sulfanyl group,10a,17 we explored the possibility of accessing PhSO2CF2S-containing derivatives by reacting S-nucleophiles with reagent I (Scheme 6). The transformation proceeded smoothly with structurally diverse thiophenol derivatives (10a–10c) including the 2-benzothiazolethiol 10c under mild reaction conditions (50 °C, no base). Interestingly, in the case of the 4-mercaptophenol 10b, complete selectivity was observed for the formation of a S-CF2SO2Ph bond over a C(sp2)–CF2SO2Ph bond.
 | ||
| Scheme 4 Electrophilic (phenylsulfonyl)difluoromethylation of electron-rich (hetero)arenes. Reaction performed on a 0.3 mmol scale. Isolated yields are given. aA combined isolated yield was provided and the ratio of regioisomers was determined using 19F NMR of the crude reaction mixture using α,α,α-trifluoroacetophenone as an internal standard. The C3-regioisomer of 5d was isolated with traces of the C4-regioisomer. bReactions performed using 3 equivalents of 4 in DMSO, 50 °C, 16 h, under argon. c5l was isolated with 13% of 4l remaining. | ||
 | ||
| Scheme 5 Isolated yields are given. aReaction performed on a 0.1 mmol scale. bReaction performed on a 0.15 mmol scale for 30 min instead of 1 h. cReaction performed on a 0.2 mmol scale. | ||
 | ||
| Scheme 6 Synthesis of (phenylsulfonyl)difluoromethylthio-containing molecules from thiol derivatives. Reaction conditions: 10 (3 equiv.), I (0.3 mmol, 1 equiv.), DMSO (0.15 M), 50 °C, 16 h, under argon. Isolated yields are given. | ||
To further showcase the synthetic utility of reagent I, the straightforward synthesis of a PhSO2CF2-analog of Lidocaine 12, an anesthetic, was achieved in 63% overall yield after the following sequence: (phenylsulfonyl)difluoromethylation of the commercially available 2,6-xylidine (3k, 84% yield), followed by amidation with the chloroacetyl chloride and reaction with diethylamine under basic conditions in a one pot process (75% yield, Scheme 7). Pleasingly, this fluorinated group was then converted into the valuable HCF2 moiety.5 Indeed, in the presence of Mg and using a catalytic amount of I2, the difluoromethylated product 13 was obtained in 80% yield starting from 12.
 | ||
| Scheme 7 Synthesis of PhSO2CF2-Lidocaine and the post-functionalization reaction. | ||
In summary, a straightforward synthesis of a bench-stable PhSO2CF2 reagent I was developed. This transition metal-free protocol was convenient and efficiently scalable and did not require any use of ozone depleting fluorinated reagents. Pleasingly, access to unprecedented fluorinated scaffolds was possible under simple and practical reaction conditions, as illustrated by the synthesis of PhSO2CF2-containing aniline derivatives and electron-rich (hetero)arenes in moderate to excellent yields. Moreover, reagent I was also applied to the functionalization of C(sp3) centers and for the preparation of valuable PhSO2CF2S-containing derivatives. The PhSO2CF2 residue was efficiently converted into the high-value added HCF2 group, which highlighted the versatility of this fluorinated group. The late-stage functionalization of bioactive molecules into their corresponding PhSO2CF2-containing analogs further demonstrated the synthetic utility of the novel reagent, which is particularly appealing in the quest for interesting scaffolds for medicinal chemistry. This newly-designed electrophilic source is distinguished by its straightforward and practical synthesis and its wide scope, thus offering a new platform to introduce the PhSO2CF2 motif into various skeletons. We believe that this original tool will enlarge the current toolbox of electrophilic reagents, will open new routes for functionalizing complex molecules and will be useful for the discovery of new bioactive molecules.
This work has been partially supported by the University of Rouen Normandy, INSA Rouen Normandy, the Centre National de la Recherche Scientifique (CNRS), the European Regional Development Fund (ERDF), Labex SynOrg (ANR-11-LABX-0029), the Carnot Institute I2C, the graduate school for research XL-Chem (ANR-18-EURE-0020 XL CHEM), and the Region Normandy. E. N. and T. B. thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 758710). E. N. thanks the Region Normandy for a doctoral fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (d) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed; (e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed.
- D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- For a selection of reviews, see: (a) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (c) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed; (d) G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941 CrossRef CAS PubMed; (e) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (f) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS PubMed; (g) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (h) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed; (i) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (j) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC; (k) H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Green Chem., 2018, 20, 1662 RSC; (l) Y. Pan, ACS Med. Chem. Lett., 2019, 10, 1016 CrossRef CAS PubMed; (m) L. Ruyet and T. Besset, Beilstein J. Org. Chem., 2020, 16, 1051 CrossRef CAS PubMed; (n) F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486 RSC.
- (a) J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626 CrossRef CAS; (b) M. D. Martínez, L. Luna, A. Y. Tesio, G. E. Feresin, F. J. Durán and G. J. Burton, J. Pharm. Pharmacol., 2016, 68, 233 CrossRef PubMed; (c) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (d) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325 CrossRef CAS PubMed; (e) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797 CrossRef CAS PubMed; (f) Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, J. Med. Chem., 2019, 62, 5628 CrossRef CAS PubMed.
- For a selection of reviews, see: (a) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed; (b) J. Hu, J. Fluorine Chem., 2009, 130, 1130 CrossRef CAS; (c) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (d) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476 CrossRef PubMed; (e) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. Am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214 RSC; (f) R. Jia, X. Wang and J. Hu, Tetrahedron Lett., 2021, 75, 153182 CrossRef CAS ; for selected examples, see: ; (g) Y.-M. Su, Y. Hou, F. Yin, Y.-M. Xu, Y. Li, X. Zheng and X.-S. Wang, Org. Lett., 2014, 16, 2958 CrossRef CAS PubMed; (h) G. Yin, M. Zhu and W. Fu, Heterocycl. Commun., 2017, 23, 275 CAS; (i) N. Surapanich, C. Kuhakarn, M. Pohmakotr and V. Reutrakul, Eur. J. Org. Chem., 2012, 5943 CrossRef CAS.
- H. Eto, Y. Kaneko, S. Takeda, M. Tokizawa, S. Sato, K. Yoshida, S. Namiki, M. Ogawa, K. Maebashi, K. Ishida, M. Matsumoto and T. S. Asaoka, Chem. Pharm. Bull., 2001, 49, 173 CrossRef CAS PubMed.
- (a) J. Pepin, C. Guern, F. Milord and P. J. Schechter, Lancet, 1987, 330, 1431 CrossRef; (b) J. E. Wolf Jr, D. Shander, F. Huber, J. Jackson, C.-S. Lin, B. M. Mathes and K. Schrode, the Eflornithine HCl Study Group, Int. J. Dermatol., 2007, 46, 94 CrossRef PubMed.
- B. Bennett, A. Matagne, P. Michel, M. Leonard, M. Cornet, M.-A. Meeus and N. Toublanc, Neurotherapeutics, 2007, 4, 117 CrossRef CAS PubMed.
- (a) W. Zhang, J. Zhu and J. Hu, Tetrahedron Lett., 2008, 49, 5006 CrossRef CAS; (b) Z. He, T. Luo, M. Hu, Y. Cao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 3944 CrossRef CAS PubMed; (c) Z. He, M. Hu, T. Luo, L. Li and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 11545 CrossRef CAS PubMed.
- X. Wang, G. Liu, X.-H. Xu, N. Shibata, E. Tokunaga and N. Shibata, Angew. Chem., Int. Ed., 2014, 53, 1827 CrossRef PubMed.
- (a) C. Zhang, Org. Biomol. Chem., 2014, 12, 6580 RSC and references therein: ; (b) G. K. S. Prakash, C. Weber, S. Chacko and G. A. Olah, Org. Lett., 2007, 9, 1863 CrossRef CAS PubMed.
- P. Franzmann, S. B. Beil, D. Schollmeyer and S. R. Waldvogel, Note that disulfide 1a was easily prepared from 2-phenylaniline after a four-step synthesis (no purification) with several grams (upto 50 g) by following the procedure depicted, Chem. – Eur. J., 2019, 25, 1936 CrossRef CAS PubMed.
- PhSO2CF2H was prepared in two steps from thiophenol when not purchased, see the ESI†.
- For more details, see the ESI†.
- For a similar selectivity in the case of trifluoromethylation with the Umemoto reagent, see: (a) T. Umemoto and S. Ishihara, Tetrahedron Lett., 1990, 31, 3579 CrossRef CAS; (b) T. Umemoto, S. Ishihara and K. Adachi, J. Fluorine Chem., 1995, 74, 77 CrossRef CAS; (c) Y. Macé, B. Raymondeau, C. Pradet, J.-C. Blazejewski and E. Magnier, Eur. J. Org. Chem., 2009, 1390 CrossRef.
- (a) H.-Y. Xiong, X. Pannecoucke and T. Besset, Chem. – Eur. J., 2016, 22, 16734 CrossRef CAS PubMed; (b) X. Xiao, Z.-T. Zheng, T. Li, J.-L. Zheng, T. Tao, L.-M. Chen, J.-Y. Gu, X. Yao, J.-H. Lin and J.-C. Xiao, Synthesis, 2020, 197 CAS; (c) T. Besset and T. Poisson, in Emerging Fluorinated Motifs: Synthesis, Properties and Applications, ed. D. Cahard and J.-A. Ma, Wiley-VCH, Weinheim, 2020, 16, 449–475 Search PubMed; (d) E. Ismalaj, D. Le Bars and T. Billard, Angew. Chem., Int. Ed., 2016, 55, 4790 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2084628 and 2084629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04737j |
| This journal is © The Royal Society of Chemistry 2021 |