
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot gram-scale synthesis of virucidal heparin-mimicking polymers as HSV-1 inhibitors†
Vahid
Ahmadi
 *a,
Chuanxiong
Nie
a,
Ehsan
Mohammadifar
a,
Katharina
Achazi
a,
Stefanie
Wedepohl
a,
Yannic
Kerkhoff
a,
Stephan
Block
a,
Klaus
Osterrieder
bc and
Rainer
Haag
*a
*a,
Chuanxiong
Nie
a,
Ehsan
Mohammadifar
a,
Katharina
Achazi
a,
Stefanie
Wedepohl
a,
Yannic
Kerkhoff
a,
Stephan
Block
a,
Klaus
Osterrieder
bc and
Rainer
Haag
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustrasse 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bInstitut für Virologie, Robert von Ostertag-Haus, Zentrum für Infektionsmedizin Freie Universität Berlin, Robert-von-Ostertag-Str. 7-13, 14163 Berlin, Germany
cDepartment of Infectious Diseases and Public Health, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Kowloon Tong, Hong Kong
First published on 21st October 2021
Abstract
A straightforward and gram-scale synthesis method was developed to engineer highly sulfated hyperbranched polyglycerol bearing sulfated alkyl chains. The compounds with shorter alkyl chains showed multivalent virustatic inhibition against herpes simplex virus type 1 (HSV-1), similar to heparin. In contrast, the compound with the longest alkyl chains irreversibly inhibited the virus.
Herpes simplex virus type 1 (HSV-1) infections are common and affect approximately 70–90% of the adult population worldwide.1 Although HSV-1 is a well-studied virus, it remains a major public health concern because vaccines are unavailable and common antiviral drugs such as acyclovir, the most commonly prescribed medication, show limited efficacy.2 HSV-1 entry into the host cell is initiated by electrostatic interaction between negatively charged heparan sulfates (HSs) located on the host cell surface, and positively charged domains of the viral envelope glycoprotein B (gB) and glycoprotein C (gC).3 Protein-polyelectrolyte interactions are dominated by counterion release, i.e., the positively charged patches become multivalent counterions of the polyelectrolyte, resulting in counterions being released from the polyelectrolyte (driving force), a process that increases entropy.4 The finding that heparin, which is a soluble derivative of HS, shows an inhibitory effect for a variety of viruses by interacting with their surface, has led to the development of numerous heparin-mimetic compounds.5–9 Despite many trials, researchers have encountered limitations that are inherent to these compounds, such as their high anticoagulant activity, their complicated synthesis, and their mechanism of virus inhibition, which is virustatic rather than virucidal.10 The compounds reversibly bind to the virus, which prevents viral attachment and, consequently, entry into the host cell. However, the dilution effect in vivo causes dissociation of the binding complexes, reversing the binding and ultimately leading to virus infection.11 Virucidal compounds, on the other hand, interact with viruses and physically render them non-infectious, for example by disrupting their envelope, thus preventing infection even upon dilution in body fluids. Development of new antiviral compounds that address these challenges is of major interest, and one must strive to design antiviral materials that are biocompatible, virucidal, and easily scalable. One way to develop a virucidal compound is to combine electrostatic and hydrophobic interactions while minimizing the potential toxicity that can be caused by increasing hydrophobicity.12 By developing gold-based nanoparticles containing 11-mercapto-undecansulfonate moieties, Stellacci et al.10 first developed the virucidal effect of hydrophobic sulfonated gold nanoparticles. As part of our approach, we are using polymeric cores to address issues concerning gold nanoparticle health risks, synthetic challenges, and economic aspects.
Current antiviral therapies are less effective over time due to virus mutations, which obviously calls for the development of broad-spectrum antiviral alternatives.13 Furthermore, due to the difference in how viruses replicate, there is little hope of developing broad-spectrum drugs with intracellular approaches. Thus, extracellular antivirals – which usually come in the form of entry inhibitors – are of importance. To inhibit viruses effectively, virus–inhibitor interactions must be stronger than those between viruses and cells.14 Monovalent inhibitors are therefore ineffective since they do not completely block receptor sites and are unable to compete with viral proteins for binding. As a result, for designing potent inhibitors, it is important to consider multivalent interactions. Utilizing polymers is then of great interest and have been used in the development of antiviral compounds over the past decades.15 Hyperbranched polyglycerol (hPG) offers a multifunctional scaffold with high biocompatibility and hydrophilicity that can be used in numerous biomedical applications, ranging from drug delivery to pathogen inhibition.16 Having found that sulfated hPG (hPGS) exhibits similar bioactivity to heparin,17 our group developed various antiviral compounds with different architectures.8,9,18 Although all of these compounds exhibit impressive antiviral properties including inhibition of HSV at picomolar concentrations,9 none are virucidal.
Here, we rationally designed a novel class of virucidal compounds based on a one-pot approach towards hPGS-bearing alkyl chains. These compounds irreversibly inhibit HSV-1 infection, and we highlight their ease of synthesis as well as their robust activity. In a one-pot, four-step synthesis approach, we used anionic ring-opening polymerization to synthesize hPG, which we functionalized with a hydrophobic alkyl chain and finally sulfated for the electrostatic interaction with the virus (Scheme 1 and Scheme S2, ESI†). It has been demonstrated that hPG is more effectively functionalized in a one-pot reaction when a glycidol derivative is used.19 We, therefore, synthesized, and characterized separately, glycidols containing hydrophobic alkyl chains of different lengths (Scheme S1, ESI†).
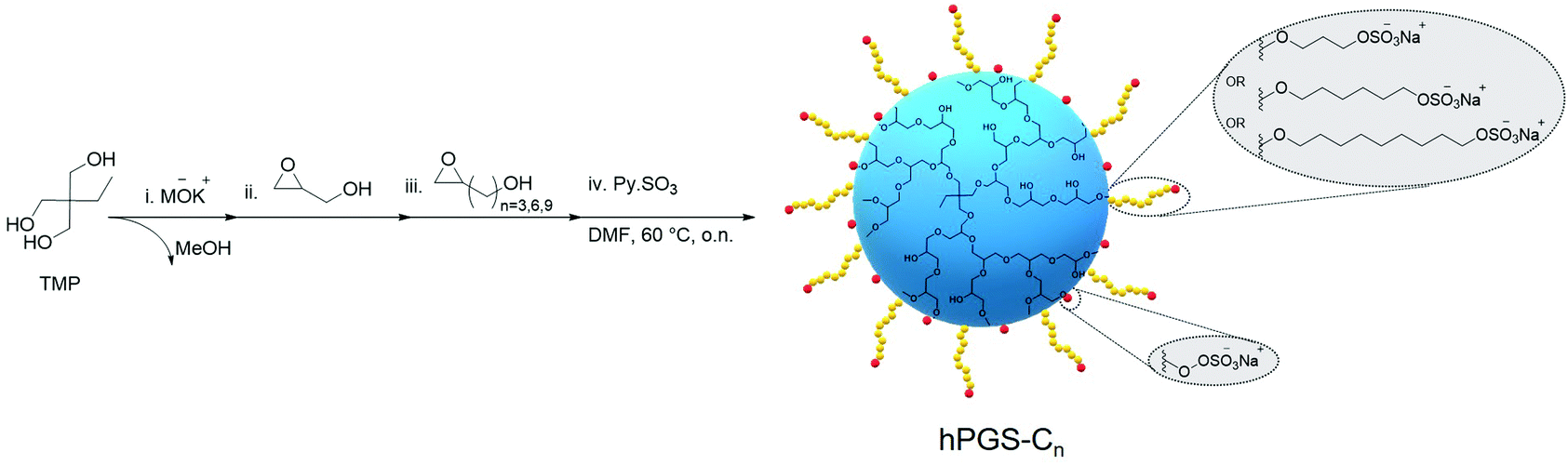 | ||
| Scheme 1 One-pot synthesis route of hPGS-Cn. | ||
Briefly, the hydrophilic core of hPG was first synthesized solvent-free from the trimethylolpropane (TMP) initiator via anionic ring-opening polymerization of glycidol, following previously described procedures.20 Monomer-to-initiator ratios were adjusted for all polymerizations to yield an hPG core of approximately 5 kDa (Table S1 and Fig. S1, ESI†). However, no quenching was performed after addition of the monomer, and the reaction continued until all of the glycidol was consumed as evidenced by the disappearance of the corresponding peaks at 2.6 ppm and 3.7 ppm in 1H-NMR spectra (Fig. S2, ESI†). The hPG cores were then functionalized by adding glycidol derivatives of varying alkyl chain length to form hPG-C3, hPG-C6, and hPG-C9. Using the integral peak ratio of alkyl chain protons in 1H-NMR at 1.2–2.1 ppm to hPG backbone protons at 3.6–4.6 ppm, the degree of functionalization was calculated to be around 40% for all compounds (Fig. S3 and eqn S2–S4, ESI†). Then, in the same reactor, dimethylformamide was added, and a reaction with sulfur trioxide pyridine complex was performed to convert the hydroxyl groups to sulfate groups following a previously described protocol.21 In a previous study,22 we had found that the degree of sulfation (DS) affects inhibitory effects; therefore, the ratios were adjusted in order to obtain highly sulfated compounds. The final products were purified using tangential flow filtration (TFF) followed by freeze-drying, resulting in hPGS-C3, hPGS-C6, and hPGS-C9 with over 90% DS as calculated by elemental analysis (Table S2 and eqn S5, S6, ESI†). As a control, a sulfated hPG without an alkyl chain, namely hPGS-C0, with the same degree of sulfation and molecular weight was synthesized according to available protocols.21,23Table 1 provides a summary of the results.
| Compound | M n [kDa] | PDI [—] | Degree of functionalization [%] | M n [kDa] | M n [kDa] | Degre of sulfation [%] | Zeta potential in 10 mM PB buffer [mV] | CC50 [mg mL−1] | IC50 [μg mL−1] | IC50 [nM] | SI [—]d | Inhibition mechanism | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A549 | VeroE6 | HBE | ||||||||||||
| a Molecular weight of polymeric core obtained by GPC. b Molecular weight after functionalization obtained by GPC. c Molecular weight after sulfation calculated by elemental analysis and 1H NMR. d Selectivity index (SI = CC50/IC50) calculated for VeroE6 cells. | ||||||||||||||
| hPGS–C9 | 3.8 | 1.7 | 38 | 6.7 | 12.1 | 97 | −35.5 ± 2.3 | 3.90 | 4.76 | 1.49 | 0.189 ± 0.054 | 15.6 | 25200 | Virucidal |
| hPGS–C6 | 3.9 | 1.8 | 39 | 6.6 | 11.5 | 93 | −35.6 ± 1.2 | 12.68 | 23.93 | 20.19 | 0.256 ± 0.089 | 22.2 | 93500 | Virustatic |
| hPGS–C3 | 3.9 | 1.8 | 39 | 6.7 | 10.6 | 96 | −33.1 ± 2.1 | 25.93 | 13.31 | 37.68 | 5.098 ± 1.048 | 480.9 | 2600 | Virustatic |
| hPGS–C0 | 4.6 | 1.7 | 0 | — | 11.3 | 91 | −32.4 ± 1.4 | 13.93 | 6.97 | 10.47 | 2.297 ± 0.695 | 203.3 | 3000 | Virustatic |
Using two human lung cell lines, A549 and 16HBE14o-, as well as a standard cell line for HSV-1 propagation, Vero E6, we performed CCK-8 assays to determine the viability of the cells after treatment with the sulfated polymers (Fig. 1 and Fig. S4, ESI†). The half-maximal cytotoxic concentration (CC50) for each compound was calculated for each cell line (Table 1). At concentrations of up to 1000 μg mL−1, none of the compounds showed discernable cytotoxicity (Fig. 1 and Fig. S4, ESI†), with the exception of 16HBE14o-cells. For this cell line, the compound with the longest alkyl chain, hPGS-C9, exerted an effect on viability at a concentration of 1000 μg mL−1. At the highest concentration tested (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg mL−1), cell viability was reduced in all cell lines for all tested compounds, with hPGS-C9 impairing viability most. Taking the CC50 values (Table 1) into account, alkyl chains have a small effect on the toxic properties of the synthesized compounds, and, as predicted, cells are more sensitive to compounds with longer the alkyl chains.
000 μg mL−1), cell viability was reduced in all cell lines for all tested compounds, with hPGS-C9 impairing viability most. Taking the CC50 values (Table 1) into account, alkyl chains have a small effect on the toxic properties of the synthesized compounds, and, as predicted, cells are more sensitive to compounds with longer the alkyl chains.
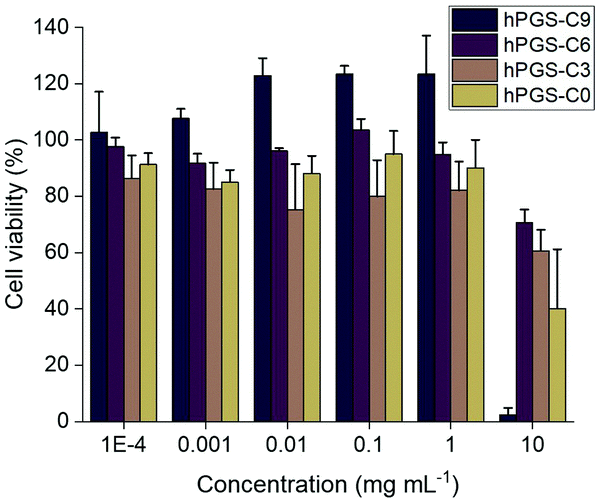 | ||
| Fig. 1 Cytotoxicity profile of the synthesized inhibitors determined by a CCK-8 assay using Vero E6 cells. The cell viability of compound treated cells is normalized to the cell viability of non-treated cells that was set to 100% cell viability. | ||
Plaque reduction assays using Vero E6 cells were used to determine the compounds’ antiviral activity against HSV-1. Herein, virions were pre-incubated with the compounds at different concentrations and then titrated on Vero cells to determine the ratio of inhibited virions. The dose–response curves obtained from the experiment are shown in Fig. 2a, and the respective half-maximal inhibitory concentrations (IC50) are shown in Table 1. As expected, the synthesized heparan-mimetic compounds exhibited considerable antiviral activity, with IC50 values in the nanomolar range. We found that the IC50 value decreases with increasing alkyl chain length. hPGS-C9 exhibited the strongest inhibitory effect (IC50 = 15.6 nM), while hPGS-C6, hPGS-C3, and hPGS-C0 showed IC50 values of 22.2, 480.9, and 203.3 nM, respectively. Hydrophobicity therefore seems to play an important role in the inhibition of HSV-1. The compounds were then evaluated for their inhibition mechanism. For this purpose, we used a virucidal assay in which compounds with a concentration of 1 mg mL−1 were pre-incubated with virus (approx. 105 PFU). The solutions were then diluted five times by a factor of 10 and each dilution was titrated by plaque assay to determine the number of active virions present. When the inhibition is remained even after dilution below its IC50, it is considered as virucidal inhibition, implying that the virus was irreversibly inactivated.10 Otherwise, it is considered as virustatic inhibition, which refers to reversible inhibition of the virus. We found that the compounds with shorter alkyl chains, hPGS-C6 and hPGS-C3, as well as the one without an alkyl chain, hPGS-C0, are solely virustatic, whereas hPGS-C9 showed virucidal properties (Fig. 2b). Therefore, an increase in alkyl chain length and therefore in hydrophobicity induce a strong virucidal effect, suggesting that sufficient alkyl chain length is causing rupture of the viral envelope. It should be noted that longer alkyl chains (>C9), despite their possibly higher inhibitory activities, were not considered in this study, due to reports indicating that longer chains (≥C10) exert a significantly toxic effect on eukaryotic cells, including Vero E6 cells.5 Next, cellular infection assays were performed to examine the inhibitory activity of viral replication (Fig. 3). In a pre-infection assay, the cells were treated with the compounds at different concentrations for 45 minutes and then infected for 48 hours with HSV-1 expressing green fluorescent protein (GFP). To evaluate if the compounds still have an effect when cells are already infected, a post-infection assay was carried out. For the post-infection assay, cells were first infected for 1h prior to addition of the compounds. The total time of infection with GFP-tagged HSV-1 in this post-infection assay was 48 hours as well. The results of the pre- and post-infection experiments indicate that all compounds are highly effective as an inhibitor (Fig. S5b and c, ESI†). Nevertheless, the pre-infection assays also revealed a clear decrease in the number of infected cells for all compounds with a concentration higher than 10 μg mL−1 (Fig. S5b, ESI†), probably because the compounds are capable of inhibiting the infection by progeny viruses, thereby reducing viral transmission between the cells. This also explains why the antiviral activity was lower in the post-infection when compared to the pre-infection assay: after the first cycle of infection, large amounts of virions were produced, resulting in a higher viral load compared to that in the pre-infection assay. Furthermore, the selectivity index (SI) of each compound was calculated by dividing the CC50 and IC50 values (Table 1). hPGS-C9, for example, demonstrated a remarkable SI of 25200, which made it effective at blocking and destroying viruses without causing serious damage to host cells. This means that it only ruptures the membranes of the virus, not the cells. Observations regarding the high SI value of the compound suggest that the surface curvature of biosystems could account for its selective rapture of the viral membrane rather than that of the cell. As HSV-1 viruses are about 200 nm in size, their curvature and strain are higher than cells, which are about micrometres in size. More importantly, unlike viruses that contain only genetic information, cells have self-repairing mechanisms that can tolerate and repair damaged membranes.24 HS-mimetic compounds, such as heparin and other sulfated polymers, have inherent anticoagulant properties. Due to their ability to activate antithrombin and turn it into a blood-clotting proteinase inhibitor, these compounds prolong blood clotting and are thus limited in concentration as virus inhibitors in clinical use.25 We therefore determined ex vivo clotting times by measuring the activated partial thromboplastin time (APTT) of plasma treated with our inhibitors at concentrations ranging from 0.5 to 1000 μg mL−1.
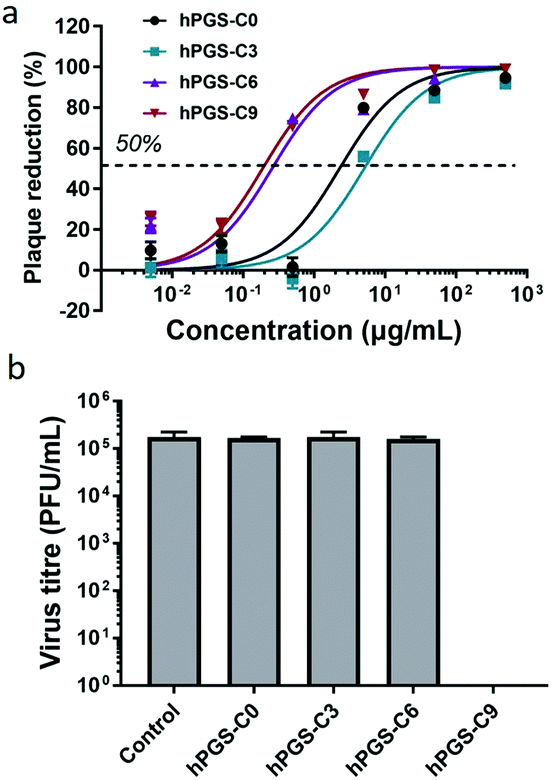 | ||
| Fig. 2 (a) Concentration-dependent inhibition of HSV-1 infection on Vero E6 cells. (b) Virucidal potency of the synthesized inhibitors against HSV-1. Data are expressed as mean ± SD (n = 4). | ||
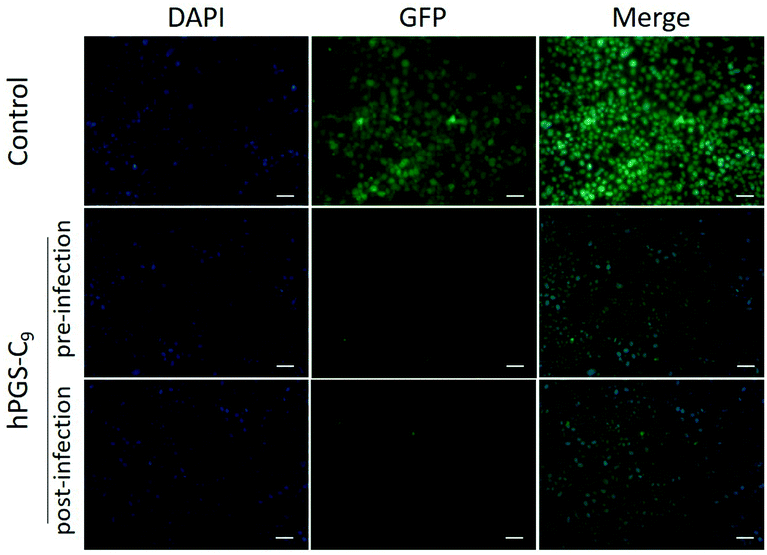 | ||
| Fig. 3 Fluorescent microscopy images of HSV-1 infected cells treated with hPGS-C9 (100 μg mL−1). Images from cells treated with other compounds are shown in Fig. S4 (ESI†). Scale bar: 20 μm. Cell nuclei are marked in blue, and infected cells in green. | ||
As a control we used commercially available heparin (Fig. S6, ESI†). Comparing the APTT of untreated human plasma with a mean of 31 seconds to the inhibitor-treated plasma, no changes were observed up to a concentration of 5 μg mL−1. A similar concentration of heparin, however, led to a significant increase in the APTT of more than 500 seconds. At a concentration of 25 μg mL−1, hPGS-C9, hPGS-C6, and hPGS-C3 started to show mild effects on the APTT, whereas hPGS-C0 increased the APTT more than fivefold. Considering the results for the alkyl-chain-functionalized polymers, hPGS-C9, hPGS-C6, and hPGS-C3, one can observe that the length of the alkyl chain interferes with coagulation, as anticoagulant activity decreases with the length of the alkyl chain. This effect can be explained by a less dense distribution of sulfate groups in the longest-chained polymer, hPGS-C9, when compared to hPGS-C6 and hPGS-C3 with their shorter chains and to hPGS-C0 that does not carry side chains at all.
In conclusion, we have developed a method to synthesize antiviral compounds in a one-pot reaction at gram scale. Our approach was to rationally design the compounds in a synergistic approach by using alkyl chains for hydrophobic interactions and sulfate groups for electrostatic interactions. In studies against HSV-1, compounds with shorter chain lengths showed virustatic inhibition similar to that of heparin, while the compound with the longest chain length, hPGS-C9, showed an irreversible viricidal effect. These compounds may also be used against other heparan sulfate-binding viruses. Furthermore, future studies into adapting the same one-pot approach, but with other comonomers with different functional groups, may allow this technique to produce gram-scale hPG-based virucidal compounds that are effective against other viruses as well.
This work was supported by SFB 1449. We thank Elisa Quaas for performing cell viability assays. We gratefully acknowledge assistance from the Core Facility BioSupraMol funded by DFG and from Ben Allen, who assisted us with the language polishing of the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, ed. A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley and K. Yamanishi, Cambridge University Press Copyright © Cambridge University Press, Cambridge, 2007 Search PubMed.
- K. Kłysik, A. Pietraszek, A. Karewicz and M. Nowakowska, Curr. Med. Chem., 2020, 27, 4118–4137 CrossRef PubMed.
- A. M. Agelidis and D. Shukla, Future Virol., 2015, 10, 1145–1154 CrossRef CAS PubMed.
- K. Achazi, R. Haag, M. Ballauff, J. Dernedde, J. N. Kizhakkedathu, D. Maysinger and G. Multhaup, Angew. Chem., Int. Ed., 2021, 60, 3882–3904 CrossRef CAS PubMed.
- I. S. Donskyi, W. Azab, J. L. Cuellar-Camacho, G. Guday, A. Lippitz, W. E. S. Unger, K. Osterrieder, M. Adeli and R. Haag, Nanoscale, 2019, 11, 15804–15809 RSC.
- E. Lee, M. Pavy, N. Young, C. Freeman and M. Lobigs, Antiviral Res., 2006, 69, 31–38 CrossRef CAS PubMed.
- P. Dey, T. Bergmann, J. L. Cuellar-Camacho, S. Ehrmann, M. S. Chowdhury, M. Zhang, I. Dahmani, R. Haag and W. Azab, ACS Nano, 2018, 12, 6429–6442 CrossRef CAS PubMed.
- E. Mohammadifar, V. Ahmadi, M. F. Gholami, A. Oehrl, O. Kolyvushko, C. Nie, I. S. Donskyi, S. Herziger, J. Radnik, K. Ludwig, C. Böttcher, J. P. Rabe, K. Osterrieder, W. Azab, R. Haag and M. Adeli, Adv. Funct. Mater., 2021, 31, 2009003 CrossRef CAS PubMed.
- P. Pouyan, C. Nie, S. Bhatia, S. Wedepohl, K. Achazi, N. Osterrieder and R. Haag, Biomacromolecules, 2021, 22, 1545–1554 CrossRef CAS PubMed.
- V. Cagno, P. Andreozzi, M. D’Alicarnasso, P. Jacob Silva, M. Mueller, M. Galloux, R. Le Goffic, S. T. Jones, M. Vallino, J. Hodek, J. Weber, S. Sen, E.-R. Janeček, A. Bekdemir, B. Sanavio, C. Martinelli, M. Donalisio, M.-A. Rameix Welti, J.-F. Eleouet, Y. Han, L. Kaiser, L. Vukovic, C. Tapparel, P. Král, S. Krol, D. Lembo and F. Stellacci, Nat. Mater., 2018, 17, 195–203 CrossRef CAS PubMed.
- B. Shogan, L. Kruse, G. B. Mulamba, A. Hu and D. M. Coen, J. Virol., 2006, 80, 4740–4747 CrossRef CAS PubMed.
- J. Said, E. Trybala, E. Andersson, K. Johnstone, L. Liu, N. Wimmer, V. Ferro and T. Bergström, Antiviral Res., 2010, 86, 286–295 CrossRef CAS PubMed.
- X. Huang, W. Xu, M. Li, P. Zhang, Y. S. Zhang, J. Ding and X. Chen, Matter, 2021, 4, 1892–1918 CrossRef CAS.
- S. T. Jones, J. Mater. Sci., 2020, 55, 9148–9151 CrossRef CAS PubMed.
- R. H. Bianculli, J. D. Mase and M. D. Schulz, Macromolecules, 2020, 53, 9158–9186 CrossRef CAS.
- H. Frey and R. Haag, Rev. Mol. Biotechnol., 2002, 90, 257–267 CrossRef CAS PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- C. Nie, P. Pouyan, D. Lauster, J. Trimpert, Y. Kerkhoff, G. P. Szekeres, M. Wallert, S. Block, A. K. Sahoo, J. Dernedde, K. Pagel, B. B. Kaufer, R. R. Netz, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2021, 60, 15870–15878 CrossRef CAS PubMed.
- F. Paulus, D. Steinhilber, P. Welker, D. Mangoldt, K. Licha, H. Depner, S. Sigrist and R. Haag, Polym. Chem., 2014, 5, 5020–5028 RSC.
- M. Wallert, J. Plaschke, M. Dimde, V. Ahmadi, S. Block and R. Haag, Macromol. Mater. Eng., 2021, 306, 2000688 CrossRef CAS.
- H. Türk, R. Haag and S. Alban, Bioconjugate Chem., 2004, 15, 162–167 CrossRef PubMed.
- B. Ziem, W. Azab, M. F. Gholami, J. P. Rabe, N. Osterrieder and R. Haag, Nanoscale, 2017, 9, 3774–3783 RSC.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- S. K. Y. Tang and W. F. Marshall, Science, 2017, 356, 1022–1025 CrossRef CAS PubMed.
- S. T. Olson, B. Richard, G. Izaguirre, S. Schedin-Weiss and P. G. W. Gettins, Biochimie, 2010, 92, 1587–1596 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc04703e |
| This journal is © The Royal Society of Chemistry 2021 |