
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pressure-induced superelastic behaviour of isonicotinamide†‡
Eleanor C. L.
Jones
 *a,
Suse S.
Bebiano
ab,
Martin R.
Ward
a,
Luis M.
Bimbo
acd and
Iain D. H.
Oswald
*a
*a,
Suse S.
Bebiano
ab,
Martin R.
Ward
a,
Luis M.
Bimbo
acd and
Iain D. H.
Oswald
*a
aStrathclyde Institute of Pharmacy & Biomedical Sciences (SIPBS), University of Strathclyde, 161 Cathedral Street, G4 0RE, Glasgow, UK. E-mail: eleanor.jones@strath.ac.uk; iain.oswald@strath.ac.uk
bEPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation, University of Strathclyde, Technology Innovation Centre, 99 George Street, Glasgow, G1 1RD, UK
cDepartment of Pharmaceutical Technology, Faculty of Pharmacy, University of Coimbra, Coimbra, Portugal
dCNC – Center for Neuroscience and Cell Biology, and CIBB – Center for Innovative Biomedicine and Biotechnology, Rua Larga, University of Coimbra, 3004-504 Coimbra, Portugal
First published on 15th October 2021
Abstract
Dynamic organic crystals have come to the fore as potential lightweight alternatives to inorganic actuators providing high weight-to-force ratios. We have observed pressure-induced superelastic behaviour in Form I of isonicotinamide. The reversible single-crystal to single-crystal transformation exhibited by the system is an important component for functioning actuators. Crucially, our observations have enabled us to propose a mechanism for the molecular movement supported by Pixel energy calculations, that may pave the way for the future design and development of functioning dynamic crystals.
Materials that convert energy into a mechanical response have a wealth of potential applications in, for example, electronics, energy harvesting, gating and even in simulation of muscle contractions.1 For the materials to be of use, there is a requirement to remain flexible and resilient when strain is applied, whether it be thermal or driven by shear stress. Crystals that exhibit a mechanical response when subjected to external perturbation can be defined as dynamic. These effects can either be restorative (i.e. bending or twisting) or disintegrative (i.e. cracking or fragmentation of the crystal).1 Previous studies have witnessed changes in shape during polymorphic transitions, attributed to martensitic transitions.2,3 Martensitic transitions are a form of cooperative structure transition, by which there is a coordinated displacement of molecules in a crystalline material. Ditert-butyl [1]benzothieno[3,2-b]benzothiophene (diBu-BTBT) possesses this behaviour on heating to 345 K. This reversible process includes a change in colour from blue to yellow that was attributed to the disorder of the tertiary butyl side groups that impacted the molecular packing.2 Simpler molecular systems, such as hexamethylbenzene (HMB), have also shown martensitic behaviour on heating.3 The mechanism of the transition was attributed to the change in the packing of the molecules. Significantly, the crystal was calculated to be capable of providing a mechanical force of 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times its own weight, placing it firmly in the realm for use as thermal actuators.3–5 There are few studies to compare the magnitude, however, in their recent review, Naumov et al. have highlighted the potential of thermally actuated organic crystals in comparison to other materials, possessing a greater maximum force per work output than nano-muscles or ceramic piezoelectrics.1
000 times its own weight, placing it firmly in the realm for use as thermal actuators.3–5 There are few studies to compare the magnitude, however, in their recent review, Naumov et al. have highlighted the potential of thermally actuated organic crystals in comparison to other materials, possessing a greater maximum force per work output than nano-muscles or ceramic piezoelectrics.1
Not all single-crystal to single-crystal transitions, where changes in crystal dimensions are observed, are martensitic, as the rate of transition plays a factor. In 7,7,8,8-tetracyanoquinodimethane-p-bis(8-hydroxyquinolinato) copper(II) (CuQ2-TCNQ), the transformation between Form II to Form I results in a 100% increase in crystal length via a cooperative movement. The movement is one of the classifications of a martensitic transition, however, the rate of change favoured a nucleation and growth mechanism instead (>0.01 mm s−1).6
These examples demonstrate transitions that are induced by temperature; however, this is not the only method by which a system can be perturbed. Phase transitions in organic or organometallic materials are now routinely surveyed using pressure. Pressure can strongly impact on the intermolecular interactions, e.g. hydrogen bonding, and also the packing of molecules, hence could be used as a tool to explore the changes imparted in the studies above.7–9 Of particular interest to us, is how smaller molecules can exhibit pressure-induced phase transformations. The previous study by Takamizawa and Takasaki investigated the effect of pressure, as well as temperature, on the martensitic transition of tetrabutyl-n-phosphonium tetraphenylborate. They were able to quantify the response to pressure on particular faces of the crystal, during transformation from the α to β form.15
Isonicotinamide is a well-known and commonly investigated compound in solid-state studies due to its hydrogen bonding behaviour. It possesses six known polymorphs, each possessing different hydrogen-bonding patterns but of particular interest is that the most thermodynamically stable form (Form I) is the only structure that interacts as dimers, with Forms II to VI forming hydrogen bonded chains.10–13
Our initial investigations of isonicotinamide at high pressure used Raman spectroscopy to identify any molecular (e.g. conformation) or environmental changes around the isonicotinamide molecules, e.g. hydrogen bonding (Fig. S1 in ESI‡). It was during this investigation that we noticed that isonicotinamide Form I exhibits pressure-induced dynamic behaviour beyond 4.3 GPa. Crystals that were square became thinner and more elongated whilst maintaining their single crystal-like nature (Fig. 1). Due to crystal confinement in the cell, we did not see the change to the third dimension hence cannot rule out bending of the crystal on compression.
 | ||
| Fig. 1 Microscopy image of Form I isonicotinamide at (a) 4.33 GPa and (b) Form I′ at 4.98 GPa showing the single crystal transformation. Scale bar represents 200 μm. | ||
The crystals exhibit an elastic behaviour with the crystals regaining their original shape as pressure is released. This behaviour allows us to define this polymorphic form as possessing superelastic properties, rarely witnessed in organic crystals;1 terepthalamide is one example. In this material, the application of shear stress, via needle pressure, transforms the α-form to the β-form via the bending of the crystal. Internally, the molecules rotate to accommodate the change in the external form of the crystal.14 Notably, the hydrogen bonding motif remained the same in the new phase with the recovery of the α-form after removal of the stressor. In isonicotinamide, the observation of the superelasticity in the crystal was not limited to one observation. A further nine crystals of varying dimensions were loaded into a membrane-driven Diamond Anvil Cell (mDAC) to see whether the aspect ratio of the crystals would have an impact on the superelastic behaviour (Fig. S2 and Table S1 in ESI‡). Five out of nine crystals showed an increase in aspect ratio on compression with one indicating a reduction (crystal 8) and a further 3 crystals that showed no change. During this process, we noted a slight delay of the phase transformation indicating that kinetics will be playing a part.
The single-crystal to single-crystal phase transition opened the possibility of studying the transition using diffraction techniques to gain a molecular understanding of this remarkable physical change. To perform the diffraction experiment, we loaded a single crystal of isonicotinamide Form I into a Merrill–Bassett DAC equipped with Boehler–Almax diamonds, a steel gasket (250 μm hole), ruby as the pressure marker and petroleum ether 35:60 as the pressure-transmitting medium. Form I isonicotinamide crystallises with one molecule in the asymmetric unit in monoclinic P21/c with unit cell dimensions: a = 10.229(3) Å, b = 5.7538(16) Å, c = 10.095(3) Å, β = 97.277(18)° (Table S2, ESI‡).10 On application of pressure, isonicotinamide displays anisotropic behaviour with the cell lengths and volume decreasing monotonically. Each dimension decreasing by 10.25%, 1.50% and 5.44% for the a-, b-, and c-axis, respectively to 4.33 GPa. (Fig. 2).16 This behaviour is similar to other simple organics e.g. aniline and α-PABA.7,9,17 We are able to fit the data to a 3rd order Birch–Murnaghan Equation of State, with a bulk modulus (K0) of 10.6(7) GPa, V0 = 589(5) and K′ = 7(15), which is typical for organic solids (Fig. S3 in ESI‡).18,19
 | ||
| Fig. 2 Variation in (a) normalised unit cell lengths and (b) unit cell volume of isonicotinamide Form I and Form I′ as a function of pressure. Different symbols indicate the two different crystallites used in the pressure study. The area highlighted in grey shows data collected after the transition at 4.98 GPa. Inset shows the cell volumes at 3.99 and 4.00 GPa where the overlap occurs on the main figure. | ||
At 4.98 GPa, there are clear changes in unit cell parameters from 4.33 GPa. The new phase was solved and remains as monoclinic P21/c. The cell parameters for the new phase (designated Form I′) are: a = 13.149(8) Å, b = 3.4103(10) Å, c = 10.173(2) Å, β = 93.11(4)°. Diffraction data provided the molecular level detail of the changes over the phase transition despite the increased mosaicity after the transition. The new phase indicates a major structural rearrangement where molecules in every second layer of the chain rotate substantially causing a change to the environment of the pyridine ring, confirming the difference indicated in the Raman spectra (Fig. S1 in ESI‡). Fig. 3 shows a comparison between the structure of isonicotinamide before and after the transition with molecules superimposed to show the differences between the structures. The Crystal Packing Similarity function in Mercury identified three out of fifteen molecules were in common with each other, with a root mean square of 0.422, signifying a substantial difference in structures.20
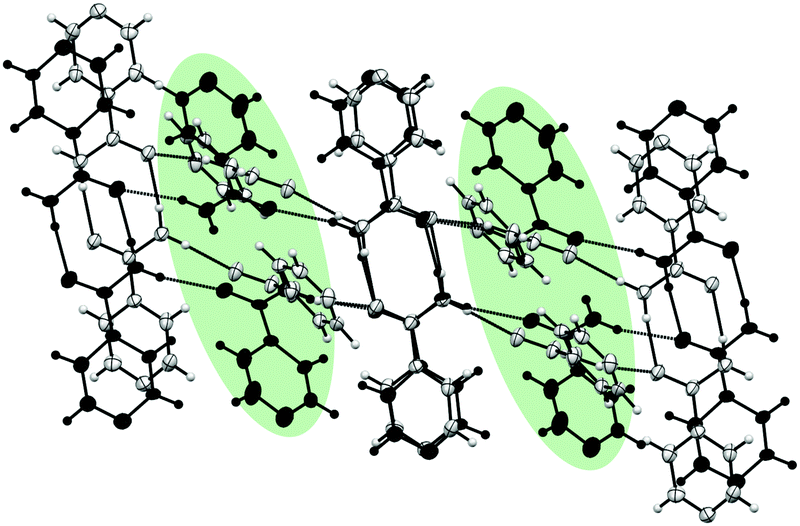 | ||
| Fig. 3 Structural differences between Form I at 4.33 GPa (grey) and the new phase at 4.98 GPa (black). Rotational differences in every other layer is highlighted in green. | ||
To understand what has to happen to the structure to cause this change, we propose a mechanism as detailed in Fig. 4. On compression, the principle axis of strain as calculated by PASCal21 indicates that greatest compression is predominantly between the a- and c-axis as shown by the indicatrix (Fig. S5a, ESI‡) which corresponds to the direction perpendicular to the hydrogen bonded sheets (Fig. S5b, ESI‡). From the perspective of the phase transition, this compression will affect the hydrogen-bonded chains and, in particular, the inter-dimer interactions. In Fig. 4, we have maintained the orientation of the top and bottom dimers which enables the comparison between the orientations of the central sets of dimers. In Form I, the direction of interactions of the central dimers are into the plane of the page whilst in Form I′ they are substantially more across the page. As we move over the transition, we witness a lateral shift to the right of the upper dimer molecules relative to the bottom dimer (indicated by the wire frame). We speculate that this movement induces a rotation of the molecules in every second dimer that forces a change in hydrogen-bonded partner for those molecules. The molecules highlighted in green rotate (about an axis though the N–C–O atoms of the molecule) and hydrogen bond to each other forming a new dimer. In Fig. 4, an orange and blue molecule rotate anticlockwise allowing the formation of a hydrogen bond between them; essentially swapping the hydrogen-bonding partners. This exchange of partners elongates the hydrogen-bonded network, hence extending the length of the crystal (Fig. S6 in ESI‡).
 | ||
| Fig. 4 Structural differences between Form I and Form I′ isonicotinamide at 4.33 and 4.98 GPa, respectively. We focus on dimers coloured in blue and orange and their movement on compression to the new phase, as indicated by the green arrows. After the transition, molecules highlighted in green hydrogen bond to each other forming a new dimer. Additional views are found in Fig. S4 (ESI‡). | ||
So what instigates the change in the structure? We have used Pixel calculations to follow the changes in energy of different intermolecular interactions in the structures. A summary of the interaction energies at each pressure is detailed the ESI‡ (Tables S4 and S5). In both Form I and I′, isonicotinamide forms amide dimers (N–H⋯O) that are positioned over an inversion centre.10 This interaction is the strongest with a total energy of −62.4 kJ mol−1 in Form I and −58.4 kJ mol−1 in Form I′ (Fig. S7a and Table S5; Int. 1, ESI‡). This reduction in favourability is due to the increased repulsion at these higher pressures. The dimers link through hydrogen bonds involving the second hydrogen of the amide to the oxygen of the neighbouring molecule to form chains in the structure along the c-axis and b-axis, respectively for Forms I and I′ (Fig. S7a and Table S5; Int. 2, ESI‡). The dimers are rotated by 68.73° with respect to each other in Form I, whilst this changes to be 74.99° in Form I′. Overall, the energy of these interactions are similar however, the distribution of each of the terms changes to be slightly more dispersive in character that reflects the need for better packing to the detriment of hydrogen bonding (Table S5 in ESI‡).
In both forms, the interaction between the layers is via weaker hydrogen bonding through dispersive CH···N interactions (Fig. S7a, Table S5 and Int. 4, Int. 5, ESI‡). It is here that we notice a change in the interaction energies and these provide a clue to the phase transition. The pyridine dimer interaction (Int. 4, ESI‡) provides some stability to the structure (−13.6 kJ mol−1) but this interaction becomes repulsive as pressure is applied such that the total energy is reduced to −10.4 kJ mol−1. Over the phase transition, we observe a relieving of the repulsive energy contribution to this interaction from 44.3 to 19.6 kJ mol−1, which is a significant change relative to the other intermolecular interactions present and is the result of better packing of the new phase. In addition to this, Interaction 5 also shows a considerable change in the repulsive contribution from 27.1 to 19 kJ mol−1. Therefore, it appears that the repulsion between the molecules contributes substantially to this phase transition, but the question remains why the crystal remains intact over the phase transition?
In solid-state polymerisation reactions, it has been speculated by Kaupp that the ability of a molecule to undergo a reaction is dependent on the ability of the molecule to move in the crystal structure.22 From void analysis of Form I isonicotinamide, we are able to observe where there is space for the molecules to move given a particular probe radius (0.5 Å) (Fig. S8, ESI‡).23 When viewed down the b-axis, the void space is between the pyridine moieties of the molecules. This is important because the location of these voids facilitates the rotation of the molecules during the phase transformation, which is driven by the reduction in the volume as indicated by the lattice enthalpy as a function of pressure (Fig. S9, ESI‡).
In conclusion, we have demonstrated that pressure can be used as a stimulus for superelasticity, opening up new areas for exploration. Isonicotinamide Form I undergoes a single-crystal to single-crystal superelastic transition at 4.98 GPa that has enabled us to propose a possible molecular mechanism by which the transition occurs. The nature of the hydrogen-bonding and void space in the crystal structure poses a question as to how universal a mechanism like this is. At the time of investigation there were five structures in the Cambridge Structural Database that possessed the necessary hydrogen bonding and packing that could exhibit pressure-induced superelastic behaviour, including the isostructural iota-form of nicotinamide.24–28 Despite exceeding a practically relevant pressure regime, the insights that we provide here may, in the future, be used to design organic solids through crystal engineering to exhibit superelastic properties.
We gratefully acknowledge the EPSRC for funding (IDHO & MRW EP/N015401/1) and the University of Strathclyde for financial support (EJ & SSB). The authors would like to thank Colin R. Pulham (University of Edinburgh) for reviewing the manuscript. The authors would like to acknowledge that this work was carried out in the CMAC National Facility supported by UKRPIF (UK Research Partnership Fund) award from the Higher Education Funding Council for England (HEFCE) (Grant Ref: HH13054).
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- P. Naumov, D. P. Karothu, E. Ahmed, L. Catalano, P. Commins, J. Mahmoud Halabi, M. B. Al-Handawi and L. Li, J. Am. Chem. Soc., 2020, 142, 13256–13272 CrossRef PubMed.
- H. Chung, D. Dudenko, F. Zhang, G. D’Avino, C. Ruzié, A. Richard, G. Schweicher, J. Cornil, D. Beljonne, Y. Geerts and Y. Diao, Nat. Commun., 2018, 9, 278 CrossRef PubMed.
- L. Li, P. Commins, M. B. Al-Handawi, D. P. Karothu, J. M. Halabi, S. Schramm, J. Weston, R. Rezgui and P. Naumov, Chem. Sci., 2019, 10, 7327–7332 RSC.
- M. K. Panda, T. Runčevski, S. Chandra Sahoo, A. A. Belik, N. K. Nath, R. E. Dinnebier and P. Naumov, Nat. Commun., 2014, 5, 4811 CrossRef PubMed.
- M. K. Panda, T. Runčevski, A. Husain, R. E. Dinnebier and P. Naumov, J. Am. Chem. Soc., 2015, 137, 1895–1902 CrossRef PubMed.
- G. Liu, J. Liu, Y. Liu and X. Tao, J. Am. Chem. Soc., 2014, 136, 590–593 CrossRef PubMed.
- N. P. Funnell, A. Dawson, W. G. Marshall and S. Parsons, CrystEngComm, 2013, 15, 1047–1060 RSC.
- M. M. Nobrega, M. L. A. Temperini and R. Bini, J. Phys. Chem. C, 2017, 121, 7495–7501 CrossRef.
- T. Yan, D. Xi, Z. Ma, X. Wang, Q. Wang and Q. Li, RSC Adv., 2017, 7, 22105–22111 RSC.
- C. B. Aakeröy, A. M. Beatty, B. A. Helfrich and M. Nieuwenhuyzen, Cryst. Growth Des., 2003, 3, 159–165 CrossRef.
- J. Li, S. A. Bourne and M. R. Caira, Chem. Commun., 2011, 47, 1530–1532 RSC.
- K. S. Eccles, R. E. Deasy, L. Fábián, D. E. Braun, A. R. Maguire and S. E. Lawrence, CrystEngComm, 2011, 13, 6923–6925 RSC.
- A. I. Vicatos and M. R. Caira, CrystEngComm, 2019, 21, 843–849 RSC.
- S. Takamizawa and Y. Miyamoto, Angew. Chem., Int. Ed., 2014, 53, 6970–6973 CrossRef PubMed.
- S. Takamizawa and Y. Takasaki, Chem. Sci., 2016, 7, 1527–1534 RSC.
- B. B. Sharma, C. Murli and S. M. Sharma, J. Raman Spectrosc., 2013, 44, 785–790 CrossRef.
- A. J. Cruz-Cabeza, R. J. Davey, I. D. H. Oswald, M. R. Ward and I. J. Sugden, CrystEngComm, 2019, 21, 2034–2042 RSC.
- R. Angel, J. Gonzalez-Platas and M. Alvaro, Z. Kristallogr., 2014, 229(5), 405–419 Search PubMed.
- J. Gonzalez-Platas, M. Alvaro, F. Nestola and R. Angel, J. Appl. Crystallogr., 2016, 49, 1377–1382 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef PubMed.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef.
- G. Kaupp and M. R. Naimi-Jamal, CrystEngComm, 2005, 7, 402–410 RSC.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B, 2016, 72, 171–179 CrossRef PubMed.
- B. W. Skelton, C. Pakawatchai and A. H. White, CSD Commun., 2017 DOI:10.5517/ccdc.csd.ccpkr21.
- A. R. Kennedy, CSD Commun., 2017 DOI:10.5517/ccdc.csd.cc1pxt9q.
- C. Cohen-Addad and J.-P. Cohen-Addad, J. Chem. Soc., Perkin Trans. 2, 1978, 168–171 RSC.
- C. Guo, M. B. Hickey, E. R. Guggenheim, V. Enkelmann and B. M. Foxman, Chem. Commun., 2005, 2220–2222 RSC.
Footnotes |
| † All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/4f1f2a57-88e4-40c0-863d-72674cf41490 |
| ‡ Electronic supplementary information (ESI) available: Crystallographic Information for the Isonicotinamide structures at pressure, hydrogen-bonding parameters for each structure and pixel calculations. CCDC 2104467–2104477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04692f |
| This journal is © The Royal Society of Chemistry 2021 |