
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFragment evolution for GPCRs: the role of secondary binding sites in optimization†
Florent
Chevillard‡
a,
Ádám
Kelemen‡
b,
Jillian G.
Baker
c,
Vivien A.
Aranyodi
b,
Frank
Balzer
a,
Peter
Kolb
 *a and
György M.
Keserű
*b
*a and
György M.
Keserű
*b
aPharmaceutical Chemistry, Philipps-University Marburg, Marbacher Weg 8, Marburg 35037, Germany. E-mail: peter.kolb@uni-marburg.de; Tel: +49 6421 28 25908
bMedicinal Chemistry Research Group, Research Centre for Natural Sciences, Magyar tudósok körútja 2, Budapest 1117, Hungary. E-mail: keseru.gyorgy@ttk.hu; Tel: +36 1382 6821
cCell Signalling Research Group, School of Life Sciences, University of Nottingham, Nottingham NG7 2RD, UK
First published on 10th September 2021
Abstract
We developed a docking-based fragment evolution approach that extends orthosteric fragments towards a less conserved secondary binding pocket of GPCRs. Evaluating 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 extensions for the β1- and β2-adrenergic receptors we synthesized and tested 112 bitopic molecules. Our results confirmed the positive contribution of the secondary binding pocket to both potency and selectivity optimizations.
000 extensions for the β1- and β2-adrenergic receptors we synthesized and tested 112 bitopic molecules. Our results confirmed the positive contribution of the secondary binding pocket to both potency and selectivity optimizations.
With more than 30% of approved drugs, G protein-coupled receptors (GPCRs) belong to one of the major drug target classes.1 Endogenous GPCR ligands are typically binding in the conserved orthosteric binding pocket (OBP) available from the extracellular side of the cell membrane, and it is a highly tractable pocket for synthetic compounds. Fragment-based approaches, however, might explore both orthosteric and potential allosteric sites and therefore provide easy-to-develop starting points for subsequent fragment-to-lead optimizations. Thanks to the increasing number of available X-ray structures,2 medicinal chemistry teams can implement structure-based strategies for these optimizations. Here we present a general structure-based protocol for the evolution of aminergic GPCR fragment hits exemplified on the β1- and β2-adrenergic receptors.
We have previously investigated the ligand space of the β2-adrenergic receptor (β2AR) with two of our approaches for structure-based FBLD. First, we have grown fragments emerging from an in vitro screen with our in silico structure-aware tool-box PINGUI.3 Second, utilizing our database of easily synthesizable molecules SCUBIDOO,4 we implemented a matrix-based strategy for potency optimization.5 Here, we merge these two strategies and apply them to a selectivity-driven task. Since fragments usually bind to the strongest hot spots, typically located in the orthosteric pocket, we started from fragments bound in the OBP of both the β1-adrenergic receptor (β1AR) and β2AR with approximately equal affinity and attempted to grow each of them such that the resulting molecule would prefer either the β1AR or the β2AR. Using every building block in multiple molecules, this approach leads to “molecular matrices” and facilitates direct comparisons of optimized fragments.5,6 As we worked on two receptors, there are two such matrices, one for the β1AR and one for the β2AR. Our concept was based on the expectation that the less conserved nature of the allosteric secondary binding pockets (SBPs) available in the extracellular vestibule of G protein-coupled receptors (GPCRs) would result in selective bitopic (i.e. occupying both OBP and SBP) compounds.7 In addition to the potentially improved selectivity, the increased number of interactions anticipates improvements in the binding affinity of the generated bitopic compounds. Using two sets of fragments specifically designed for the β1 and β2 SBPs, our further goal was identifying the role of the SPB in both potency and selectivity optimizations. Comparative analysis of the corresponding optimization matrices revealed that SBP-binding moieties mostly improved the binding affinity and to a smaller extent also played a part in compound selectivity. Furthermore, mapping the corresponding SBPs with a limited number of fragments provided detailed information on the activity landscape of the targets and hints about the main selectivity drivers for both of the receptors.
The orthosteric binding pocket of β1AR and β2AR is embedded in the transmembrane intrahelical space and binds their endogenous ligands, i.e. adrenaline and noradrenaline. This site is highly similar between both receptors: only one residue differs in the OBP, F3597.35 in the β1AR is changed to Y3087.35 in the β2AR (Fig. S1, ESI†). Both receptors harbor an allosteric secondary binding pocket (SBP) located in the extracellular vestibule9 containing three divergent residues: V3607.36, D3567.32 and I1182.64 in the β1AR are replaced with I3097.36, K3057.32 and H932.64, respectively, in the β2AR. The last two have drastic physiochemical property differences which makes the SBP a sound target area for exploring its role in the selectivity between the two receptors.8
First, we selected the core OBP binding fragment set. Docking 387707 primary amine fragments (cf. ESI,† Methods) to the OBP of both the β1AR and β2AR was done using FRED.9 This yielded 238 fragments which were then carefully visually inspected for their binding modes and interactions formed with the receptors (e.g. direct interactions with D1133.32 and N3127.39 were mandatory). This evaluation resulted in 32 fragments that were subjected to synthetic feasibility and availability checks. Finally, 21 OBP fragments were ordered and tested experimentally (Table S1 and Fig. S2, S4, ESI†).
In this study we combined the structure-based evolution of the core fragments with the power of SAR-driven optimization using the concept of ligand efficiency (LE = pKD/number of non-hydrogen atoms in the molecule).10 Out of the 21 fragments tested, 13 (62%) showed a ligand efficiency (LE) above 0.4, which is a remarkably high fraction (see ESI,† Table SI_assays.csv). The five fragments (A1–A5) with the best LE (Fig. S3, ESI†) and no significant preference for either target were selected for further growing. Interestingly, some compounds (e.g.A11 and A12) showed a more noticeable fold selectivity of 9.8 and 9.3, respectively, suggesting that the OBP is involved in ligand selectivity. Curiously, none of the assayed fragments showed higher affinity for the β1AR than for the β2AR.
For the selection of suitable fragments with affinity towards the secondary binding site, we first defined surrogates. Considering that reductive alkylation was selected as a robust reaction for growing the orthosteric fragments, our surrogates were derived from aldehyde building blocks by replacing the aldehyde moiety with a primary amine group. This procedure allows us to mimic the interactions of the final product during the initial docking calculation in which the surrogate is placed inside the SBP. The 7893 surrogates (cf. ESI,† Methods) were individually docked to the SBP of both receptors. For each receptor, the top 1000 surrogates according to FRED score were kept after ensuring that they did not overlap with the predicted core fragment position in the OBP. As we intended to explore the receptor-specific affinities and the selectivity, surrogates ranked favorably in both receptors were removed, yielding a set of 598 preferred surrogates for each receptor (i.e. 402 surrogates were found in both receptors in the top 1000 ranked molecules).
For each OBP fragment to be grown, the corresponding derivative products were generated by reacting it in silico with each of the 598 surrogates selected in the previous step using the specified chemistry of reductive alkylation. Docking-derived binding modes and receptor interactions were carefully inspected visually, furthermore ensuring pose fidelity, i.e. that the binding modes of the SBP surrogate and the core OBP fragment did overlay consistently with the pose of the corresponding product. This analysis left 13 and 16 derivatives for the β1AR and β2AR, respectively. All the key steps of the computational workflow are presented in Fig. 1.
 | ||
| Fig. 1 Workflow depicting all required steps to select the building blocks and build the final molecular matrices. | ||
Next, synthesis of bitopic compounds was realized by the robust reductive alkylation reaction (Fig. S15, ESI†), which shows broad substrate and functional group tolerance and is only little sensitive to reaction parameters.11 Moreover, reductive alkylation results in a protonable amine in the molecule, which is a precondition for binding to the βARs. One hundred twelve out of the 145 designed bitopic products (Fig. S5–S12, ESI†) were synthesized successfully (62% and 93% of compounds designed for β1AR and β2AR, respectively) and were submitted to biological assays.
All 112 synthesized molecules were assessed for affinity in the 3H-CGP12177 whole cell binding assay (Fig. 2 and Fig. S5–S9 and SI_assays.csv for summary data, ESI†). Fig. 2 suggests that the core compounds behaved differently when combined with the SBP fragments. For example, core fragment A4 retained affinity for both receptors when the β2-SBP bitopic compounds were examined, however when the same core was combined with the β1-SBP fragment extensions, a loss in affinity was observed in many cases (Fig. 2). When looking at both matrices together, only one out of the 29 SBP fragments (3%) did not yield any active compound against its designed target receptor (B24), while 18 SBP fragments (62%) showed at least one product improving the initial affinity of its core OBP fragment (Fig. 2 and Fig. S13, S14, ESI†). Overall, the accuracy for the β1-SBP bitopic compounds (i.e. compounds with improved affinity against β1AR) was 34%. It was 23% for β2-SBP bitopic compounds. In the β2AR-specific matrix (Fig. S10, ESI†), when compared to their respective core OBP fragments, 15 out of the 60 (25%) synthesized compounds showed an increase in affinity against the β2AR and 35 compounds (58%) showed a decrease in affinity against the β1AR. Compound A1B15 (KD = 35 nM) displayed the highest affinity gain of 69-fold of any compound. The addition of certain SBP-fragments resulted in a lower affinity for all cores e.g.B16, B17 and B18. In contrast, for the β1AR matrix (Fig. S11, ESI†), addition of B1 to core A1 caused the largest increase in affinity – an increase of 9.1 and 27-fold over the core compound A1 at the β1AR and β2AR, respectively. For the β2AR matrix, it was the addition of fragment B15 that resulted in the greatest increase of affinity compared to the core alone. Thus, the addition of B15 increased the affinity of core A1 by 27- and 69-fold (molecule A1B15), 20- and 21-fold (A3B15) and 13- and 11-fold (A5B15) at the β1AR and β2AR, respectively. The molecule with the highest overall affinity of 7 nM at the β2AR was A4B27, however with only marginal increases of the affinity at the β1- and β2AR (2.5- and 5-fold, respectively) over the core fragment.
 | ||
| Fig. 2 Bubble plot representation of the fold selectivity and the fold affinity improvement ratio for the core OBP fragments and the SBP fragments in the β1AR. Ratio below 1 indicates a property deterioration while a ratio above 1 indicates a property gain. The vertical red line separates the β1AR matrix (i.e. compounds designed for higher β1-affinity/selectivity) (left) from those designed for the β2AR matrix (right). | ||
In the β2AR-specific matrix, compound A4B14 showed the most pronounced selectivity for the β2AR (14-fold). Curiously, it was one of the compounds designed for the β1AR, A1B05, that showed the highest absolute β2AR-selectivity overall (i.e. from either matrix). Compound A4 yielded the series with highest β2 preference (maximum fold selectivity = 14.1, mean fold selectivity = 8.2). This can be explained by the fact that A4 was the core OBP fragment with the highest initial fold selectivity (4.5). Interestingly, compound B21, which contains a carboxyl moiety (i.e. predicted to interact with K3057.32 or H932.64 in the β2AR), yielded three products with appreciable selectivity (fold selectivity >10). These outcomes indicate that some degree of selectivity can be achieved via the SBP by targeting K3057.32 or H932.64, but that the initial OBP contribution is vital for the total achievable selectivity.
While it has to be admitted that the overall selectivity trends are moderate, the analysis of several pairs still provides illuminating details about the molecular origins of affinity and selectivity between the β1AR and β2AR. Compounds A1B14 and A1B17 (Fig. S18, ESI†) share the same building block in the OBP, and both are mildly β2-selective. The building blocks in the SBP, B14 and B17, differ by the heteroatoms in the five-membered ring (triazole vs. thiophene) and the distance between the ring and the carboxyl group (one carbon vs. direct attachment). The shorter fragment yields the less potent compounds in both receptors, indicating that the interaction entertained by the carboxyl group lies further away from the OBP. This is likely to be close to position 2.64, which is a His in the β2AR, thus also explaining the slight preference of the compound for this receptor. Comparative analysis of the corresponding matrix revealed that we have basically two situations to consider. In the first one, small changes of the OBP-binding fragment influenced the affinity of the bitopic compounds (compound A1B15versusA2B15). Here we hypothesize that the changed binding mode of the orthosteric fragment points the growing vector to a different part of the SBP. In the other case, changes of the SBP-binding fragment position the identical OBP-binding fragment differently (compound A1B14versusA1B17), which in turn influences the affinity of the bitopic compounds. This highlights the complexity of fragment growing procedures, where subtle changes, such as a slight increase in steric constraints, might have a tremendous impact on the resulting binding mode. This observation also emphasizes the utility of the matrix approach in FBDD optimization programs.
In terms of selectivity, an instructive pair is A1B18 and A1B21 (Fig. 3, Fig. S19, ESI†). In the docking poses, the hydroxy group on the phenyl ring of B18 interacts with the conserved Tyr7.43, while the carboxyl group can’t quite reach to position 2.64 and thus presumably interacts with a water molecule. This explains the relatively similar affinities in the β1AR and β2AR. When B21 is installed, there are two changes: first, the hydroxy group is missing, second, there is an additional carbon between phenyl ring and carboxyl group. This, similar to what was observed for building blocks B14 and B17, pushes the carboxyl group more towards residue 2.64. The resulting favorable interaction with the positively charged His at this position in the β2AR seems to compensate for the loss of the interaction with Tyr7.43. Conversely, given that residue 2.64 is an Ile in the β1AR, the resulting proximity between a charged moiety and an apolar side chains results in complete loss of binding of A1B21 to this receptor. These results together with other pairs identified in the β1AR and β2AR matrices suggest that the selectivity of bitopic compounds is influenced by both of the pockets i.e. a more complicated picture than the one implicated previously.7,12,13 Our analysis revealed that the optimization of selectivity needs the identification of a suitable combination of OBP- and SBP-binding fragments. The matrix-based methodology suggested here can contribute to effectively finding such combinations.
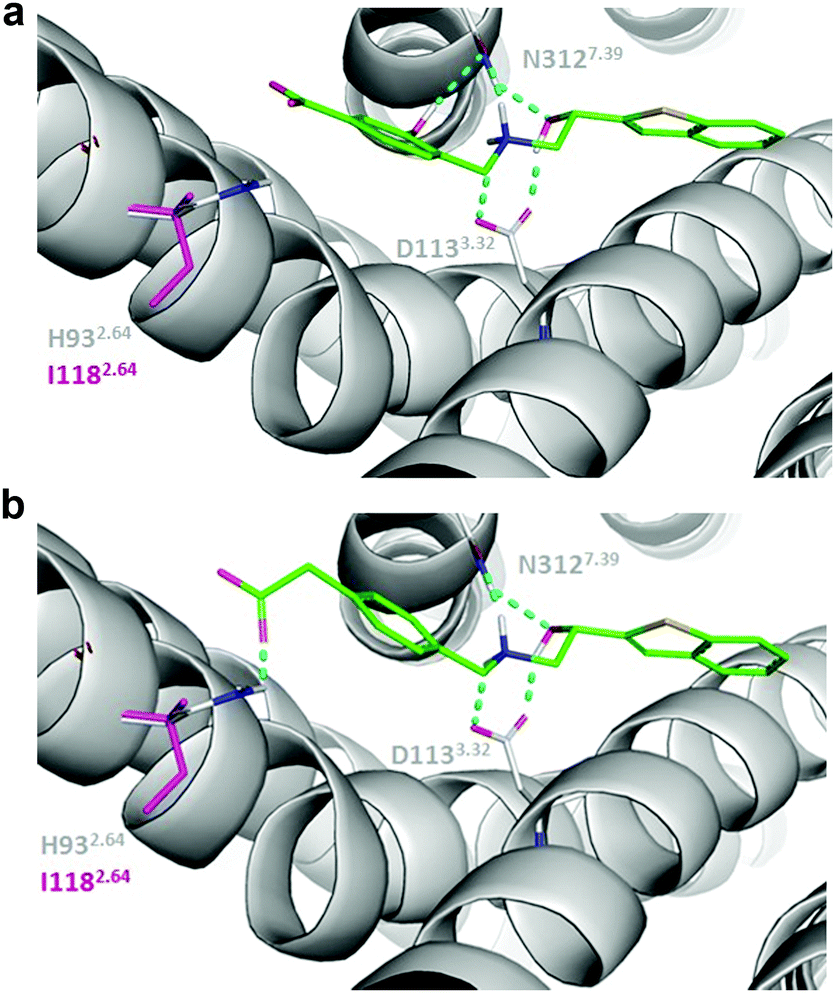 | ||
| Fig. 3 Binding mode prediction of (a) A1B18, (b) A1B21 in the β1AR (residues in magenta) and the β2AR (residues in light grey) binding sites. Ligands are shown with green sticks and polar contacts are represented with cyan dashed lines. | ||
Although we explored SAR and selectivity trends effectively, we achieved mild selectivity for our β2AR designs at best. What could have been the reasons for this limited selectivity? The primary reason is certainly that the two receptors have very similar binding pockets. The OBP is virtually the same except for the Phe to Tyr change at position 7.35, and substantial exploitable differences occur only at the far end of the SBP. A second reason might be that we used a homology model of the human β1AR, as the X-ray structures available at the start of this project were exclusively from turkey. Last, but not least, selectivity in this receptor system might not only be due to direct ligand–receptor interactions. Different water networks, polarization effects and subtle conformational changes of the receptor upon ligand binding, to name just a few, might play substantial roles. However, these phenomena are clearly beyond our approach, which used docking to rigid receptors.
Our study yielded insights into structure–activity and structure–selectivity relationships and demonstrated the power of using optimization matrices as a design concept that ensures efficient resource usage while maximizing the information obtained from experiments. Paired with high hit rates, such methods can support hit expansion in the lead discovery projects. Investigating the impact of the orthosteric and secondary binding pockets on the binding affinity and selectivity, we found that the optimization is influenced by both sites. While binding affinity was less sensitive to the SBP interactions, our data suggests that the optimization of selectivity needs the identification of bitopic compounds constructed from suitable OBP and SBP pairs. This goal can be achieved efficiently by the optimization matrix approach used here. Providing detailed SAR information, this strategy maps both of the binding pockets effectively and suggests viable growing vectors for optimizing OBP-bound fragment starting points.
The authors are grateful to the GLISTEN (CM1207) and ERNEST (CA18133) COST Actions facilitating their collaboration. A. K., V. A. A. and G. M. K. were supported by the National Brain Research Program (2017-1.2.1-NKP-2017-00002). P. K. thanks the German Research Foundation DFG for Emmy Noether fellowship KO4095/1-1 (also supporting F. C.) and Heisenberg professorships KO4095/4-1 and KO4095/5-1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schiöth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16(12), 829–842 Search PubMed.
- A. J. Kooistra, S. Mordalski, G. Pándy-Szekeres, M. Esguerra, A. Mamyrbekov, C. Munk, G. M. Keserű and D. E. Gloriam, Nucleic Acids Res., 2021, 49, 335–343 Search PubMed.
- F. Chevillard, H. Rimmer, C. Betti, E. Pardon, S. Ballet, N. van Hilten, J. Steyaert, W. E. Diederich and P. Kolb, J. Med. Chem., 2018, 61, 1118–1129 Search PubMed.
- F. Chevillard and P. Kolb, J. Chem. Inf. Model., 2015, 55, 1824–1835 Search PubMed.
- F. Chevillard, S. Stotani, A. Karawajczyk, S. Hristeva, E. Pardon, J. Steyaert, D. Tzalis and P. Kolb, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11496–11501 Search PubMed.
- J. Galambos, A. Bielik, M. Krasavin, Z. Orgován, G. Domány, K. Nógrádi, G. Wágner, G. T. Balogh, Z. Béni, J. Kóti, Z. Szakács, A. Bobok, S. Kolok, M. L. Mikó-Bakk, M. Vastag, K. Sághy, J. Laszy, A. S. Halász, O. Balázs, K. Gál, I. Greiner, Z. Szombathelyi and G. M. Keserű, J. Med. Chem., 2017, 60(6), 2470–2484 Search PubMed.
- M. Michino, T. Beuming, P. Donthamsetti, A. H. Newman, J. A. Javitch and L. Shi, Pharmacol. Rev., 2015, 67, 198–213 Search PubMed.
- A. E. Wakefield, J. S. Mason, S. Vajda and G. M. Keserű, Sci. Rep., 2019, 9, 6180 Search PubMed.
- M. Mcgann, J. Chem. Inf. Model., 2011, 51, 578–596 Search PubMed.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 Search PubMed.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 Search PubMed.
- R. O. Hutchins, N. R. Natale and I. M. Taffer, Chem. Commun., 1978, 1088–1089 Search PubMed.
- A. H. Newman, T. Beuming, A. K. Banala, P. Donthamsetti, K. Pongetti, A. LaBounty, B. Levy, J. Cao, M. Michino and R. R. Luedtke, et al. , J. Med. Chem., 2012, 55, 6689–6699 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc04636e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |