
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUse of 3D domain swapping in constructing supramolecular metalloproteins
Shun
Hirota
 *,
Tsuyoshi
Mashima
and
Naoya
Kobayashi
*,
Tsuyoshi
Mashima
and
Naoya
Kobayashi
Division of Materials Science, Graduate School of Science and Technology, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: hirota@ms.naist.jp
First published on 29th October 2021
Abstract
Supramolecules, which are formed by assembling multiple molecules by noncovalent intermolecular interactions instead of covalent bonds, often show additional properties that cannot be exhibited by a single molecule. Supramolecules have evolved into molecular machines in the field of chemistry, and various supramolecular proteins are responsible for life activities in the field of biology. The design and creation of supramolecular proteins will lead to development of new enzymes, functional biomaterials, drug delivery systems, etc.; thus, the number of studies on the regulation of supramolecular proteins is increasing year by year. Several methods, including disulfide bond, metal coordination, and surface–surface interaction, have been utilized to construct supramolecular proteins. In nature, proteins have been shown to form oligomers by 3D domain swapping (3D-DS), a phenomenon in which a structural region is exchanged between molecules of the same protein. We have been studying the mechanism of 3D-DS and utilizing 3D-DS to construct supramolecular metalloproteins. Cytochrome c forms cyclic oligomers and polymers by 3D-DS, whereas other metalloproteins, such as various c-type cytochromes and azurin form small oligomers and myoglobin forms a compact dimer. We have also utilized 3D-DS to construct heterodimers with different active sites, a protein nanocage encapsulating a Zn–SO4 cluster in the internal cavity, and a tetrahedron with a designed building block protein. Protein oligomer formation was controlled for the 3D-DS dimer of a dimer-monomer transition protein. This article reviews our research on supramolecular metalloproteins.
 Shun Hirota | Shun Hirota received his PhD from the Graduate University for Advanced Studies in Japan in 1995. After postdoctoral studies at the Institute for Molecular Science in Japan and Emory University in the US, he joined Nagoya University as an assistant professor in 1996 and became an associate professor at Kyoto Pharmaceutical University in 2002. He was invited to Nara Institute of Science and Technology as a full professor in 2007. His research interests include structures, functions, and reaction mechanisms of metalloproteins. |
 Tsuyoshi Mashima | Tsuyoshi Mashima received his PhD in chemistry from Osaka University under the supervision of Prof. Takashi Hayshi in 2018. He undertook postdoctoral studies at the Eindhoven University of Technology with Prof. Luc Brunsveld from 2018 to 2021. He moved to Nara Institute of Science and Technology as an assistant professor in the group of Prof. Shun Hirota in 2021. His research interests include proteins-assembly, DNA-nanotechnology, supramolecular chemistry and metalloproteins. |
 Naoya Kobayashi | Naoya Kobayashi received his PhD from Shinshu University in Japan in 2016. He engaged postdoctoral studies at the Institute for Molecular Science and the Exploratory Research Center on Life and Living Systems in Japan from 2017 to 2021. He joined Nara Institute of Science and Technology as an assistant professor in 2021. His research interests are on protein design and engineering. |
1. Introduction
In recent years, various methods have been attempted to construct supramolecular proteins.1–7 Research on constructing artificial supramolecular proteins to impart properties that cannot be exhibited by a single molecule, such as molecular association and dissociation, has attracted increasing attention. Methods for creating supramolecular proteins are rapidly developing, where structural control is advantageous for functional control. The introduction of a bridging site to the protein surface by amino acid mutation has been widely used to connect proteins; this method includes disulfide bond8,9 and metal coordination.10–13 Attempts have also been made to utilize interfacial interactions between proteins for the construction of supramolecular proteins. Two proteins in which each protein forms homo-oligomers are connected as a fusion protein, resulting in the formation of structured supramolecular proteins,14–17 and computer design has been used to optimize the interfacial interactions between the two proteins.18,19We have been studying the phenomenon of 3D domain swapping (3D-DS), in which the same structural region is exchanged three-dimensionally between molecules of the same protein (Fig. 1). The unit structure of domain-swapped oligomers is essentially identical to the corresponding monomer structure, except for the hinge region that connects the exchanged regions. The first 3D-DS structure was reported in 1994 for a dimer of diphtheria toxin.20 Since then, 3D-DS has been reported to occur in many natural proteins and suggested to be related to protein deposition diseases;21 for example, a mutant of α1-antitrypsin22—the serpin (serine protease inhibitory protein) involved in serpin disease—and β2 microglobulin23 that causes dialysis amyloidosis have been shown to involve 3D-DS, as well as human prion protein24 and cystatin C.25 The number of proteins known to exhibit 3D-DS has been increasing, and over 600 3D-DS structures have been deposited in the Protein Data Bank.26 Most swapped domains are at either the N- or C-terminus and the swapped domains are diverse in their primary and secondary structures, suggesting that almost any protein may undergo 3D-DS.27 3D-DS has also been applied to create functional protein molecules.28–31 On the other hand, the metal site is essential to the function of metalloproteins, and new strategies for constructing supramolecular metalloproteins are particularly useful for creating functional proteins.32–36 We have shown that various metalloproteins can exhibit 3D-DS and constructed various supramolecular metalloproteins.37,38 This review introduces 3D-DS for producing supramolecular metalloproteins.
 | ||
| Fig. 1 Schematic view of 3D-DS. The protein example is diphtheria toxin (PDB ID: monomer, 1MDT; dimer, 1DDT). The magenta and cyan structural regions are exchanged between molecules, resulting in the formation of a 3D-DS dimer. | ||
2. Preparation of supramolecular proteins using covalent bonds and surface interactions
2.1. Disulfide bond
The simplest method for connecting proteins is to introduce a cysteine (Cys) to the protein surface and form an intermolecular disulfide bond via the sulfur atom of the Cys residue. When a Cys was introduced to the surface of the homohexameric protein Hcp1, which forms a ring structure, a tube structure with stacked rings was constructed by intermolecular disulfide bonds (Fig. 2A).8 The hexamer of the Hcp1 mutant reversibly converted between the tube and ring structures by controlling the formation of disulfide bonds with a redox reaction.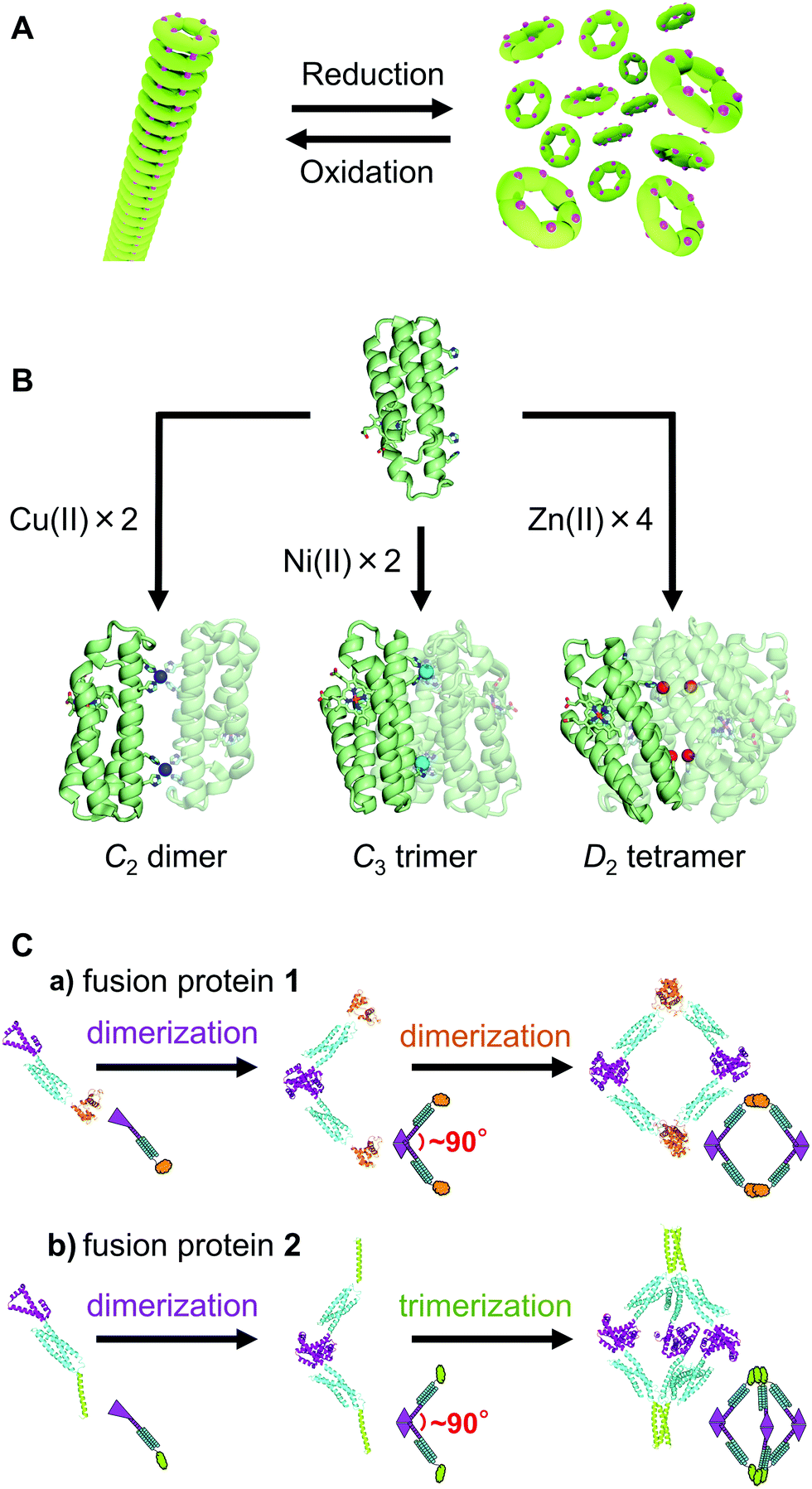 | ||
| Fig. 2 Schematic views of supramolecular construction. (A) Conversion between tube and ring structures with a redox reaction. Ring-shaped protein hexamers are connected through a disulfide bond in the tube structure. The magenta dots on the rings represent the Cys residues. (B) Dimer, trimer, and tetramer constructed by Cu2+ (blue), Ni2+ (cyan), and Zn2+ (red) ion coordination to a cyt cb562 mutant, respectively. (C) Design of fusion proteins 1 and 2 and construction of their assemblies: (a) tetramer, (b) hexamer. Adapted with permission from ref. 16. Copyright 2019 American Chemical Society. | ||
A two-dimensional crystal without lattice defects has been constructed by introducing Cys to the surface of the tetrameric protein RhuA and forming a disulfide bond network between molecules,9 where the flexibility of the disulfide bond allowed the crystal to reduce the defects. The arrangement of the protein molecules in this two-dimensional crystal was changed cooperatively by gentle mixing with a pipette and sedimentation.
2.2. Metal coordination
Since a metal and its ligand form a bond in a fixed orientation, metal coordination is widely used in metal–organic frameworks.39,40 The metal coordination bond shows a specific coordination structure depending on the metal ion, and the bond can form and cleave depending on the presence of metal ions; thus, it is also effective to use the metal coordination bond to control the formation of supramolecular proteins. A supramolecule of a cytochrome (cyt) b562 mutant with a periodic structure was obtained by chemically modifying the surface of the protein so that the haem-protein interaction occurs intermolecularly.41 Two- and three-dimensional supramolecules of cyt cb562 (a cyt b562 mutant protein with a haem immobilized on the 4-helix bundle protein moiety by introducing two Cys residues to the polypeptide)42 have been constructed by introducing metal coordination sites to the surface of the protein. Metal binding sites were formed between the cyt cb562 molecules at the protein surfaces with the introduced histidine(s) (His), bis-His clamp(s), and/or an aspartate (Asp). When Cu2+, Ni2+, and Zn2+ were coordinated to this site, dimers, trimers, and tetramers were obtained, respectively (Fig. 2B).10,43 Additionally, when two Zn2+-binding sites with different affinities were introduced to the surface of cyt cb562, one-dimensional spiral nanotubes and two- or three-dimensional protein sequence crystals were obtained, depending on the pH and Zn2+ concentration of the solution.11A metal binding site comprising two bis-His clamps was constructed between the surface of glutathione-S-transferase (GST) molecules by introducing two consecutive His (bis-His clamps), where nanorings were constructed through Ni2+ ion coordination.12 A chaperonin GroEL mutant appended with zwitterionic merocyanine units, was assembled one-dimensionally utilizing the coordination of Mg2+ ions.13 ATP hydrolysis induced conformational changes in the chaperonin units, which in turn generated a mechanical force that led to the disassembly of the tube and release of the guest molecules.
2.3. Surface–surface interaction
Protein surface–surface interactions are also useful for the construction of supramolecular proteins. When constructing supramolecular proteins with surface–surface interactions, fusion proteins that link subunits of different homo oligomeric proteins are frequently used. Using a fusion protein (WA20-foldon) in which the artificial dimeric protein WA20 and the trimeric protein foldon were connected through a 5-residue linker, a barrel structure hexamer, a tetrahedral cage 12-mer, and various other shaped supramolecular proteins were constructed.14 A uniform icosahedral 60-meric supramolecular protein, TIP60, which contained 20 regular-triangle-like pores on the surface, symmetrically self-assembled from fusion proteins of a pentameric Sm-like protein and a dimeric MyoX-coil domain.44,45 A supramolecular protein was constructed using a fusion protein in which a coiled-coil tetrameric peptide and a trimeric protein were linked with a relatively short peptide.15 When the number of residues in the peptide linker was four, a cage structure could be selectively obtained, but when the number of linker residues was two, the ideal octamer was not producible due to the short linker, and the sizes varied. A flexible linker consisting of several amino acids is convenient for obtaining a protein oligomer with the most stable structure, although it is often difficult to control the supramolecular protein to a fixed structure.A fusion protein (fusion protein 1) was constructed by linking two dimeric proteins with a three-helix bundle protein and self-assembled mainly into tetramers (Fig. 2C).16 Replacing one of the dimeric proteins of fusion protein 1 with a trimeric protein (fusion protein 2) resulted mainly in hexamers. According to ab initio structural models reconstructed from the small-angle X-ray scattering (SAXS) data, the tetramer of 1 and hexamer of 2 adopted quadrangle and cage-like structures, respectively, although high-speed atomic force microscopy observations indicated that the tetramer and hexamer exhibited conformational dynamics. These results show that the method of utilizing three-helix bundle-linked fusion proteins is useful in the construction of protein nanostructures.
A dodecameric fusion protein in which a dimeric and a trimeric protein are linked by an α-helix linker has been shown to self-assemble into a stable cage structure with a diameter of 16 nm.17 In linking two types of oligomeric proteins with an α-helix linker, the interaction between the fusion proteins utilizes the interaction of the original oligomeric proteins and is relatively strong. However, the structure of the α-helix linker may fluctuate greatly, and precise design is required to obtain a uniform supramolecular structure. A 24-subunit, 13-nm diameter complex with octahedral symmetry and a 12-subunit, 11 nm dimeter complex with tetrahedral symmetry were constructed with trimeric protein building blocks using computational design.46 A computationally designed transmembrane monomer, homodimer, trimer, and tetramer have been shown to localize to bacterial and mammalian cell membranes.18 Amino acid residues on the protein surface have been computationally designed to construct a protein-inorganic hybrid material.19 These results indicate that computer design is a promising tool for supramolecular protein design.
3. Preparation of supramolecular metalloproteins utilizing 3D domain swapping
3.1. 3D domain swapping of native metalloproteins
Our first report of 3D-DS addressed horse cyt c in 2010.47 Cyt c was reported to polymerize in 1962, but the polymerization mechanism remained unknown.48 To solve the polymerization mechanism, we made small oligomers of horse cyt c by treating its monomer with ethanol, subsequently freeze-drying the solution, redissolving the protein residue, and heating the obtained high-order oligomers at 37 °C for 40 min to dissociate them into small oligomers.47 We purified the small oligomers (dimer, trimer, and tetramer) by size exclusion chromatography and found that the dimer and trimer exhibit 3D-DS structures in which the C-terminal α-helix is exchanged between protomers (dimer, PDB ID: 3NBS and trimer, PDB I D: 3NBT).47 We also performed SAXS and differential scanning calorimetry analyses on the purified small-size oligomers and deduced that the nature of cyt c polymerization is successive 3D-DS (in other words, ‘runaway domain swapping’ or ‘propagated domain swapping’) (Fig. 3).47 Similar successive domain swapping has been reported for several proteins, including RNase A,49 serpin,50 cystatin C,51 T7 endonuclease I,52 β2-microglobulin,23 and γD-crystallin,53 indicating that successive 3D-DS occurs in various proteins. Successive 3D-DS has also been achieved in a three-helix bundle protein by removing the loop between the second and third helices.54 | ||
| Fig. 3 Schematic view of cyt c polymerization: (A) monomer crystal structure (PDB: 1HRC),59 (B) model structure of open monomer, (C) model structure of open dimer, (D) dimer crystal structure (PDB: 3NBS),47 (E) model structure of open trimer, (F) trimer crystal structure (PDB: 3NBT),47 and (G) model structure of open tetramer. Met80 is highlighted in yellow. Sphere models are shown below each structure. Adapted with permission from ref. 47. Copyright 2010 National Academy of Sciences. | ||
The binding properties of cyt c to its partner proteins, cyt bc1 complex and cyt c oxidase, and the cell membrane, may not be affected by 3D-DS, since the interface of the subunits in the 3D-DS dimer is different from the putative binding sites of cyt c to the partner proteins and cell membrane.55–57 On the other hand, the reaction properties of cyt c are altered by 3D-DS. Met and His are coordinated to the haem iron in the cyt c monomer.58,59 In contrast, haem-coordinating Met80 disassociates from the haem iron in the cyt c 3D-DS dimer and trimer; thus, external ligands, such as the cyanide ion, bind more easily to the haem iron of oligomers than to that of the monomer.60 Peroxides may also bind to the haem iron of cyt c oligomer more easily, making the peroxidase activity of the cyt c dimer higher than that of the monomer,61 which is consistent with the fact that the peroxidase activity of cyt c increases when Met80 dissociates from the haem iron, which may be relevant to apoptosis.38,62–64 3D-DS induces other changes in the protein properties of cyt c. The cyt c 3D-DS oligomers bound more strongly than the monomer to anionic phospholipid-containing vesicles and to the outer membrane of HeLa cells, where the damage to HeLa cells by the cyt c oligomer was proportional to the membrane binding.65 The decreased stability of the cyt c 3D-DS dimer compared to the monomer has been utilized to construct amyloid fibrils; amyloid fibrils were obtained by laser trapping the cyt c 3D-DS dimer, whereas it was relatively difficult to obtain amyloid fibrils from the monomer.66
3D-DS cyt c oligomers dissociate to monomers through exothermic processes.47 The enthalpy change ΔH of oligomer dissociation is decreased by ∼20 kcal mol−1 when the oligomer is elongated by one functional (monomer structure) unit.47 The ΔH of Met coordination to the haem in cyt c has been estimated to be −18 kcal mol−1,67 indicating that Met coordination contributes greatly to the ΔH (−20 kcal mol−1 per monomer) of cyt c oligomer dissociation. According to theoretical calculations,68 the stability of the horse cyt c 3D-DS dimer decreases slightly compared to that of its monomer with a total free energy increase of 25 kcal mol−1. The dimer-to-monomer dissociation temperature (61.0 °C) was similar between wild-type (WT) and M80A human cyt c, although Met80 coordination to the haem iron contributed to the stabilization of the monomer (ΔH = −16 kcal mol−1 in human cyt c).69 The activation enthalpy values for the dissociation of 3D-DS dimers to monomers in WT and M80A cyt c were similar and relatively large (WT, 120 ± 10 kcal mol−1; M80A, 110 ± 10 kcal mol−1). Similarly, the activation heat capacity change for the dissociation of yeast iso-1-cyt c 3D-DS dimers to monomers was large.70 These results reveal that a large protein structural change takes place in cyt c upon the dissociation of 3D-DS dimers to monomers regardless of the Met80-haem iron bond, whereas Met80 coordination stabilizes the monomer.
Various metalloproteins can also form oligomers by 3D-DS (Fig. 4). For example, negatively charged Pseudomonas aeruginosa (PA) cyt c551 and positively charged Hydrogenobacter thermophilus (HT) cyt c552 both belong to the c-type cyt protein family, exhibit similar monomer structures (three long α-helices),71,72 and can form 3D-DS dimers by a similar treatment to cyt c 3D-DS using ethanol,73,74 although the N-terminal region containing the N-terminal α-helix and haem is exchanged between protomers in the dimers (PA cyt c551, PDB ID: 3X39; HT cyt c552, PDB ID: 3VYM; Fig. 4), whereas the C-terminal α-helix is exchanged in the cyt c 3D-DS dimer.47 In PA cyt c551 and HT cyt c552, a relatively strong hydrogen bond network is formed at the loop containing the haem-coordinating Met, whereas an omega loop is formed at the corresponding region in cyt c. Thus, in PA cyt c551 and HT cyt c552, the C-terminal α-helix following this loop may be relatively rigid and interact relatively strongly with the rest of the protein during 3D-DS, resulting in formation of the 3D-DS dimer exchanging the N-terminal region.73 When we inserted three Gly residues into the hinge loop of WT HT cyt c552, high-order oligomers were obtained by the 3D-DS treatment, presumably due to the decreased steric hindrance between the HT cyt c552 structural (functional) units in 3D-DS (Fig. 5).75 For PA cyt c551 and WT HT cyt c552, the hinge loop of 3D-DS is short, only 3 residues; thus, it is difficult for the WT of both proteins to form high-order oligomers.73,74 In addition, two 3D-DS dimers (major and minor) were obtained for the HT cyt c552 mutant with three Gly inserted (Fig. 5).75 We were able to separate the two dimers by ion exchange chromatography owing to the elongation of the structural units in the major dimer. The N-terminal region was exchanged in the major dimer (PDB ID: 5AUR), similar to the WT HT cyt c552 dimer, whereas the C-terminal region was exchanged in the minor dimer (PDB ID: 5AUS), similar to the horse cyt c dimer, demonstrating that HT cyt c552 and presumably other c-type cyts can undergo both N- and C-terminal 3D-DS. Similar dual 3D-DS has been reported for RNase A.76–78 These results show that 3D-DS may occur at various protein sites, where the swapping region depends on the stability and/or strength of the interaction between the swapping region and the rest of the protein.
 | ||
| Fig. 4 Schematic view of 3D-DS of metalloproteins. Puzzles (cyclic oligomers, polymers, small oligomers, and compact dimers) are constructed using a glue (swapping domain) to connect pieces (proteins). | ||
 | ||
| Fig. 5 Schematic view of the introduction of three Gly residues into the hinge loop of HT cyt c552 and the formation of 3D-DS dimers: major (PDB ID: 5AUR, red and green) and minor dimers (PDB ID: 5AUS, red and blue). The pink arrow in the WT monomer structure represents the insertion position. The hinge loop is shown in orange, and the inserted residues (Gly19Gly20Gly21) are shown in pink. Adapted with permission from ref. 75. Copyright 2015 Royal Society of Chemistry. | ||
Similar to cyt c, cyt c555 from the hyperthermophilic bacterium Aquifex aeolicus (AA) converts into a ligand-binding protein by 3D-DS.74 AA cyt c555 is a hyperstable c-type cyt that possesses a six-coordinate haem and a unique additional long 310-α-310 helix containing the haem-coordinating Met61.79 It forms a 3D-DS dimer by exchanging the region containing the 310-α-310 and C-terminal helices between molecules (PDB ID: 3X15).74 The overall protein structure of the AA cyt c555 3D-DS dimer corresponds well to that of the monomer, except for the hinge region (Val53–Lys57). The coordination of Met61 in the extra 310-α-310 helix to the haem iron is also observed in the dimer as in the monomer, but Met61 originates from the other protomer to which the haem belongs, similar to 3D-DS dimers of other bacterial c-type cyts.73–75 The 3D-DS dimer maintains thermostability (dissociation temperature to monomers, 92 °C) and pH stability (pH 2.2–11.0), apparently owing to the conservation of the overall secondary and tertiary structures of the monomer in the 3D-DS dimer. Similar to the cyt c 3D-DS dimer,60,61 external ligands bound to the haem iron of the AA cyt c555 3D-DS dimer in solution, such as CN− and CO in the ferric and ferrous states, respectively, whereas these ligands do not bind to the monomer under the same conditions,75 although Met61 was coordinated to the haem iron in the crystal structure of the 3D-DS dimer.74 The hinge region is located close to Met61 in AA cyt c555. Thus, the haem coordination structure may be perturbed by 3D-DS in AA cyt c555, allowing external ligands to bind to the haem iron in the dimer.
The property of c-type cyt may change by 3D-DS, depending on the distance between the active site and hinge loop. In horse cyt c and AA cyt c555, the C-terminal region is exchanged between molecules by 3D-DS, and the active site structure and properties are changed.47,75 On the other hand, PA cyt c551 and HT cyt c552 mainly exchange the N-terminal region by 3D-DS, and the active site structure and properties are not changed significantly,73,74 although these proteins exhibit tertiary structures similar to those of horse cyt c and AA cyt c555, all belonging to the same protein family. The hinge loop is located relatively close to the active site haem in horse cyt c and AA cyt c555, and external ligands may bind to the haem iron of the 3D-DS dimer but may not bind (or may bind weakly) to the monomer.47,60,74 On the other hand, the hinge loop is located relatively far from the haem in PA cyt c551 and HT cyt c552, and the redox potential is not changed significantly by 3D-DS.73,74 Additionally, cyt cb562, Shewanella violacea (SV) cyt c5, and Aquifex aeolicus cyt c′ exhibit 3D-DS, in which the hinge loop is located relatively far from the haem.80–82 In these proteins, the properties of the monomer and dimer are similar. These results indicate that the active site structure and properties of a haem protein may be changed by 3D-DS when the hinge loop is located close to the haem active site, but not when the hinge loop is located far from it.
Other haem proteins may also undergo 3D-DS. Myoglobin (Mb) was reported to form dimers but not polymers more than half a century ago.83 The nature of the Mb dimer remained unknown until our study,84 which revealed that the Mb dimer exhibits a 3D-DS structure in which the loop located between the E- and F-helices that sandwiches the haem converts to a helical structure connecting the E- and F-helices, forming one long α-helix (PDB ID: 3VM9; Fig. 6A). Although the active site structures of the monomer and 3D-DS dimer of cyt c were different,47 the active site structure of the Mb dimer was similar to that of the monomer.84 The Mb 3D-DS dimer dissociated to monomers upon heating at 70 °C for 5 min, indicating that the dimer was less stable than the monomer,84 similar to cyt c and other c-type cyts.47,73,74 The EF-loop in the monomer converted to a part of a long α-helix, structurally showing the difficulty of Mb polymerization by 3D-DS, which has been observed in horse cyt c with a relatively long hinge loop.47 Since the loop at the hinge region in the Mb monomer converts to a helix in the 3D-DS dimer, we mutated Gly80 in the hinge region to Ala (a high helix propensity residue)85 to stabilize the helical structure in the hinge region, thereby stabilizing the 3D-DS dimer.86 Additional Ala mutations at positions 81 and 82 (G80A/G81A/G82A Mb) in the hinge loop stabilized the 3D-DS dimer more (PDB ID: 6LTM; Fig. 6B).86 As the helical propensity of the residue at position 80 in the hinge region was increased in a series of Mb mutants, the 3D-DS tendency correspondingly increased.86 These results demonstrate that a few mutations in the hinge loop can control 3D-DS and dimer formation in Mb, providing new ideas to create protein oligomers using 3D-DS.
 | ||
| Fig. 6 Schematic view of the design and three-dimensional structures of Mb 3D-DS dimers. (A) WT homodimer (PDB ID: 3VM9).84 (B) Stabilized dimer with Ala mutation in the hinge region (PDB ID: 6LTM).86 (C) Dimer with metal binding sites in the hinge region (PDB ID: 7DGK).98 Adapted with permission from ref. 98. Copyright 2021 Elsevier. (D) Heterodimer with different active sites (PDB ID: 3WYO).99 Adapted with permission from ref. 99. Copyright 2015 Wiley-VCH Verlag Gmbh & Co. | ||
In addition to haem proteins, we found that a β-sheet copper protein azurin from Alcaligenes xylosoxidans may undergo 3D-DS by treating Cu(I)–azurin with 2,2,2-trifluoroethanol at pH 5.0, subsequently freeze-drying the solution, and redissolving the protein residue at pH 7.0.87 The N-terminal region containing three β-strands was exchanged between protomers in the 3D-DS dimer (PDB ID: 6L1V; Fig. 4). The copper coordination structure was tetrahedrally distorted in the dimer, similar to that in the monomer; however, the Cu–O(Gly45) bond length was longer in the dimer (monomer, 2.46–2.59 Å; dimer, 2.98–3.25 Å). The ratio of the absorbance of the dimer at 460 nm to that at ∼620 nm (Abs460/Abs618 = 0.113) was higher than that of the monomer (Abs460/Abs622 = 0.067) and the EPR A‖ value of the dimer (5.85 mT) was slightly smaller than that of the monomer (5.95 mT), indicating slightly more rhombic copper coordination for the dimer in solution. The redox potential of the azurin 3D-DS dimer was 342 ± 5 mV vs. NHE, which was 50 mV higher than that of the monomer. These results show that 3D-DS slightly perturbed the active site structure and properties of azurin.
The aforementioned examples demonstrate that the native sequences of various types of metalloproteins can undergo 3D-DS upon treatment with alcohol. The swapping domain could be the N-terminal domain, the C-terminal domain, or both domains, depending on the protein and the flexibility of the loop connecting the swapping region. In the next section, we show that 3D-DS of metalloproteins occurs during folding in vitro and in vivo.
3.2. Mechanism of 3D-DS
3D-DS oligomers were obtained when cyt c was refolded from the denatured state.88 The structure of the 3D-DS dimer obtained by refolding was similar to that of the dimer obtained by the ethanol treatment. The number of the 3D-DS oligomers increased when the concentration of cyt c during refolding was increased, indicating that intermolecular interactions between cyt c molecules during folding lead to 3D-DS (Fig. 7A).88 At the early stage of cyt c folding, the N- and C-terminal regions form α-helices, and the two α-helices interact hydrophobically to fold into a monomer.89,90 Almost no oligomer was obtained by refolding a cyt c mutant in which a hydrophobic residue in the N- or C-terminal α-helix was replaced with Gly.88 3D-DS dimers were also obtained by refolding cyt c from the molten globule state.91 More oligomers were obtained by refolding from the molten globule state in the presence of chaotropic anions than in the presence of kosmotropic anions. Approximately 11% of cyt c was obtained as oligomers when refolding from the molten globule state containing 125 mM ClO4−, whereas approximately 25% of cyt c was dimerized in the molten globule state containing the same amount of ClO4−, according to SAXS measurements.91 These results indicate that cyt c molecules already interacted intermolecularly in the molten globule state and that a certain amount of oligomers in the molten globule state converted to 3D-DS oligomers upon folding; thus the intermolecular interaction necessary for 3D-DS may already exist in the molten globule state. It has also been shown that 3D-DS of RNase A occurs during refolding from the urea- or guanidinium ion-denatured state.92 These results indicate that 3D-DS oligomers can form during protein folding, especially at high protein concentrations and in the presence of chaotropic anions. | ||
| Fig. 7 Schematic view of cyt c 3D-DS dimerization by protein folding in vitro and in vivo. (A) Formation of 3D-DS dimer by intermolecular interactions during the folding process. Adapted with permission from ref. 88. Copyright 2013 American Chemical Society. (B) Formation of 3D-DS dimer by interaction with intracellular membrane. Adapted with permission from ref. 82. Copyright 2019 Elsevier. | ||
A slow phase (time constant of ∼3 s) for cyt c folding has been reported at high protein concentrations,93 whereas a transient cyt c dimer has been detected in its refolding kinetics by SAXS measurements.94 In addition to the formation of 3D-DS oligomers during the folding of cyt c at relatively high protein concentrations, a slow 4–5 s phase in the haem coordination structural change was observed in addition to a 400–500 ms fast phase during folding in the presence of 1.17 M guanidine hydrochloride.88 The ratio of the amplitude of the slow phase to that of the fast phase increased when the protein concentration was increased, indicating that the slow phase was an intermolecular process. We attributed the slow phase to the intermolecular ligand exchange process specific to 3D-DS during cyt c folding,88 whereas the fast phase has been attributed to the intramolecular ligand exchange process.95,96 These results show the importance of considering the formation of 3D-DS oligomers when folding a protein at high protein concentrations.
3D-DS oligomers of a c-type cyt may form during folding in cells depending on the surface charge of the c-type cyt.82 Horse cyt c and HT cyt c552 are positively charged, whereas PA cyt c551 and SV cyt c5 are negatively charged. 3D-DS oligomers of horse cyt c and HT cyt c552 were found to form in E. coli cells that expressed the corresponding proteins,82,97 and the HT cyt c552 dimer obtained directly from the E. coli cell exhibited a 3D-DS structure similar to that obtained by the ethanol treatment. In contrast, no oligomer was detected from the E. coli cells that expressed PA cyt c551 or SV cyt c5, although these proteins can undergo 3D-DS.73,74,82 Additionally, the amount of oligomers of positively charged horse cyt c and HT cyt c552 obtained from its folding in vitro increased upon the addition of negatively charged liposomes, whereas the amount of oligomers of negatively charged PA cyt c551 or SV cyt c5 obtained from the folding did not change significantly upon liposome addition.82 These results indicate that the protein surface charge affects the oligomerization of c-type cyts in cells; thus, we propose the following mechanism for the 3D-DS of a c-type cyt in cells (Fig. 7B).82 A c-type cyt polypeptide (with a signal peptide) is transported from the cytoplasm to the periplasm by secretory proteins (Fig. 7B-a). Subsequently, the signal peptide is cleaved from the polypeptide, and another c-type cyt peptide is transported to the periplasm (Fig. 7B-b). When the unfolded apo c-type cyt molecule is positively charged, it may assemble on the surface of a negatively charged membrane before haem incorporation in the periplasm of bacteria, allowing unfolded c-type cyt molecules to concentrate at the membrane surface (Fig. 7B-b), whereas the unfolded apo c-type cyt molecule will not concentrate at the membrane when it is negatively charged. When a haem is inserted into an apo protein by cyt c maturation (Ccm) proteins, the produced holo protein may interact intermolecularly with another apo protein and form a transient holo-apo complex (Fig. 7B-c). According to mass measurements,97 the apo protein in the holo-apo complex obtained from the E. coli cell did not contain the signal peptide, supporting the hypothesis that the holo-apo complex forms at the periplasm. At the final stage, another haem is inserted into the apo protein of the holo-apo complex, resulting in the formation of a 3D-DS dimer (Fig. 7B-d). Instead, the holo–holo complex may interact with another apo protein before folding into a 3D-DS dimer, resulting in the formation of a higher-order oligomer when the holo–holo complex interacts with the negatively charged membrane.
The amount of HT cyt c552 oligomers formed in E. coli cells decreased by decreasing the protein stability with amino acid mutation, indicating that 3D-DS of HT cyt c552 decreases in cells when the protein stability decreases. Additionally, the HT cyt c552 oligomers of the destabilized A5F/M11V/Y32F/Y41E/I76V mutant formed in E. coli cells did not contain an apo protein, while the oligomers of WT HT cyt c552 formed in E. coli cells contained apo proteins.97 These results support the hypothesis that 3D-DS oligomers of HT cyt c552 are formed by stabilization of the transient oligomer containing the apo protein before haem attachment in the cells. If the transient oligomer is not highly stable, it may dissociate into monomers before becoming a 3D-DS oligomer, and exceedingly stable proteins may be disadvantaged in forming 3D-DS oligomers in cells.97
3.3. Construction of unique metalloprotein dimers
We constructed various Mb and c-type cyt dimers utilizing 3D-DS. For example, stable Mb dimers with metal binding sites were obtained by shifting the His position and introducing two Ala residues to the hinge region (K78H/G80A/H82A and K79H/G80A/H81A Mbs) (Fig. 6C).98 The Ala residues were inserted to stabilize the dimer structures, and no negative peak related to the dimer-to-monomer dissociation was observed in the differential scanning calorimetry thermograms of K78H/G80A/H82A and K79H/G80A/H81A Mbs below the denaturation temperature, indicating that the two mutants were stable in the dimer state. Metal ions bound to the sites containing the introduced His in the dimers. Co2+-Bound and Ni2+-bound K78H/G80A/H82A Mb exhibited octahedral metal-coordination structures, where His78, His81, Glu85, and three H2O/OH− molecules coordinated to the metal ion. On the other hand, Co2+-bound and Zn2+-bound K79H/G80A/H81A Mb exhibited tetrahedral metal-coordination structures, where His79, His82, Asp141, and a H2O/OH− molecule coordinated to the metal ion. The Co2+-bound site exists deep inside the protein in the K79H/G80A/H81A Mb dimer, which may allow unique tetrahedral coordination of the Co2+ ion, whereas the Co2+-bound site is on the surface of the K78H/G80A/H82A Mb dimer. These results show that we can utilize 3D-DS to construct artificial metalloproteins.We also constructed a Mb heterodimer with different active site coordination. In the WT Mb 3D-DS dimer,84 four salt bridges are identified at the interface of the protomers. These bridges resulted from electrostatic interactions between positively and negatively charged residues, which held the Mb protomers together. Two Mb surface mutants were constructed by modifying the charges of the residues at the salt bridge of the 3D-DS dimer; two positively charged residues were replaced with two negatively charged residues in one mutant, whereas two negatively charged residues were replaced with two positively charged residues in the other mutant (Fig. 6D). Additionally, the haem site of one of the two Mb mutants was modified so that it underwent 3D-DS with the other active-site unmodified mutant through the introduced salt bridges, resulting in the formation of a stable heterodimer with bis-His and His/H2O active sites (PDB ID: 3WYO) (Fig. 6D).99 The two sites in the 3D-DS heterodimer exhibited different reactivities upon reduction with the mild agent ascorbic acid; the His/H2O coordinate haem was reduced, while the bis-His site remained oxidized. Reduction of the Mb heterodimer with dithionite or ascorbic acid and subsequent exposure to air resulted in the formation of a heterodimer that contained both an oxy haem and a ferric bis-His-coordinated haem.
We also constructed a heterodimer of c-type cyt utilizing 3D-DS.100 As noted above, PA cyt c551 and HT cyt c552 both belong to the c-type cyt protein family and exhibit similar monomer structures.71,72 We designed two chimeric proteins (PA cyt c551–HT cyt c552 comprising 21 N-terminal residues of PA cyt c551 and 61 C-terminal residues of HT cyt c552; HT cyt c552–PA cyt c551 comprising 19 N-terminal residues of HT cyt c552 and 61 C-terminal residues of PA cyt c551) in which the N-terminal regions were genetically exchanged between proteins.100 By treating these chimeric proteins together with a similar method using ethanol for 3D-DS of horse cyt c, a heterodimer with the N-terminal domains exchanged between the chimeric proteins was obtained owing to the favourable interaction between the natural domains.100 By further mutating the haem coordination structure of one of the two chimeric proteins, a 3D-DS heterodimer with different haem active sites (His/Met and His/H2O) was formed (Fig. 8),100 similar to the case of the Mb 3D-DS heterodimer. These results demonstrate that 3D-DS can be used to construct supramolecular proteins, and dimers with different or additional metal coordination and function can be designed.
 | ||
| Fig. 8 Schematic view of the design of a c-type cyt heterodimer (left) and three-dimensional structures of PA cyt c551 (PDB ID: 351C; green, middle top), HT cyt c552 (PDB ID: 1YNR; brown, right top), and the c-type cyt heterodimer (PDB ID: 5XED; green and brown, right bottom). Adapted with permission from ref. 100. Copyright 2017 Wiley-VCH Verlag Gmbh & Co. | ||
3.4. Construction of unique protein assemblies
Supramolecular haem proteins were constructed utilizing 3D-DS. For example, cyt cb562 formed a 3D-DS dimer in which the two helices in the N-terminal region of one protomer interacted with the other two helices in the C-terminal region of the other protomer in the dimer (PDB ID: 5AWI; Fig. 9).80 The haem coordination structure in the dimer was similar to that in the monomer. In addition, three cyt cb562 3D-DS dimers formed a unique nanocage with a Zn-SO4 cluster inside the cavity in the crystal, where the Zn–SO4 cluster consisted of fifteen Zn2+ and seven SO42− ions. The cage structure was stabilized by the coordination of the amino acid side chains of the dimers to the Zn2+ ions and connection of the two four-helix bundle units through a conformation-adjustable hinge loop. | ||
| Fig. 9 Cage structure constructed of three cyt cb562 domain-swapped dimers (PDB ID: 5AWI): (A and B) The overall structure of the cage, (C and D) enlarged views of the Zn2+ and SO42− ions in the internal cavity. B and D are 90°-rotated views of A and C, respectively. The three dimers forming the cage are shown in combinations of green and light-green, blue-green and cyan, and red and pink, respectively. The coordination bonds between Zn2+ and SO42− ions are shown as black dashed lines. The Zn2+ ions are shown as spheres, and the SO42− ions are shown as stick models. Adapted with permission from ref. 80. Copyright 2015 Royal Society of Chemistry. | ||
A building block protein (BBP) was constructed based on hyperthermostable AA cyt c555 to force intermolecular interactions; circular permutation that cleaved the protein at the 3D-DS hinge loop74 and α-helical linker insertion at the connection of the original N- and C-terminal α-helices were performed.101 BBP was expressed as a monomer in E. coli, whereas it formed oligomers as large as ∼40 mers with a relatively large amount of trimers when refolded at high protein concentrations. BBP molecules formed a 3D-DS trimer, with the N-terminal region of a BBP molecule interacting intermolecularly with the C-terminal region of another BBP molecule, resulting in a triangle-shaped structure with an edge length of 68 Å (PDB ID: 5Z25) (Fig. 10).101 Additionally, four trimers assembled into a unique tetrahedron in the crystal (Fig. 10). These results demonstrate that the circular permutation connecting the original N- and C-terminal α-helices with an α-helical linker considerably enhances 3D-DS, and this method is useful for the construction of protein assemblies.
 | ||
| Fig. 10 Schematic view of the BBP design and its trimer and tetrahedron formations (PDB ID: 5Z25). N- and C-terminal regions of AA cyt c555 are depicted in magenta and green, respectively. The α-helical linker is depicted in orange. | ||
Protein oligomer formation has also been controlled by utilizing 3D-DS for a dimer–monomer transition protein, Allochromatium vinosum (AV) cyt c′.81 AV cyt c′ is a homodimeric protein in its native form, in which its protomer exhibits a four-helix bundle structure containing a covalently bound five-coordinate haem as a gas binding site.102 AV cyt c′ exhibits a unique reversible dimer–monomer transition according to the absence and presence of CO. In the oxidized form, the sixth coordination site of AV cyt c′ is occupied by the side chain of Tyr16, which has been suggested to be a trigger for the dimer-to-monomer transition upon CO binding to the haem.103 AV cyt c′ formed a tetramer comprising one 3D-DS dimer subunit and two monomer subunits (PDB ID: 5GYR).81 Similar to the cyt cb562 3D-DS dimer, two helices in the N-terminal region of one protomer interacted with the other two helices in the C-terminal region of the other protomer in the AV cyt c′ dimer (Fig. 11).81 The haem environments of the 3D-DS dimer were similar to that of the native dimer. Interestingly, high-order oligomers formed in the solution mainly containing 3D-DS dimers of AV cyt c′ and then dissociated to 3D-DS dimers upon the addition of CO (Fig. 11). These results demonstrate that 3D-DS can be used to construct and control supramolecular metalloprotein formation.
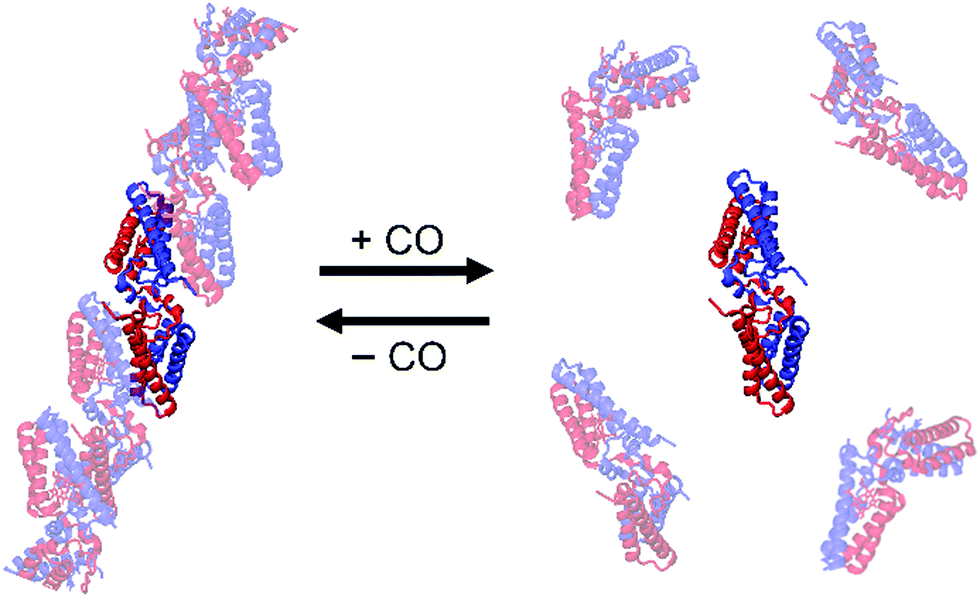 | ||
| Fig. 11 Schematic view of the oligomer association and dissociation of AV cyt c′ 3D-DS dimers controlled by binding of CO to the haem of AV cyt c′. The protomers in the dimer are depicted in red and blue. Adapted with permission from ref. 81. Copyright 2017 Wiley-Blackwell. | ||
4. Conclusions and future prospects
Various methods have been applied to construct protein oligomers; some utilize a single bond, whereas some utilize a protein surface. Owing to the development of computational science, successful examples of de novo protein oligomer design are accumulating. Since the first 3D-DS structure on diphtheria toxin was reported in 1994, the number of reports on 3D-DS has increased rapidly. Almost any protein has the potential to form a 3D-DS structure, but few proteins exhibit 3D-DS in its native structure. As mentioned above, the moderate protein stability may prevent proteins from forming oligomers.Several attempts have been made to control the orientation of 3D-DS. For example, 3D-DS formation by loop deletion and insertion of a polyproline rod to the outer-surface protein A (OspA) from Borrelia, which is composed of sequential 21 antiparallel β-strands, has been reported.104 The rigid nature of the polyproline rod enabled precise control of the interdomain distance and orientation. It was shown that 3D-DS propensity may depend on the amino acid composition of the hinge loop by investigating the 3D-DS of loop-deletion mutants of a non-3D-DS β-sheet protein monellin.105 Additionally, a hydrophobic five-residue cystatin motif has been reported to drive 3D-DS in β-hairpin proteins.106 Although further studies are necessary, precise control of 3D-DS will allow construction of functional supramolecular metalloproteins.
Protein structures are complex, but this very complexity enables us to construct unique structures. In 3D-DS, some intramolecular interactions are replaced with intermolecular interactions, but most of the intramolecular interactions are retained. Thus, 3D-DS is suitable for constructing unique supramolecular metalloproteins since the constructed oligomers may maintain the function of the metalloprotein monomer. We have shown that 3D-DS may be used to construct unique metalloprotein dimers and assemblies; for example, 3D-DS heterodimers with different active sites by designing the subunit interface or using chimeric proteins, a triangle-shaped structure and a tetrahedron by designing a building block protein, and oligomers of which formation was regulated by carbon monoxide addition by utilizing the 3D-DS dimer of a dimer–monomer transition protein. If we could control the 3D-DS orientations of metalloproteins that build up these supramolecular proteins, it may lead to the development of functional supramolecular metalloproteins. However, the difficulty in controlling the orientation between proteins in 3D-DS is an obstacle to its use for supramolecular protein design, especially for new enzymes, functional biomaterials, and drug delivery systems. One way to solve this problem may be the use of computer design. We believe that 3D-DS would be a powerful method to design various unique protein oligomers in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grants-in-Aid from JSPS for Scientific Research (B) (No. JP21H02060 (S. H.)) and by JST CREST, Japan (No. JP20338388 (S. H.)).Notes and references
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- Y. Bai, Q. Luo and J. Liu, Chem. Soc. Rev., 2016, 45, 2756–2767 RSC.
- G. Yang, L. Wu, G. Chen and M. Jiang, Chem. Commun., 2016, 52, 10595–10605 RSC.
- T. O. Yeates, Annu. Rev. Biophys., 2017, 46, 23–42 CrossRef CAS PubMed.
- S. L. Kuan, F. R. G. Bergamini and T. Weil, Chem. Soc. Rev., 2018, 47, 9069–9105 RSC.
- C. J. Wilson, A. S. Bommarius, J. A. Champion, Y. O. Chernoff, D. G. Lynn, A. K. Paravastu, C. Liang, M. C. Hsieh and J. M. Heemstra, Chem. Rev., 2018, 118, 11519–11574 CrossRef CAS PubMed.
- I. W. Hamley, Biomacromolecules, 2019, 20, 1829–1848 CrossRef CAS PubMed.
- E. R. Ballister, A. H. Lai, R. N. Zuckermann, Y. Cheng and J. D. Mougous, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3733–3738 CrossRef CAS PubMed.
- Y. Suzuki, G. Cardone, D. Restrepo, P. D. Zavattieri, T. S. Baker and F. A. Tezcan, Nature, 2016, 533, 369–373 CrossRef CAS PubMed.
- J. B. Bailey, R. H. Subramanian, L. A. Churchfield and F. A. Tezcan, Methods Enzymol., 2016, 580, 223–250 CAS.
- J. D. Brodin, X. I. Ambroggio, C. Tang, K. N. Parent, T. S. Baker and F. A. Tezcan, Nat. Chem., 2012, 4, 375–382 CrossRef CAS PubMed.
- Y. Bai, Q. Luo, W. Zhang, L. Miao, J. Xu, H. Li and J. Liu, J. Am. Chem. Soc., 2013, 135, 10966–10969 CrossRef CAS PubMed.
- S. Biswas, K. Kinbara, T. Niwa, H. Taguchi, N. Ishii, S. Watanabe, K. Miyata, K. Kataoka and T. Aida, Nat. Chem., 2013, 5, 613–620 CrossRef CAS PubMed.
- N. Kobayashi, K. Yanase, T. Sato, S. Unzai, M. H. Hecht and R. Arai, J. Am. Chem. Soc., 2015, 137, 11285–11293 CrossRef CAS PubMed.
- A. Sciore, M. Su, P. Koldewey, J. D. Eschweiler, K. A. Diffley, B. M. Linhares, B. T. Ruotolo, J. C. Bardwell, G. Skiniotis and E. N. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8681–8686 CrossRef CAS PubMed.
- T. Miyamoto, Y. Hayashi, K. Yoshida, H. Watanabe, T. Uchihashi, K. Yonezawa, N. Shimizu, H. Kamikubo and S. Hirota, ACS Synth. Biol., 2019, 8, 1112–1120 CrossRef CAS PubMed.
- Y. T. Lai, D. Cascio and T. O. Yeates, Science, 2012, 336, 1129 CrossRef CAS PubMed.
- P. Lu, D. Min, F. DiMaio, K. Y. Wei, M. D. Vahey, S. E. Boyken, Z. Chen, J. A. Fallas, G. Ueda, W. Sheffler, V. K. Mulligan, W. Xu, J. U. Bowie and D. Baker, Science, 2018, 359, 1042–1046 CrossRef CAS PubMed.
- H. Pyles, S. Zhang, J. J. De Yoreo and D. Baker, Nature, 2019, 571, 251–256 CrossRef CAS PubMed.
- M. J. Bennett, S. Choe and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3127–3131 CrossRef CAS PubMed.
- M. J. Bennett, M. R. Sawaya and D. Eisenberg, Structure, 2006, 14, 811–824 CrossRef CAS PubMed.
- M. Yamasaki, T. J. Sendall, M. C. Pearce, J. C. Whisstock and J. A. Huntington, EMBO Rep., 2011, 12, 1011–1017 CrossRef CAS PubMed.
- C. Liu, M. R. Sawaya and D. Eisenberg, Nat. Struct. Mol. Biol., 2011, 18, 49–55 CrossRef CAS PubMed.
- K. J. Knaus, M. Morillas, W. Swietnicki, M. Malone, W. K. Surewicz and V. C. Yee, Nat. Struct. Biol., 2001, 8, 770–774 CrossRef CAS PubMed.
- R. Janowski, M. Kozak, E. Jankowska, Z. Grzonka, A. Grubb, M. Abrahamson and M. Jaskolski, Nat. Struct. Biol., 2001, 8, 316–320 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Y. Liu and D. Eisenberg, Protein Sci., 2002, 11, 1285–1299 CrossRef CAS PubMed.
- J. M. Reis, D. C. Burns and G. A. Woolley, Biochemistry, 2014, 53, 5008–5016 CrossRef CAS PubMed.
- J. H. Ha, J. M. Karchin, N. Walker-Kopp, C. A. Castaneda and S. N. Loh, Chem. Biol., 2015, 22, 1384–1393 CrossRef CAS PubMed.
- J. M. Karchin, J. H. Ha, K. E. Namitz, M. S. Cosgrove and S. N. Loh, Sci. Rep., 2017, 7, 44388 CrossRef PubMed.
- A. Ghanbarpour, C. Pinger, R. E. Salmani, Z. Assar, E. M. Santos, M. Nosrati, K. Pawlowski, D. Spence, C. Vasileiou, X. S. Jin, B. Borhan and J. H. Geiger, J. Am. Chem. Soc., 2019, 141, 17125–17132 CrossRef CAS PubMed.
- K. Oohora and T. Hayashi, Curr. Opin. Chem. Biol., 2014, 19, 154–161 CrossRef CAS PubMed.
- S. Hirota and Y. W. Lin, J. Biol. Inorg. Chem., 2018, 23, 7–25 CrossRef CAS PubMed.
- K. A. Cannon, J. M. Ochoa and T. O. Yeates, Curr. Opin. Struct. Biol., 2019, 55, 77–84 CrossRef CAS PubMed.
- J. K. Leman, B. D. Weitzner, S. M. Lewis, J. Adolf-Bryfogle, N. Alam, R. F. Alford, M. Aprahamian, D. Baker, K. A. Barlow, P. Barth, B. Basanta, B. J. Bender, K. Blacklock, J. Bonet, S. E. Boyken, P. Bradley, C. Bystroff, P. Conway, S. Cooper, B. E. Correia, B. Coventry, R. Das, R. M. De Jong, F. DiMaio, L. Dsilva, R. Dunbrack, A. S. Ford, B. Frenz, D. Y. Fu, C. Geniesse, L. Goldschmidt, R. Gowthaman, J. J. Gray, D. Gront, S. Guffy, S. Horowitz, P. S. Huang, T. Huber, T. M. Jacobs, J. R. Jeliazkov, D. K. Johnson, K. Kappel, J. Karanicolas, H. Khakzad, K. R. Khar, S. D. Khare, F. Khatib, A. Khramushin, I. C. King, R. Kleffner, B. Koepnick, T. Kortemme, G. Kuenze, B. Kuhlman, D. Kuroda, J. W. Labonte, J. K. Lai, G. Lapidoth, A. Leaver-Fay, S. Lindert, T. Linsky, N. London, J. H. Lubin, S. Lyskov, J. Maguire, L. Malmstrom, E. Marcos, O. Marcu, N. A. Marze, J. Meiler, R. Moretti, V. K. Mulligan, S. Nerli, C. Norn, S. O'Conchuir, N. Ollikainen, S. Ovchinnikov, M. S. Pacella, X. Pan, H. Park, R. E. Pavlovicz, M. Pethe, B. G. Pierce, K. B. Pilla, B. Raveh, P. D. Renfrew, S. S. R. Burman, A. Rubenstein, M. F. Sauer, A. Scheck, W. Schief, O. Schueler-Furman, Y. Sedan, A. M. Sevy, N. G. Sgourakis, L. Shi, J. B. Siegel, D. A. Silva, S. Smith, Y. Song, A. Stein, M. Szegedy, F. D. Teets, S. B. Thyme, R. Y. Wang, A. Watkins, L. Zimmerman and R. Bonneau, Nat. Methods, 2020, 17, 665–680 CrossRef CAS PubMed.
- C. Lv, X. Zhang, Y. Liu, T. Zhang, H. Chen, J. Zang, B. Zheng and G. Zhao, Chem. Soc. Rev., 2021, 50, 3957–3989 RSC.
- S. Hirota, J. Inorg. Biochem., 2019, 194, 170–179 CrossRef CAS PubMed.
- S. Hirota and S. Nagao, Bull. Chem. Soc. Jpn., 2021, 94, 170–182 CrossRef CAS.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed.
- K. Oohora, N. Fujimaki, R. Kajihara, H. Watanabe, T. Uchihashi and T. Hayashi, J. Am. Chem. Soc., 2018, 140, 10145–10148 CrossRef CAS PubMed.
- P. D. Barker, E. P. Nerou, S. M. Freund and I. M. Fearnley, Biochemistry, 1995, 34, 15191–15203 CrossRef CAS PubMed.
- E. N. Salgado, R. A. Lewis, S. Mossin, A. L. Rheingold and F. A. Tezcan, Inorg. Chem., 2009, 48, 2726–2728 CrossRef CAS PubMed.
- N. Kawakami, H. Kondo, Y. Matsuzawa, K. Hayasaka, E. Nasu, K. Sasahara, R. Arai and K. Miyamoto, Angew. Chem., Int. Ed., 2018, 57, 12400–12404 CrossRef CAS PubMed.
- J. Obata, N. Kawakami, A. Tsutsumi, E. Nasu, K. Miyamoto, M. Kikkawa and R. Arai, Chem. Commun., 2021, 57, 10226–10229 RSC.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. Andre, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- S. Hirota, Y. Hattori, S. Nagao, M. Taketa, H. Komori, H. Kamikubo, Z. Wang, I. Takahashi, S. Negi, Y. Sugiura, M. Kataoka and Y. Higuchi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12854–12859 CrossRef CAS PubMed.
- E. Margoliash and J. Lustgarten, J. Biol. Chem., 1962, 237, 3397–3405 CrossRef CAS.
- S. Sambashivan, Y. Liu, M. R. Sawaya, M. Gingery and D. Eisenberg, Nature, 2005, 437, 266–269 CrossRef CAS PubMed.
- M. Yamasaki, W. Li, D. J. Johnson and J. A. Huntington, Nature, 2008, 455, 1255–1258 CrossRef CAS PubMed.
- M. Wahlbom, X. Wang, V. Lindstrom, E. Carlemalm, M. Jaskolski and A. Grubb, J. Biol. Chem., 2007, 282, 18318–18326 CrossRef CAS PubMed.
- Z. Guo and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8042–8047 CrossRef CAS PubMed.
- P. Das, J. A. King and R. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10514–10519 CrossRef CAS PubMed.
- N. L. Ogihara, G. Ghirlanda, J. W. Bryson, M. Gingery, W. F. DeGrado and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1404–1409 CrossRef CAS PubMed.
- S. R. Solmaz and C. Hunte, J. Biol. Chem., 2008, 283, 17542–17549 CrossRef CAS PubMed.
- K. Sakamoto, M. Kamiya, M. Imai, K. Shinzawa-Itoh, T. Uchida, K. Kawano, S. Yoshikawa and K. Ishimori, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12271–12276 CrossRef CAS PubMed.
- H. Kobayashi, S. Nagao and S. Hirota, Angew. Chem., Int. Ed., 2016, 55, 14019–14022 CrossRef CAS PubMed.
- R. E. Dickerson, T. Takano, D. Eisenberg, O. B. Kallai, L. Samson, A. Cooper and E. Margoliash, J. Biol. Chem., 1971, 246, 1511–1535 CrossRef CAS.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CrossRef CAS PubMed.
- A. D. Nugraheni, S. Nagao, S. Yanagisawa, T. Ogura and S. Hirota, J. Biol. Inorg. Chem., 2013, 18, 383–390 CrossRef CAS PubMed.
- Z. Wang, T. Matsuo, S. Nagao and S. Hirota, Org. Biomol. Chem., 2011, 9, 4766–4769 RSC.
- V. E. Kagan, V. A. Tyurin, J. Jiang, Y. Y. Tyurina, V. B. Ritov, A. A. Amoscato, A. N. Osipov, N. A. Belikova, A. A. Kapralov, V. Kini, I. I. Vlasova, Q. Zhao, M. Zou, P. Di, D. A. Svistunenko, I. V. Kurnikov and G. G. Borisenko, Nat. Chem. Biol., 2005, 1, 223–232 CrossRef CAS PubMed.
- N. A. Belikova, Y. A. Vladimirov, A. N. Osipov, A. A. Kapralov, V. A. Tyurin, M. V. Potapovich, L. V. Basova, J. Peterson, I. V. Kurnikov and V. E. Kagan, Biochemistry, 2006, 45, 4998–5009 CrossRef CAS PubMed.
- L. C. Godoy, C. Munoz-Pinedo, L. Castro, S. Cardaci, C. M. Schonhoff, M. King, V. Tortora, M. Marin, Q. Miao, J. F. Jiang, A. Kapralov, R. Jemmerson, G. G. Silkstone, J. N. Patel, J. E. Evans, M. T. Wilson, D. R. Green, V. E. Kagan, R. Radi and J. B. Mannick, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2653–2658 CrossRef CAS PubMed.
- S. Junedi, K. Yasuhara, S. Nagao, J. Kikuchi and S. Hirota, ChemBioChem, 2014, 15, 517–521 CrossRef CAS PubMed.
- K. I. Yuyama, M. Ueda, S. Nagao, S. Hirota, T. Sugiyama and H. Masuhara, Angew. Chem., Int. Ed., 2017, 56, 6739–6743 CrossRef CAS PubMed.
- P. George, S. C. Glauser and A. Schejter, J. Biol. Chem., 1967, 242, 1690–1695 CrossRef CAS.
- N. Yoshida, M. Higashi, H. Motoki and S. Hirota, J. Chem. Phys., 2018, 148, 025102 CrossRef PubMed.
- S. Hirota, N. Yamashiro, Z. Wang and S. Nagao, J. Biol. Inorg. Chem., 2017, 22, 705–712 CrossRef CAS PubMed.
- L. J. McClelland, H. B. Steele, F. G. Whitby, T. C. Mou, D. Holley, J. B. Ross, S. R. Sprang and B. E. Bowler, J. Am. Chem. Soc., 2016, 138, 16770–16778 CrossRef CAS PubMed.
- Y. Matsuura, T. Takano and R. E. Dickerson, J. Mol. Biol., 1982, 156, 389–409 CrossRef CAS PubMed.
- C. Travaglini-Allocatelli, S. Gianni, V. K. Dubey, A. Borgia, A. Di Matteo, D. Bonivento, F. Cutruzzolà, K. L. Bren and M. Brunori, J. Biol. Chem., 2005, 280, 25729–25734 CrossRef CAS PubMed.
- Y. Hayashi, S. Nagao, H. Osuka, H. Komori, Y. Higuchi and S. Hirota, Biochemistry, 2012, 51, 8608–8616 CrossRef CAS PubMed.
- M. Yamanaka, S. Nagao, H. Komori, Y. Higuchi and S. Hirota, Protein Sci., 2015, 24, 366–375 CrossRef CAS PubMed.
- C. Ren, S. Nagao, M. Yamanaka, H. Komori, Y. Shomura, Y. Higuchi and S. Hirota, Mol. BioSyst., 2015, 11, 3218–3221 RSC.
- Y. Liu, P. J. Hart, M. P. Schlunegger and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3437–3442 CrossRef CAS PubMed.
- Y. Liu, G. Gotte, M. Libonati and D. Eisenberg, Nat. Struct. Biol., 2001, 8, 211–214 CrossRef CAS PubMed.
- G. Gotte, A. Mahmoud Helmy, C. Ercole, R. Spadaccini, D. V. Laurents, M. Donadelli and D. Picone, PLoS One, 2012, 7, e46804 CrossRef CAS PubMed.
- M. Obuchi, K. Kawahara, D. Motooka, S. Nakamura, M. Yamanaka, T. Takeda, S. Uchiyama, Y. Kobayashi, T. Ohkubo and Y. Sambongi, Acta Crystallogr. D: Biol. Crystallogr., 2009, 65, 804–813 CrossRef CAS PubMed.
- T. Miyamoto, M. Kuribayashi, S. Nagao, Y. Shomura, Y. Higuchi and S. Hirota, Chem. Sci., 2015, 6, 7336–7342 RSC.
- M. Yamanaka, M. Hoshizumi, S. Nagao, R. Nakayama, N. Shibata, Y. Higuchi and S. Hirota, Protein Sci., 2017, 26, 464–474 CrossRef CAS PubMed.
- H. Yang, M. Yamanaka, S. Nagao, K. Yasuhara, N. Shibata, Y. Higuchi and S. Hirota, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 140265 CrossRef CAS PubMed.
- A. H. Van den Oord, J. J. Wesdorp, A. F. Van Dam and J. A. Verheij, Eur. J. Biochem., 1969, 10, 140–145 CrossRef CAS PubMed.
- S. Nagao, H. Osuka, T. Yamada, T. Uni, Y. Shomura, K. Imai, Y. Higuchi and S. Hirota, Dalton Trans., 2012, 41, 11378–11385 RSC.
- J. W. Bryson, S. F. Betz, H. S. Lu, D. J. Suich, H. X. Zhou, K. T. O'Neil and W. F. DeGrado, Science, 1995, 270, 935–941 CrossRef CAS PubMed.
- S. Nagao, A. Suda, H. Kobayashi, N. Shibata, Y. Higuchi and S. Hirota, Chem. – Asian J., 2020, 15, 1743–1749 CrossRef CAS PubMed.
- R. N. Cahyono, M. Yamanaka, S. Nagao, N. Shibata, Y. Higuchi and S. Hirota, Metallomics, 2020, 12, 337–345 CrossRef CAS PubMed.
- P. P. Parui, M. S. Deshpande, S. Nagao, H. Kamikubo, H. Komori, Y. Higuchi, M. Kataoka and S. Hirota, Biochemistry, 2013, 52, 8732–8744 CrossRef CAS PubMed.
- H. Roder, G. A. Elove and S. W. Englander, Nature, 1988, 335, 700–704 CrossRef CAS PubMed.
- W. Hu, Z. Y. Kan, L. Mayne and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3809–3814 CrossRef CAS PubMed.
- M. S. Deshpande, P. P. Parui, H. Kamikubo, M. Yamanaka, S. Nagao, H. Komori, M. Kataoka, Y. Higuchi and S. Hirota, Biochemistry, 2014, 53, 4696–4703 CrossRef CAS PubMed.
- J. P. López-Alonso, M. Bruix, J. Font, M. Ribó, M. Vilanova, M. A. Jiménez, J. Santoro, C. González and D. V. Laurents, J. Am. Chem. Soc., 2010, 132, 1621–1630 CrossRef PubMed.
- J. P. Nawrocki, R. A. Chu, L. K. Pannell and Y. Bai, J. Mol. Biol., 1999, 293, 991–995 CrossRef CAS PubMed.
- D. J. Segel, D. Eliezer, V. Uversky, A. L. Fink, K. O. Hodgson and S. Doniach, Biochemistry, 1999, 38, 15352–15359 CrossRef CAS PubMed.
- G. A. Elove, A. K. Bhuyan and H. Roder, Biochemistry, 1994, 33, 6925–6935 CrossRef CAS PubMed.
- S. R. Yeh, S. Takahashi, B. Fan and D. L. Rousseau, Nat. Struct. Biol., 1997, 4, 51–56 CrossRef CAS PubMed.
- Y. Hayashi, M. Yamanaka, S. Nagao, H. Komori, Y. Higuchi and S. Hirota, Sci. Rep., 2016, 6, 19334 CrossRef CAS PubMed.
- S. Nagao, A. Idomoto, N. Shibata, Y. Higuchi and S. Hirota, J. Inorg. Biochem., 2021, 217, 111374 CrossRef CAS PubMed.
- Y. W. Lin, S. Nagao, M. Zhang, Y. Shomura, Y. Higuchi and S. Hirota, Angew. Chem., Int. Ed., 2015, 54, 511–515 CAS.
- M. Zhang, T. Nakanishi, M. Yamanaka, S. Nagao, S. Yanagisawa, Y. Shomura, N. Shibata, T. Ogura, Y. Higuchi and S. Hirota, ChemBioChem, 2017, 18, 1712–1715 CrossRef CAS PubMed.
- A. Oda, S. Nagao, M. Yamanaka, I. Ueda, H. Watanabe, T. Uchihashi, N. Shibata, Y. Higuchi and S. Hirota, Chem. – Asian J., 2018, 13, 964–967 CrossRef CAS PubMed.
- M. A. Hough and C. R. Andrew, Adv. Microb. Physiol., 2015, 67, 1–84 CrossRef CAS PubMed.
- Z. Ren, T. Meyer and D. E. McRee, J. Mol. Biol., 1993, 234, 433–445 CrossRef CAS PubMed.
- S. Shiga, M. Yamanaka, W. Fujiwara, S. Hirota, S. Goda and K. Makabe, ChemBioChem, 2019, 20, 2454–2457 CrossRef CAS PubMed.
- N. Nandwani, P. Surana, J. B. Udgaonkar, R. Das and S. Gosavi, Protein Sci., 2017, 26, 1994–2002 CrossRef CAS PubMed.
- N. Nandwani, P. Surana, H. Negi, N. M. Mascarenhas, J. B. Udgaonkar, R. Das and S. Gosavi, Nat. Commun., 2019, 10, 452 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |