
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of a C4-symmetrical inherently chiral calix[4]arene†
Luke
Hodson
 ,
Kevin J.
Visagie
,
Michael-Phillip
Smith
,
Leigh
Loots
,
David
Kuter
,
Trégen M.
Snayer
and
Gareth E.
Arnott
*
,
Kevin J.
Visagie
,
Michael-Phillip
Smith
,
Leigh
Loots
,
David
Kuter
,
Trégen M.
Snayer
and
Gareth E.
Arnott
*
Department of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1, Matieland, 7602, South Africa. E-mail: arnott@sun.ac.za
First published on 29th September 2021
Abstract
Inherently chiral calix[4]arenes with C4-symmetry are extremely rare and difficult to synthesise, severely hampering any effort to expand on their potential as chiral supramolecular catalysts and building blocks. Herein we report a reaction of a tetracarbamate calix[4]arene with NBS which results in a high yield of an inherently chiral calix[4]arenes with C4-symmetry. Furthermore, employing a chiral N-Boc proline moiety allows for separation of the diastereomers formed, thus obtaining the pure enantiomers after hydrolysis. The enantiomers could be assigned based on the CD spectra and DFT calculated values.
Calix[4]arenes have for many years drawn the attention of researchers owing to their defined three dimensional shape and ease of synthesis. An intriguing aspect of calixarenes is their modification to generate so-called inherently chiral structures, whose chirality is not associated with the more common point chirality of tetrahedral carbon atoms.1,2 There are four main strategies that give rise to inherently chiral calix[4]arenes (see Fig. 1a), although combinations of these themes can also be employed.
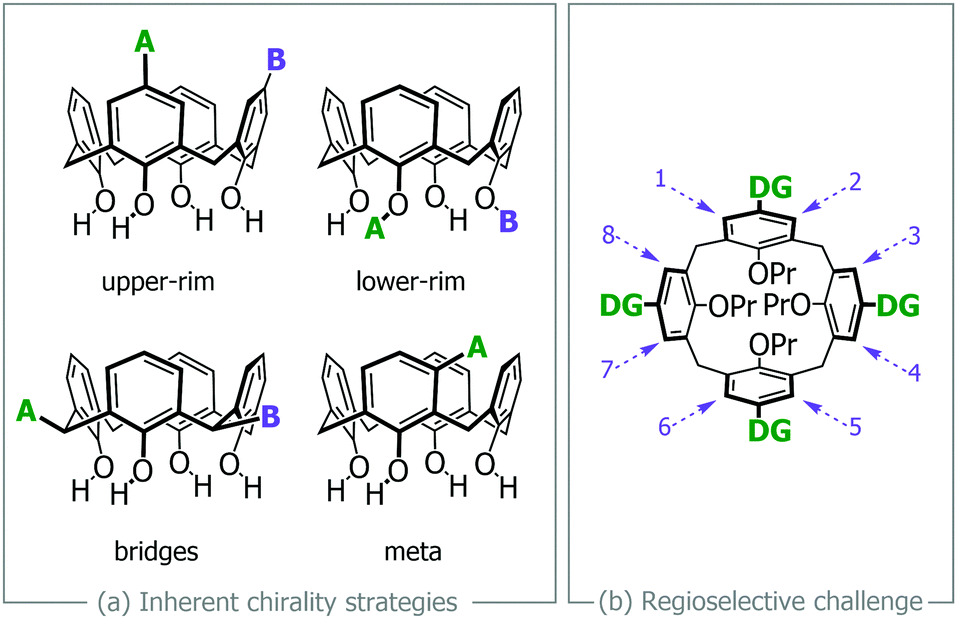 | ||
| Fig. 1 Strategies for creating inherently chiral calix[4]arene: (a) the four main ways of generating inherent chirality (A and B may be any chemically distinct group),2 (b) the regioselectivity challenge in functionalising multiple meta-positions (DG = any directing group). | ||
For us, the meta-functionalization‡ strategy has resonated as it has flavors of planar chiral ferrocenes, which have been used extensively in asymmetric catalytic processes. The synthetic pathways to meta-functionalized calixarenes has recently been reviewed,3 with the majority of these methods functionalizing only one or two of the aryl rings, resulting in chiral C1- or C2-symmetrical calix[4]arenes. The construction of chiral C4-symmetrical calixarenes, i.e. where all four aryl rings have been meta-functionalized, has only ever been achieved through the so-called fragment condensation route, first described in the early 1990s.4,5 The major problem with this method is that very low yields are obtained, typically less than 30% for the cyclisation step, notwithstanding the low yields for steps needed to synthesize the fragments themselves.
Functionalizing a calix[4]arene in the meta-position has been achieved in distinct ways such as intramolecular ring closing,6–9 the p-bromodienone route10 or directly using mercury(II) trifluoroacetate.11,12 More commonly though this is achieved through the use of directing groups (DG) placed in the para-position (i.e. upper rim) of the calix[4]arene. In the literature, the DGs used include ethers,13 amides,14,15 oxazolines16 and sulfoxides,17,18 but in the majority of these cases only one DG was attached to the calix[4]arene. The number of potential compounds and side-products grows with each additional DG, becoming a rather daunting task (see Fig. 1b). The calix[4]arene is nevertheless unlikely to be functionalized at all eight positions at once, since steric and electronic factors would make this difficult. However, if tetra-meta-functionalization could be synthetically achieved (i.e. avoiding any mono, di and tri-functionalized side-products), then only four possible isomers are possible, with two of these being inherently chiral (Fig. 2). Of interest, is that there do not appear to be any reported attempts that try to tetra-meta-functionalize with four directing groups. At most only two directing groups (situated distally) have been used, which gave mixtures of products in all cases.7,8,14 Our own research theme on inherently chiral calixarenes has had us looking for ways to generate these types of compounds. Our diastereoselective ortho-lithiation approaches are still the only successful direct method for stereoselectively forming meta-functionalized inherently chiral calix[4]arenes, but suffer from difficult synthetic protocols that do not scale up very well.16,17 We have therefore avoided any attempt to tetra-lithiate a calix[4]arene, presuming it to be a battle with a low chance of success.
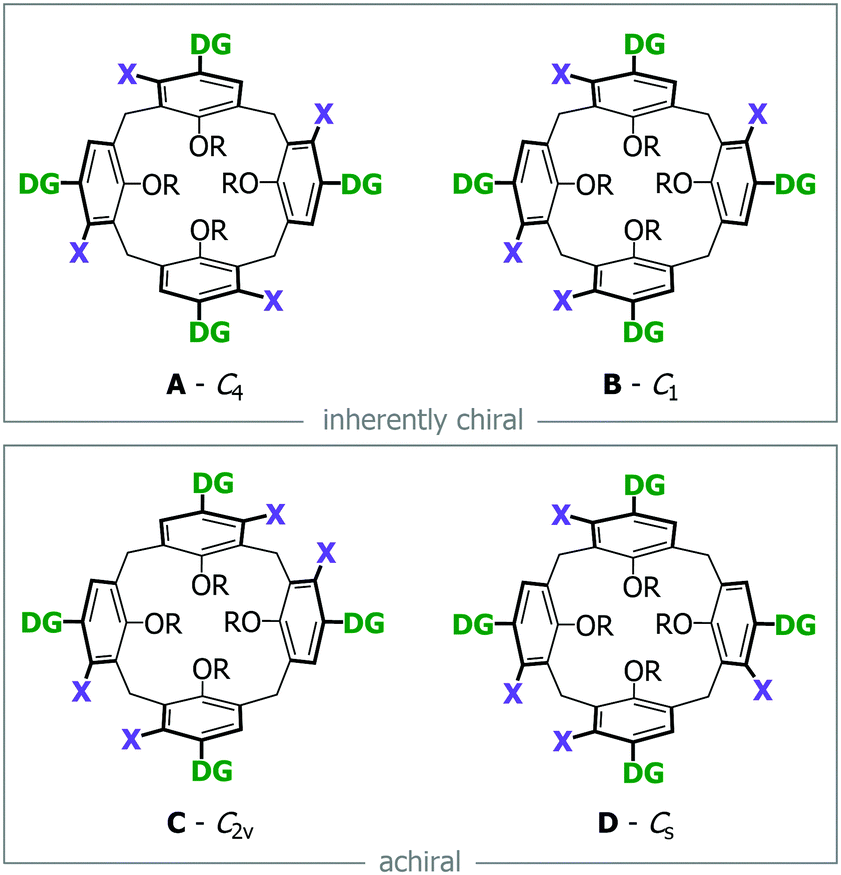 | ||
| Fig. 2 The four possible isomers via tetra-meta-functionalisation. DG = directing group; X = any electrophilic group, e.g., Br. | ||
We have recently investigated a spuriously reported palladium catalysed C–H activation route employing a carbamate group and N-bromosuccinimide (NBS), but could show that this paper was actually overstated and incorrect on a number of levels.19 Nevertheless, we wondered whether using a readily available tetra-carbamate as a directing group might allow for a facile synthesis of a C4-symmetrical inherently chiral calix[4]arene.
Our strategy involved using tetra-methylcarbamate calix[4]arene 2a as the starting material for bromination at the calix[4]arene meta-positions. Whilst 2a has been synthesized before using triphosgene,20 we synthesized it following a different route from the known tetra-amino calix[4]arene 120 and methyl chloroformate in an excellent yield (Scheme 1, R = Me). A preliminary selectivity experiment using tetra-methylcarbamate 2a, five equivalents of NBS and two equivalents of p-toluene sulfonic acid (PTSA), returned a very interesting result by TLC, showing only two compounds in a roughly 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 2). Isolation of these compounds was achieved by silica gel chromatography (41% & 15% yields); their isotopic distribution patterns in high-resolution mass spectra were identical and corresponded to the tetra-brominated targets. The room temperature 1H NMR spectrum of the more dominant product was difficult to interpret due to extremely broad signals but running the spectrum at 100 °C in DMSO-d6 neatly resolved these, gratifyingly confirming an isomer that would be consistent with C2v (achiral) or C4 (chiral) symmetry (see Fig. 3). One important feature of this 1H NMR spectrum is the two signals at δ 3.98 and 3.80 ppm corresponding to the (now) diastereotopic methylene signals on the lower rim propoxy groups (–OCH2CH2CH3), supporting the evidence of an inherently chiral isomer 3a.
:1 ratio (Scheme 2). Isolation of these compounds was achieved by silica gel chromatography (41% & 15% yields); their isotopic distribution patterns in high-resolution mass spectra were identical and corresponded to the tetra-brominated targets. The room temperature 1H NMR spectrum of the more dominant product was difficult to interpret due to extremely broad signals but running the spectrum at 100 °C in DMSO-d6 neatly resolved these, gratifyingly confirming an isomer that would be consistent with C2v (achiral) or C4 (chiral) symmetry (see Fig. 3). One important feature of this 1H NMR spectrum is the two signals at δ 3.98 and 3.80 ppm corresponding to the (now) diastereotopic methylene signals on the lower rim propoxy groups (–OCH2CH2CH3), supporting the evidence of an inherently chiral isomer 3a.
 | ||
| Scheme 1 Synthesis of the tetra-acylated calix[4]arenes 2. Reagents and conditions: for R = OMe: methyl chloroformate (8 equiv.), pyridine (8 equiv.), DCM, −10 °C – rt, 1 h; for R = Ot-Bu: Boc2O (8 equiv.), Et3N (8 equiv.), THF, 0 °C – reflux, 2 h; for R = Me: Ac2O (8 equiv.), Et3N (8 equiv.), THF, rt – reflux, 2 h. | ||
 | ||
| Scheme 2 Preliminary bromination of tetra-methyl carbamate calix[4]arene 2a. Reagents and conditions: (a) NBS (5 equiv.), PTSA (2 equiv.), DCM, −35 °C, 5 h. | ||
 | ||
| Fig. 3 1H NMR spectra for compounds 3a (major product, 400 MHz, DMSO-d6, 100 °C) and 4a (minor product, 300 MHz, CDCl3, 25 °C). | ||
The assignment of the minor product was more challenging. Its 1H NMR spectrum was consistent with an isomer with C2 or Cs symmetry, having two singlets each for the aromatic C–H, amide N–H and carbamate methyl protons (see Fig. 3). This eliminated the C1 isomer as a possibility, since a considerably more complex spectrum would be expected. Eliminating C2v and Cs was however not easy; The C2v isomer should have only one aromatic signal, but if the boat conformation was dominant, then one would expect to see two aromatic signals. To confirm this, the sample was heated to 100 °C, but still, two aromatic signals persisted. Although this suggested that this isomer could be the achiral CS symmetric calix[4]arene (D in Fig. 2), the calix[4]arene ‘bridge’ methylene signals appeared as a neat set of four doublets (a double AX system), which was inconsistent with the Cs isomer (see, purple arrows). Fortunately, 2D NMR spectroscopy resolved this issue, with the gHMBCAD spectrum revealing a clear connection between the one AX system and both CAr–H carbon atoms and the other AX system and both CAr–Br carbon atoms (see ESI† page S13 for full assignments), confirming the C2-symmetric compound 4a.
The synthesis of a C4-symmetric inherently chiral calixarene in one step was an important result and prompted us to investigate this reaction further. We reasoned that changing the methyl carbamate to a tert-butyl carbamate (Boc group), would introduce a greater steric effect that might improve the selectivity of this reaction. Tetra-Boc calix[4]arene 2b was therefore synthesised under optimum conditions, returning a quantitative yield of the desired tetracarbamate (Scheme 1, R = t-Bu). Running the bromination reaction under the same conditions gave a slightly improved yield of the major C4 isomer 3b, which nevertheless encouraged us to undertake a small optimisation study (see ESI† for full details of optimisation). In the optimisation study we found that the acid (PTSA) could be reduced to 0.2 equivalents, the optimum temperature was −42 °C and the bromine source needed to be NBS. The solvent was screened, but methylene chloride remained the preferred choice, although butanone gave similar results and could thus be considered an alternative, more environmentally friendly option. An experiment using chiral camphorsulfonic acid, unfortunately returned racemic product. Using the optimised conditions (see Scheme 3 and ESI†), a one gram scale reaction returned an 83% yield of the inherently chiral C4-symmetric calix[4]arene 3b. The method was reapplied to the tetramethylcarbamate calix[4]arene 2a where the yield obtained was 3a:4a = 51%:24%.
 | ||
| Scheme 3 Optimised synthesis of an inherently chiral calixarene. Reagents and conditions: (a) NBS (5 equiv.), PTSA (0.2 equiv.), CH2Cl2, −42 °C, 6 h; (b) conc. HCl (10%), THF, reflux, 3 h. | ||
Whilst this was an improvement on our starting point, the result still suggested that the Boc-group was more important for both yield and ratio of products. Three procedures for the removal of the Boc-groups were then examined (see ESI†), with HCl giving yields of 80–85% for tetra-bromo-tetra-amino calix[4]arene 5.
We attempted several crystallisation experiments to gain concrete evidence for the structural assignment of the C4-isomer. Fortuitously, 3b gave crystals suitable for diffraction. The solved structure (see Fig. 4 and ESI†) proved that the chiral C4-symmetric isomer 3b was the major product in these reactions. Whilst the crystal structure is in the so-called boat conformation, the high symmetry of the NMR spectra is due to the dynamic equilibrium at 100 °C.
 | ||
| Fig. 4 Crystal data for 3b: Side view; hydrogen atoms and solvent molecules omitted for clarity (see ESI† for more clarity). | ||
To establish the necessity for a carbamate directing group, the tetra acetamide calixarene analogue 2c was synthesized (Scheme 1, R = Me) using a different procedure than that reported21 giving an 87% yield (vs. 43% previously reported). For the ortho bromination reactions, solubility issues restricted the use of methylene chloride, but several reactions were carried out using both butanone and DMF with the optimised conditions. Unfortunately, all reactions resulted in the formation of several brominated products, that were difficult to isolate or characterise.
Finally, we attempted to separate the enantiomers using classical diastereomeric techniques. In this case we found that coupling the racemic calix[4]arene 5 to N-Boc-(L)-proline under standard conditions (DCC, DMAP) gave a mixture of diastereomers (top spot = 6a & bottom spot = 6b) in good yield, which could be adequately separated via preparative thin layer chromatography (Scheme 4).
 | ||
| Scheme 4 Separation of calixarene enantiomers. Reagents and conditions: (a) N-Boc-proline (6 equiv.), DCC (6 equiv.), DMAP (0.5 equiv.), CH2Cl2, rt, 13 h; (b) Ba(OH)2·8H2O (xs), tBuOH, reflux, 48 h. | ||
Removal of the chiral Boc-protected amino acid groups with barium hydroxide in tert-butanol under reflux15 returned the two separate enantiomers of 5 (Scheme 4) whose ECD spectra were obtained in acetonitrile confirming their enantiomeric nature (Fig. 5). Furthermore, an ECD spectrum for (M)-5 was calculated using DFT (PBE0/TZVP see ESI† for full details)22–24 and corresponded to the enantiomer obtained from hydrolysis of diastereomer 6a (top spot), confirming its assignment.
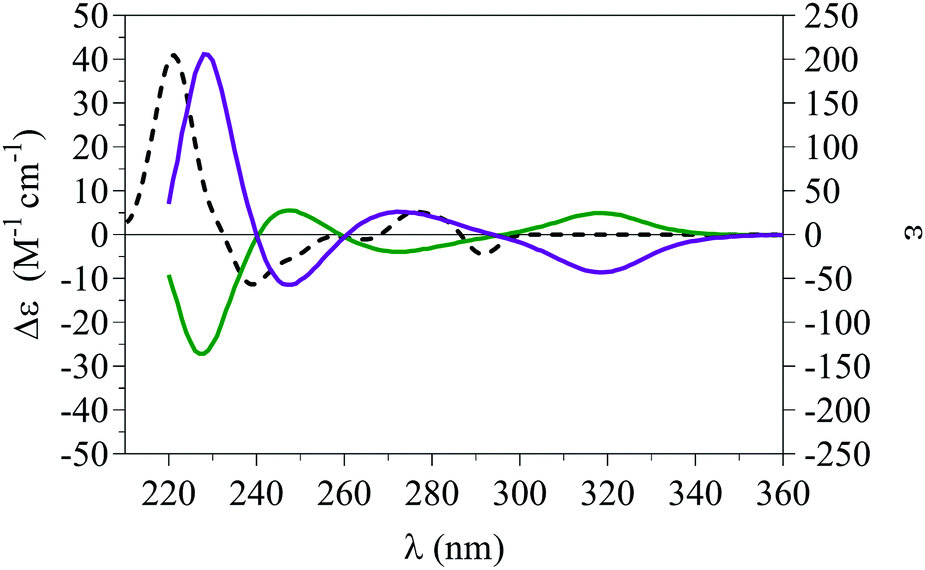 | ||
| Fig. 5 ECD spectra of (+)-5 (purple line) and (–)-5 (green line) showing the opposite cotton-effect and the computed spectrum of (M)-5 (black dashed line) corresponding to (+)-5 obtained from the diastereomer 6a (top spot). | ||
In conclusion, we have demonstrated a scalable method for synthesising racemic C4-symmetrical inherently chiral calix[4]arenes and a preliminary method on obtaining the pure enantiomeric forms. Future work will involve further developing the method to separate the enantiomers, investigating the potential for diastereoselectivity in the reaction and further functionalisation of the inherently chiral backbone to create chiral ligand-like materials suitable for asymmetric catalysis.
This work was supported by the National Research Foundation (CPRR160428163281).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- G. E. Arnott, Chem. – Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed.
- O. Kundrat and P. Lhoták, in Calixarenes and Beyond, Springer International Publishing, Cham, 2016, pp. 43–73 Search PubMed.
- G. D. Andreetti, V. Böhmer, J. G. Jordon, M. Tabatabai, F. Ugozzoli, W. Vogt and A. Wolff, J. Org. Chem., 1993, 58, 4023–4032 CrossRef CAS.
- D. K. Fu, B. Xu and T. M. Swager, J. Org. Chem., 1996, 61, 802–804 CrossRef CAS PubMed.
- R. Miao, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, J. Org. Chem., 2005, 70, 7662–7671 CrossRef CAS PubMed.
- O. G. Barton, B. Neumann, H.-G. Stammler and J. Mattay, Org. Biomol. Chem., 2008, 6, 104–111 RSC.
- W. Hüggenberg, A. Seper, I. M. Oppel and G. Dyker, Eur. J. Org. Chem., 2010, 6786–6797 CrossRef.
- M. Tlustý, H. Dvořáková, J. Čejka, M. Kohout and P. Lhoták, New J. Chem., 2020, 44, 6490–6500 RSC.
- C. Gaeta, F. Troisi, C. Talotta, T. Pierro and P. Neri, J. Org. Chem., 2012, 77, 3634–3639 CrossRef CAS PubMed.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Org. Lett., 2017, 19, 2933–2936 CrossRef PubMed.
- M. Tlustý, D. Spálovská, M. Kohout, V. Eigner and P. Lhoták, Chem. Commun., 2020, 56, 12773–12776 RSC.
- M. Mascal, R. T. Naven and R. Warmuth, Tetrahedron Lett., 1995, 36, 9361–9364 CrossRef CAS.
- W. Verboom, P. J. Bodewes, G. van Essen, P. Timmerman, G. J. van Hummel, S. Harkema and D. N. Reinhoudt, Tetrahedron, 1995, 51, 499–512 CrossRef CAS.
- Z.-X. Xu, C. Zhang, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, Org. Lett., 2007, 9, 4447–4450 CrossRef CAS PubMed.
- S. A. Herbert and G. E. Arnott, Org. Lett., 2010, 12, 4600–4603 CrossRef CAS PubMed.
- D. C. Castell, N. Lesotho, V. I. Nikolayenko and G. E. Arnott, Eur. J. Org. Chem., 2017, 4328–4333 CrossRef CAS.
- J. Holub, V. Eigner, L. Vrzal, H. Dvořáková and P. Lhoták, Chem. Commun., 2013, 49, 2798–2800 RSC.
- K. J. Visagie, L. Hodson and G. E. Arnott, S. Afr. J. Chem., 2020, 73, 15–21 CrossRef CAS.
- A. M. A. Van Wageningen, E. Snip, W. Verboom, D. N. Reinhoudt and H. Boerrigter, Liebigs Ann., 1997, 11, 2235–2245 CrossRef.
- S. Tommasone, C. Talotta, C. Gaeta, L. Margarucci, M. C. Monti, A. Casapullo, B. MacChi, S. P. Prete, A. Ladeiradearaujo and P. Neri, Angew. Chem., Int. Ed., 2015, 54, 15405–15409 CrossRef CAS PubMed.
- C. Schiel, G. A. Hembury, V. V. Borovkov, M. Klaes, C. Agena, T. Wada, S. Grimme, Y. Inoue and J. Mattay, J. Org. Chem., 2006, 71, 976–982 CrossRef CAS PubMed.
- C. Talotta, C. Gaeta, F. Troisi, G. Monaco, R. Zanasi, G. Mazzeo, C. Rosini and P. Neri, Org. Lett., 2010, 12, 2912–2915 CrossRef CAS PubMed.
- G. Concilio, C. Talotta, C. Gaeta, P. Neri, G. Monaco, R. Zanasi, D. Tedesco and C. Bertucci, J. Org. Chem., 2017, 82, 202–210 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2104244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04607a |
| ‡ The term meta-functionalization may appear odd to general practitioners, but it is a convention in the calixarene community to group functionalizations of this type together under this term. |
| This journal is © The Royal Society of Chemistry 2021 |