
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-controlled interconversion between a [c2]daisy chain and a lasso-type pseudo[1]rotaxane†
Chih-Wei
Chu
 ,
Daniel L.
Stares
and
Christoph A.
Schalley
*
,
Daniel L.
Stares
and
Christoph A.
Schalley
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 20, Berlin 14195, Germany. E-mail: c.schalley@fu-berlin.de
First published on 25th October 2021
Abstract
A light-responsive self-complementary crown ether/ammonium conjugate bearing an arylazopyrazole photoswitch as a spacer can be switched between a [c2]daisy chain (E-isomer) and a lasso-type pseudo[1]rotaxane (Z-isomer) by light.
Molecular daisy chains1 and supramolecular polymers made by them2 represent a special class in mechanically interlocked molecules (MIMs). [c2]Daisy chains have gained much attention due to their potential as molecular muscles.3,4 The self-complementary building blocks of [c2]daisy chains consist of a wheel and an axle as the binding units, and several examples have been reported using crown ethers,5–8 cyclodextrins,9,10 pillararenes11,12 and cucurbiturils13 as the wheels together with the corresponding axles. The introduction of stimuli-responsive units in the molecular design provides access to [c2]daisy chains that can be stimulated by pH,7,14,15 light9,10 and redox reactions.16 Recently, these stimuli-responsive motifs have been coupled to polymers, so that the shuttling of the macrocycle and thus the expansion/contraction can be observed at a macroscopic level.9,10,14,15
Pseudo[1]rotaxanes stand for another interesting category of MIMs. Similar to the precursors of [c2]daisy chains, they comprise a macrocycle and a threaded axle, in which the components are linked covalently. Though they exist in nature, i.e. as lasso peptides,17,18 synthetic pseudo[1]rotaxanes have been less frequently investigated compared to [c2]daisy chains.19–21 Synthetic chemists have engineered the molecular design to control the self-inclusion behaviour and the mechanical motion by external stimuli, such as pH,21,22 light23 and redox.24–26
Usually, the molecular design of a self-complementary monomer is different for making a [c2]daisy chain and a lasso-type pseudo[1]rotaxane. The “spacer” between the axle site and the wheel part should be short and rigid for making a [c2]daisy chain;8 whereas it is often longer and more flexible in the case of a lasso-type pseudo[1]rotaxane in order to allow for the backfolding of the axle into the wheel.25 Since Shinkai and coworkers presented a photoresponsive “tail-biting”, but non-threading crown ether by taking advantage of the configurational difference between E- and Z-isomers of azobenzene in 1985,27 further studies aiming at photoresponsive pseudo[1]rotaxane are rare. Using azobenzene, a light-driven molecular pump28 and gated photochromism29 have been reported.
Herein, we demonstrate the possibility of forming both a [c2]daisy chain and a lasso-type pseudo[1]rotaxane from the same building block in a controlled way. In our design, we synthesise a heteroditopic monomer 1 that bears an arylazopyrazole (AAP) to bridge a threadable macrocycle, benzo-21-crown-7, and a secondary ammonium ion (Fig. 1a). AAP is a photoswitch that isomerises reversely under UV and green light irradiation with almost quantitative photoswitching and can be easily modified to tune its photophysical properties.30–34 In this work, the rigid AAP plays an essential role in the molecular design. Firstly, the E-AAP provides sufficient distance between the crown ether and the ammonium station, so that the self-inclusion conformation is unfavourable in this state. Instead, a dimeric daisy chain (E,E-DC) is likely more preferred due to its thermodynamic stability.6,8 Secondly, the metastable Z-AAP brings the two recognition sites closer, thus a self-included lasso-type pseudo[1]rotaxane (Z-L) is expected. The formation of E,E-DC and Z-L and the light-controlled transformation are investigated by NMR, UV/vis and tandem mass spectrometry.
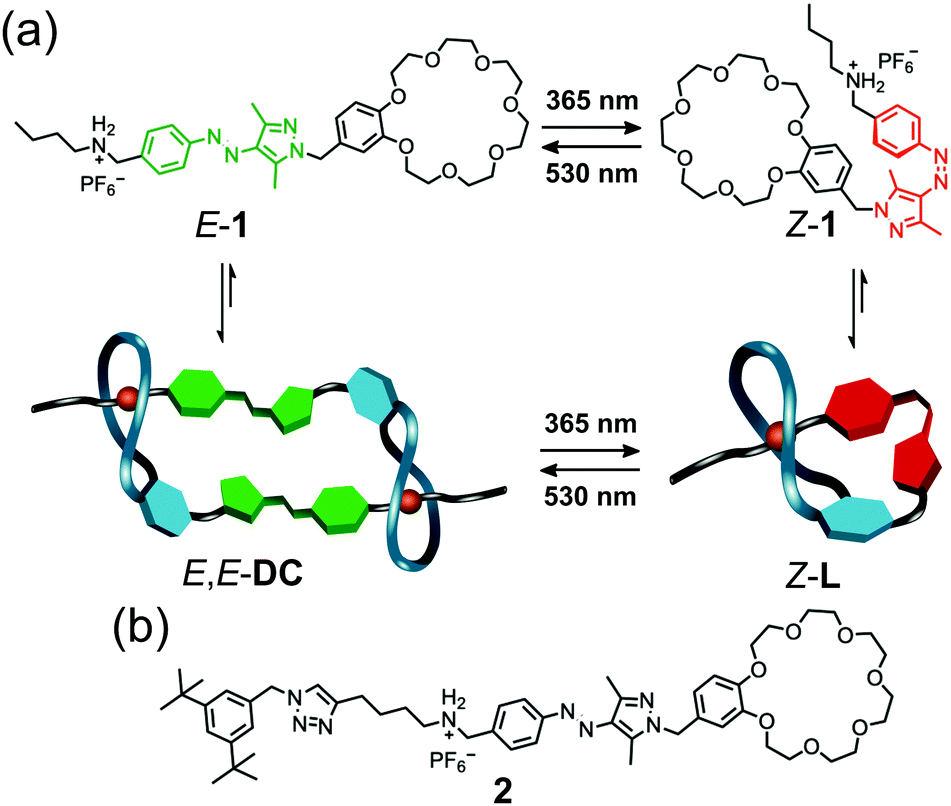 | ||
| Fig. 1 (a) Molecular structure and light-responsiveness of 1 and the corresponding [c2]daisy chain E,E-DC and pseudo[1]rotaxane Z-L. (b) Molecular structure of control compound 2. | ||
Heteroditopic monomer 1 was synthesised from the aldehyde precursor containing the AAP unit attached to benzo-21-crown-7, and n-butylamine via reductive amination, followed by protonation with HCl and counterion exchange with NH4PF6 (for synthetic procedures and characterisation data, see ESI†). NMR and UV/vis experiments were carried out to determine the photoswitching properties of 1.
In the UV/vis spectra (Fig. 2a), the characteristic absorption bands of AAP appear, namely a π–π* transition at ca. 350 nm and an n–π* transition at ca. 450 nm.30,31 When irradiating at 365 nm for 15 min, the π–π* band diminished and blue-shifted, and the n–π* band increased and red-shifted, suggesting effective E- to Z-isomerisation of 1. After 15 min of green light irradiation, not only the original spectrum was retrieved, but the absorbance at ca. 350 nm was further increased, implying the coexistence of the E- with some Z-isomer in the as-prepared sample. This feature is also observed in the 1H NMR experiments. The aromatic protons of AAP and the methylene protons between the AAP and the benzo-21-crown-7 moieties can be integrated to determine the photostationary state (PSS) at different wavelength (Fig. 2b). In the as-prepared sample, the ratio between E- and Z-isomer is approximately 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, which resonates with the UV/vis experiments. Irradiation of 1 at 365 nm for 15 min led to almost quantitative photoswitching (>98%) to the Z-isomer. Excitation of the n–π* transition of 1 led to a PSS containing >95% of E-isomer. These results indicate that 1 possesses excellent photoswitching properties comparable to literature-known AAP derivatives.32–34
:3, which resonates with the UV/vis experiments. Irradiation of 1 at 365 nm for 15 min led to almost quantitative photoswitching (>98%) to the Z-isomer. Excitation of the n–π* transition of 1 led to a PSS containing >95% of E-isomer. These results indicate that 1 possesses excellent photoswitching properties comparable to literature-known AAP derivatives.32–34
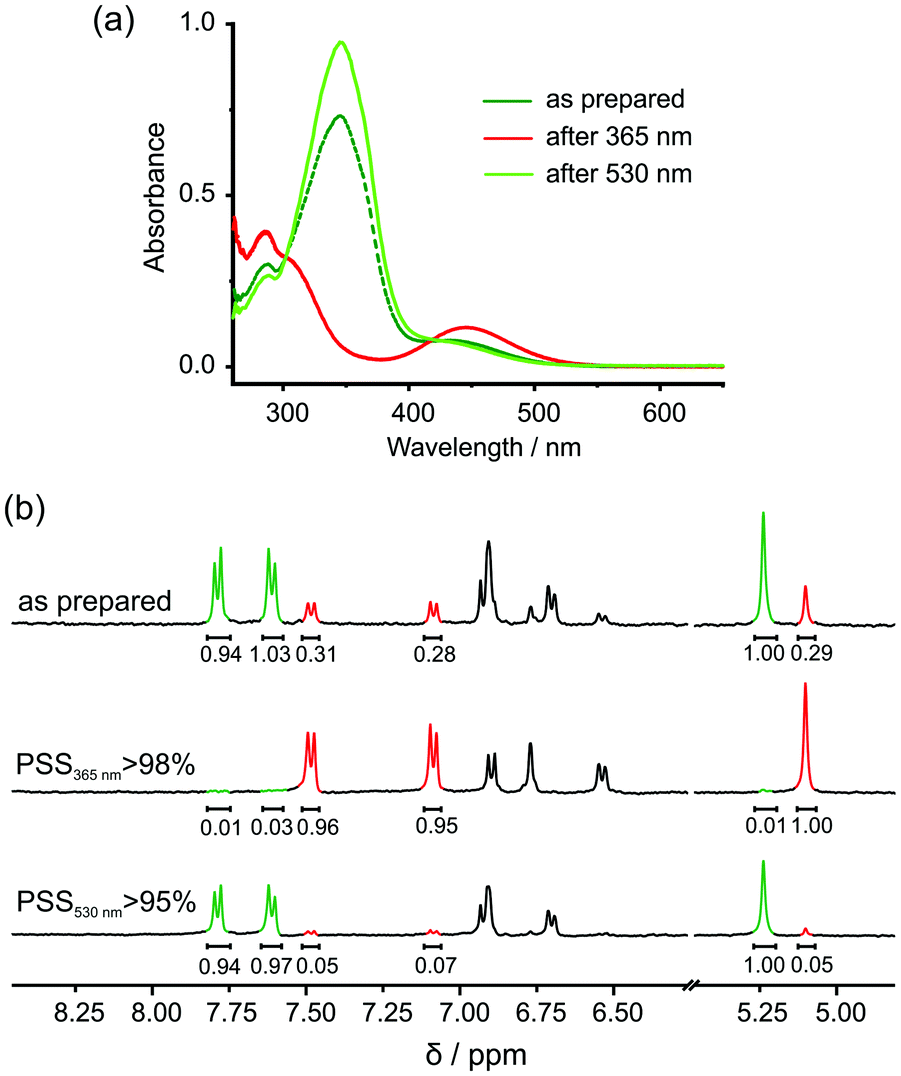 | ||
| Fig. 2 (a) UV/vis spectra of 1 upon irradiation of UV and green light. (b) PSSs (at 365 and 530 nm) of 1 determined by 1H NMR (400 MHz, 0.5 mM, DMSO-d6). | ||
Formation of E,E-DC from E-1 is investigated by 1H NMR spectroscopy. In competitive DMSO, 1 exists as an E-monomer (Fig. 3a). In CD3CN, the equilibrium between monomer and dimer shifts almost completely towards E,E-DC, as indicated by the much more complex spectrum revealing a diastereotopic splitting of the crown ether CH2 groups and of proton He (Fig. 3b) which is indicative of threading. In addition, aromatic protons at the phenyl ring of the AAP (Hc and Hd) shift upfield, and the adjacent methylene protons to the ammonium group Ha and Hb downfield. These observations can be easily understood: firstly, due to the self-complementary threading of E,E-DC, the phenyl group of the AAP is positioned atop the pyrazole group of the other and thus experiences its aromatic ring current. Secondly, the deshielding of the N–CH2 protons is due to the hydrogen bonding to the crown ether's oxygen atoms. The assignment of protons in E,E-DC is further confirmed by 1H–1H COSY NMR spectroscopy (Fig. S1, ESI†) as well as concentration scanning (Fig. S2, ESI†) and a variable temperature NMR experiment (Fig. S3, ESI†). The ratios between non-threaded E-1 and E,E-DC in CD3CN range from ca. 4:6 (1 mM) up to 8:2 (20 mM). At elevated temperatures, E,E-DC dissociates to a non-threaded E-1 due to a favourable entropy.
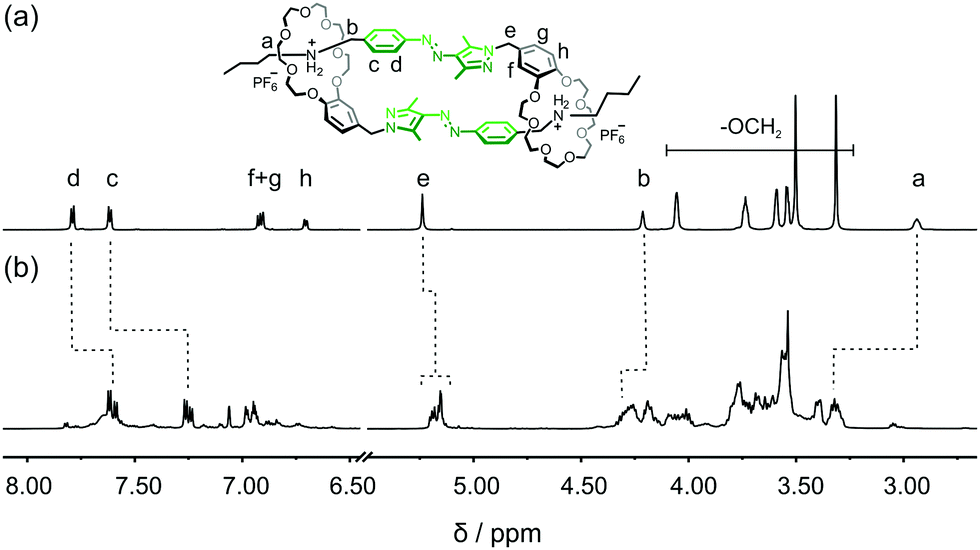 | ||
| Fig. 3 Partial 1H NMR spectra of E-1 in (a) DMSO-d6 (700 MHz, 298 K) and (b) CD3CN (700 MHz, 20 mM, 298 K). | ||
Irradiating E-1 with UV light at a concentration of 5 mM in CD3CN resulted in a mixture of Z-1 and Z-L (Fig. 4b). Unfortunately, the concentration cannot go beyond 5 mM, otherwise E-1 as well as E,E-DC were detected (Fig. S4, ESI†). We attribute this finding to an acid-mediated Z- to E-isomerisation of AAP, which has been discussed earlier in the literature35,36 and will be discussed below. Similar to E,E-DC, the CH2 signals of Z-L reveal diastereotopic splitting as particular nicely observed for He. Also consistent with a threaded structure, upfield shifting of the aromatic protons Hc and Hd and downfield shifting of the N–CH2 protons Ha and Hb were observed in Z-L. 1H–1H COSY NMR spectrum of Z-1 supports our assignment of Z-L in CD3CN (Fig. S5, ESI†).
 | ||
| Fig. 4 Partial 1H NMR spectra of Z-1 in (a) DMSO-d6 (700 MHz, 298 K) and (b) CD3CN (700 MHz, 5 mM, 298 K). | ||
The light-mediated transformations between E,E-DC and Z-L was investigated in gas phase by tandem and ion-mobility mass spectrometry (IMS). IMS allows the separation of ions based on their size which makes it an especially powerful tool to analyse photoisomers with their typically identical elemental compositions and thus identical m/z values.37–39 IMS has been used to analyse the topology of intertwined molecules40 and recently both MS and IMS utilised to identify pseudorotaxanes.25E,E-DC and Z-L have the same m/z, however their isotope patterns differ as E,E-DC is doubly charged (peak spacing of 0.5) while Z-L is singly charged (peak spacing of 1.0). Prior to UV irradiation there is one clear peak at 8.6 ms in the arrival time distribution (ATD). The corresponding mass spectrum has the peak spacing of 0.5 expected for a doubly charged ion showing E,E-DC (Fig. 5a) to be the major product. When irradiating with UV light for 5 min, the characteristic dimer peak decreases in the spectrum (Fig. 5b) whilst the ATD becomes more complex with two new major peaks at 8.4 ms and 10.9 ms. The corresponding mass spectra reveal the peak at 8.4 ms to correspond to doubly charged and that at 10.9 ms to singly charged ions, suggesting the formation of E,Z-DC and Z-L upon irradiation, respectively. It is noteworthy that there is also a trace amount of a doubly charged ion at 7.2 ms, which could potentially be Z,Z-DC. After 20 min of UV irradiation, the major product is Z-L with only trace amount of E,E- and E,Z-DC left as identified by the IMS and MS results (Fig. 5c).
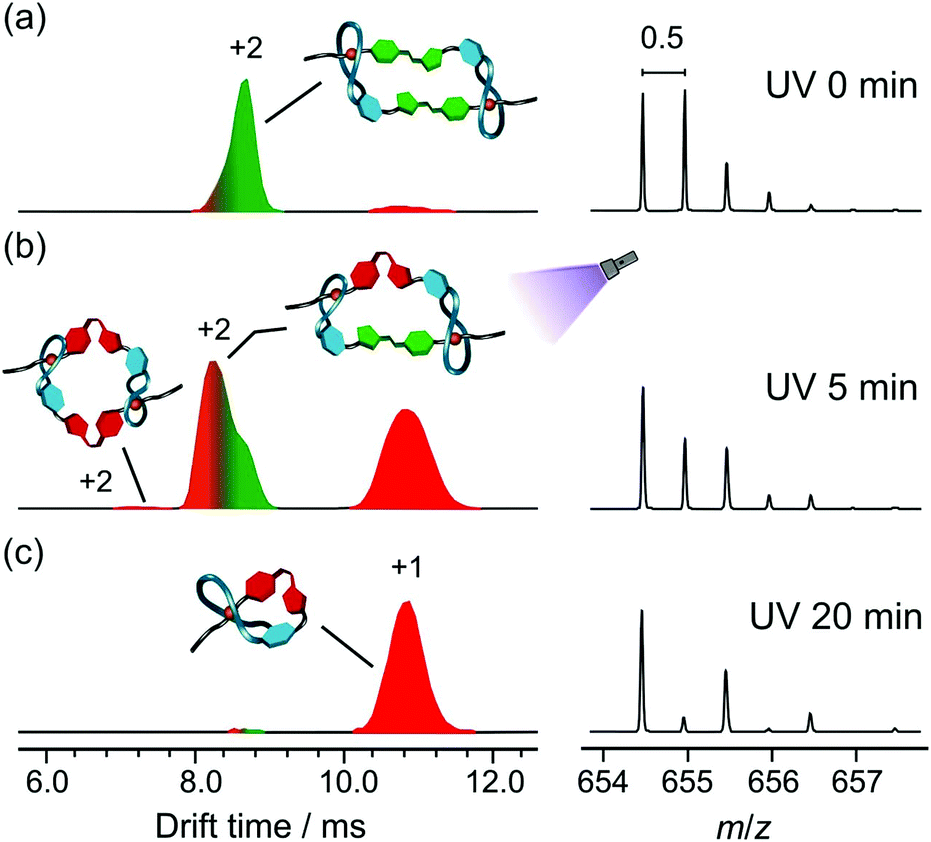 | ||
| Fig. 5 Normalised arrival time distributions (ATD) in the IMS experiments and the corresponding mass spectra after (a) 0 min, (b) 5 min and (c) 20 min of UV irradiation at 365 nm. | ||
E,E-DC and E,Z-DC were assigned based on energy resolved IMS measurements and further irradiation experiments utilising green light. Energy resolved IMS measurements of a sample containing all species (i.e. after 5 min UV irradiation) resulted in a decrease in the shoulder peak with a corresponding increase in the monomer peak whilst the initial peak is relatively unchanged (Fig. S6, ESI†). As E,Z-DC has a twisted conformation and is potentially less stable, it will more easily dissociate upon increasing collision voltage compared to E,E-DC. The same state was then also irradiated with green light (530 nm). Not only did the monomer species diminish, but the shoulder at lower drift time (8.4 ms) also gradually decreased (Fig. S7, ESI†). After irradiating at 530 nm for 10 min, an ATD similar to Fig. 5a is obtained. Z- to E-isomerisation of both Z-1, and E,Z-DC would be expected to form E,E-DC, which fits the experimental results. Notably, neither the shoulder peak nor the peak at 7 ms were increased under green light irradiation. Irradiation of the Z-1 would result in the formation of E-1 thus the only dimer expected to form would be the E,E-DC which was indeed observed. These findings support our hypothesis of photo-controlled transformations from E,E-DC to E,Z-DC and finally to Z-L under UV irradiation. The Z-isomer can reliably be switched back with green light to the E-isomer, which subsequently reassembles into the daisy chain. In addition, the Z-L shows a good thermal stability with a half-life in the range of several hours (Fig. S8, ESI†). Semi-empirical calculations and theoretical CCS values are in very good agreement with the experimental data and, for example, reflect the somewhat smaller size of the E,Z-DC as compared to that of the E,E-DC (ESI,† Section S5).
As observed in the NMR experiments (see above), the half-life of the thermal back isomerisation of Z-1 significantly decreases with increasing concentration from 16 days at 38 μM to 1.5 hours at 1 mM in acetonitrile (Fig. S16, ESI†). We hypothesise that the ammonium ion can act as an acid in organic solvents and the acid-mediated thermal back relaxation of Z-azo compounds is known both in solution35,36 and in the gas phase.39 This effect is potentially not only concentration dependent, but also solvent dependent. To further investigate the solvent effect for the Z–E switching, 2 and Boc-protected 2 were tested. Three UV/vis spectra of each sample were recorded in each solvent (Fig. S17, ESI†). Boc-protected 2 showed a great thermal stability in all tested solvents after irradiating at 365 nm for 10 min, whereas 2 exhibited a clear solvent dependence of the back-isomerisation rate. In polar solvents, such as CH3CN and DMSO, a good stability of Z-2 is observed due to the well-solvated secondary ammonium ion. In the case of non-polar solvents like CH2Cl2, CHCl3 and DCE, a fast Z–E switching is detected. The back isomerisation is more prominent because the lack of hydrogen bond acceptors in these solvent molecules makes the ammonium ion acidic enough to cause the acid-mediated Z–E isomerisation.41,42 When looking at the dielectric constant of THF, it should fit in the category of non-polar solvent. However, Z-2 showed a good stability in THF. We attribute this observation to the stabilised secondary ammonium ion by the oxygen of THF, so that the protonation-assisted Z–E switching is inhibited.
In the gas phase, the atom which is protonated plays an essential role in the ion's behaviour. It has been reported that a secondary amine can accommodate a proton better than azo groups, so that the E- and Z-isomers of the azobenzene can be generated and monitored by IMS.37,39 In our study, the ATD of 2 shifted towards shorter drift time upon UV irradiation (Fig. S9, ESI†). These two photoisomers can be resolved because the proton is situated at the secondary amine, thus the protonation-assisted Z- to E-isomerisation is inhibited. On the contrary, the Boc-protected 2 did not show a distinct difference in size upon UV irradiation (Fig. S10, ESI†). In Boc-protected 2, the preferred protonation site is the azo nitrogen adjacent to the phenyl ring, which possesses a mesomeric stabilisation through the pyrazole ring,35,36 so that a fast Z–E switching is occurring.
In conclusion, we demonstrated a proof of principle that the [c2]daisy chain made by 1 can be reversibly converted to the lasso-type pseudo[1]rotaxane under light irradiation. In our design, we employ an AAP to photo-control the distance between the crown ether and the secondary ammonium station in the thread, so that E-1 forms a self-complementary E-DC, while Z-1 self-includes to a Z-L. These observations are not only presented in solution as demonstrated by NMR and UV/vis experiments, but also investigated in gas phase by MS and IMS.
We thank Deutsche Forschungsgemeinschaft (project number 434455294) and the European Union through the NOAH project (H2020-MSCA-ITN project ref. 765297) for funding. Support of NMR and MS experiments from the BioSupraMol core facility at FU Berlin is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Jürgen and M. Marcel, Chem. Soc. Rev., 2013, 42, 44–62 RSC.
- Examples: (a) Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS PubMed; (b) M. Zhang, S. Li, S. Dong, J. Chen, B. Zheng and F. Huang, Macromolecules, 2011, 44, 9629–9634 CrossRef CAS.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- A. Goujon, E. Moulin, G. Fuks and N. Giuseppone, CCS Chem., 2019, 1, 83–96 CAS.
- P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS PubMed.
- B. Zheng, M. Zhang, S. Dong, J. Liu and F. Huang, Org. Lett., 2012, 14, 306–309 CrossRef CAS PubMed.
- C. Romuald, A. Ardá, C. Clavel, J. Jiménez-Barbero and F. Coutrot, Chem. Sci., 2012, 3, 1851–1857 RSC.
- B. Zheng, F. Klautzsch, M. Xue, F. Huang and C. A. Schalley, Org. Chem. Front., 2014, 1, 532–540 RSC.
- K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- S. Ikejiri, Y. Takashima, M. Osaki, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2018, 140, 17308–17315 CrossRef CAS PubMed.
- L. Gao, Z. Zhang, B. Zheng and F. Huang, Polym. Chem., 2014, 5, 5734–5739 RSC.
- K. Wang, C.-Y. Wang, Y. Zhang, S. X.-A. Zhang, B. Yang and Y.-W. Yang, Chem. Commun., 2014, 50, 9458–9461 RSC.
- L. Cao and L. Isaacs, Org. Lett., 2012, 14, 3072–3075 CrossRef CAS PubMed.
- A. Goujon, T. Lang, G. Mariani, E. Moulin, G. Fuks, J. Raya, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 14825–14828 CrossRef CAS PubMed.
- A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923–4928 CrossRef CAS PubMed.
- C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- F. Saito and J. W. Bode, Chem. Sci., 2017, 8, 2878–2884 RSC.
- Q. Zhou, P. Wei, Y. Zhang, Y. Yu and X. Yan, Org. Lett., 2013, 15, 5350–5353 CrossRef CAS PubMed.
- X.-S. Du, C.-Y. Wang, Q. Jia, R. Deng, H.-S. Tian, H.-Y. Zhang, K. Meguellati and Y.-W. Yang, Chem. Commun., 2017, 53, 5326–5329 RSC.
- P. Waelès, C. Clavel, K. Fournel-Marotte and F. Coutrot, Chem. Sci., 2015, 6, 4828–4836 RSC.
- C. Clavel, C. Romuald, E. Brabet and F. Coutrot, Chem. – Eur. J., 2013, 19, 2982–2989 CrossRef CAS PubMed.
- A. Saura-Sanmartin, A. Martinez-Cuezva, A. Pastor, D. Bautista and J. Berna, Org. Biomol. Chem., 2018, 16, 6980–6987 RSC.
- Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC.
- H. V. Schröder, J. M. Wollschläger and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- G. Orlandini, L. Casimiro, M. Bazzoni, B. Cogliati, A. Credi, M. Lucarini, S. Silvi, A. Arduini and A. Secchi, Org. Chem. Front., 2020, 7, 648–659 RSC.
- S. Shinkai, M. Ishihara, K. Ueda and O. Manabe, J. Chem. Soc., Perkin Trans. 2, 1985, 511–518 RSC.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed.
- M. Lohse, K. Nowosinski, N. L. Traulsen, A. J. Achazi, L. K. S. von Krbek, B. Paulus, C. A. Schalley and S. Hecht, Chem. Commun., 2015, 51, 9777–9780 RSC.
- C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS PubMed.
- J. Calbo, C. E. Weston, A. J. P. White, H. S. Rzepa, J. Contreras-García and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS PubMed.
- L. Stricker, M. Böckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem. – Eur. J., 2018, 24, 8639–8647 CrossRef CAS PubMed.
- S. Devi, M. Saraswat, S. Grewal and S. Venkataramani, J. Org. Chem., 2018, 83, 4307–4322 CrossRef CAS PubMed.
- R. S. L. Gibson, J. Calbo and M. J. Fuchter, ChemPhotoChem, 2019, 3, 372–377 CrossRef CAS.
- S. Ludwanowski, M. Ari, K. Parison, S. Kalthoum, P. Straub, N. Pompe, S. Weber, M. Walter and A. Walther, Chem. – Eur. J., 2020, 26, 13203–13212 CrossRef CAS PubMed.
- L. H. Urner, M. Schulze, Y. B. Maier, W. Hoffmann, S. Warnke, I. Liko, K. Folmert, C. Manz, C. V. Robinson, R. Haag and K. Pagel, Chem. Sci., 2020, 11, 3538–3546 RSC.
- M. S. Scholz, J. N. Bull, N. J. A. Coughlan, E. Carrascosa, B. D. Adamson and E. J. Bieske, J. Phys. Chem. A, 2017, 121, 6413–6419 CrossRef CAS PubMed.
- J. M. Wollschläger and C. A. Schalley, ChemPhotoChem, 2019, 3, 473–479 CrossRef.
- A. Kruve, K. Caprice, R. Lavendomme, J. M. Wollschläger, S. Schoder, H. V. Schröder, J. R. Nitschke, F. B. L. Cougnon and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 11324–11328 CrossRef CAS PubMed.
- H. K. Hall, J. Phys. Chem., 1956, 60, 63–70 CrossRef CAS.
- M. R. Crampton and S. D. Lord, J. Chem. Soc., Perkin Trans. 2, 1997, 369–376 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data and original NMR and mass spectra for new compounds, additional NMR and UV/Vis spectroscopic experiments and ion mobility mass spectrometric data. See DOI: 10.1039/d1cc04419b |
| This journal is © The Royal Society of Chemistry 2021 |