
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of a trigonal bipyramidal architecture with stabilisation of iron in three spin states†
Lauren L. K.
Taylor
 a,
Iñigo J.
Vitorica-Yrezabal
a,
Ivana
Borilović
ab,
Floriana
Tuna
*ab and
Imogen A.
Riddell
*a
a,
Iñigo J.
Vitorica-Yrezabal
a,
Ivana
Borilović
ab,
Floriana
Tuna
*ab and
Imogen A.
Riddell
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: imogen.riddell@manchester.ac.uk
bPhoton Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 11th October 2021
Abstract
Self-assembly and characterisation of a supramolecular trigonal bipyramidal iron cage containing an [FeIII(μ2-F)6(FeII)3]3+ star motif at its core is reported. The complex can be formed in a one step reaction using an heterotopic ligand that supports site-specific incorporation of iron in three distinct electronic configurations: low-spin FeII, high-spin FeII and high-spin FeIII, with iron(II) tetrafluoroborate as the source of the bridging fluorides. Formation of a μ2-F bridged mixed-valence FeII–FeIII star is unprecedented. The peripheral high-spin FeII centres of the mixed-valence tetranuclear star incorporated in the iron cage are highly anisotropic and engage in F-mediated antiferromagnetic exchange with the central FeIII ion.
Design approaches for the synthesis of self-assembled complexes have grown increasingly elaborate in recent years in a bid to diversify the structures generated and thus the applications of these molecular constructs.1,2 Initial approaches to metal–organic cage formation focused on the construction of capsules using symmetric, multitopic ligands in combination with a single metal ion.3,4 More recently heteroleptic5 and heterometallic6,7 systems incorporating more than one type of ligand or metal ion have gained interest as viable routes to synthesise novel architectures displaying properties not observed in their simpler analogues.2,8 To date however, examples of discrete three-dimensional structures generated from heterotopic ligands and a single metal precursor remain scarce,9 as do reports of complexes that incorporate one metal ion in a variety of spin or oxidation states.10–12 We hypothesized that ligand L (Scheme 1) that features both a pyridyl benzimidazole binding unit and a pyridine aldehyde moiety, that undergoes self-assembly reactions13 in the presence of amine and metal ion subcomponents, could give rise to novel metal–organic architectures not accessible when only one of these binding sites was incorporated. Furthermore, including pyridyl benzimidazole moieties, which favour binding of high-spin (HS) iron(II),14,15 alongside pyridyl imine coordination sites, which generally support complexation of low-spin (LS) iron(II),13,16 provided the opportunity for spin-state selective binding.
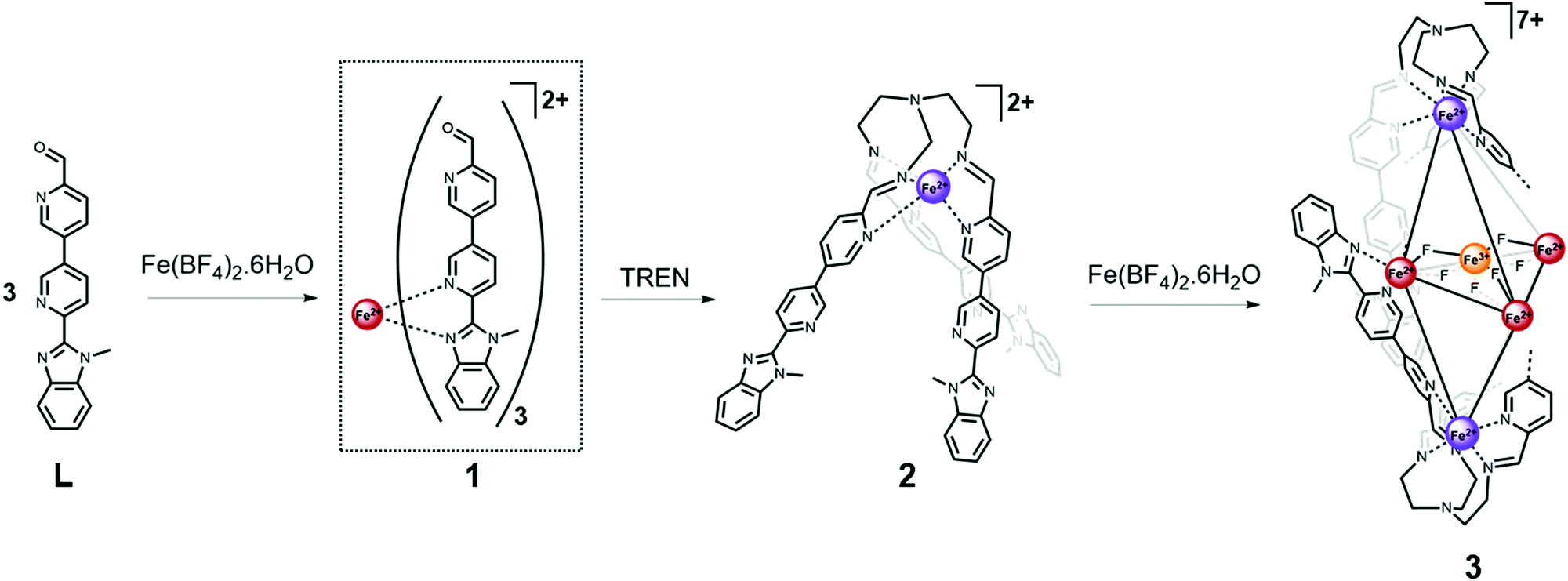 | ||
| Scheme 1 Stepwise self-assembly of the trigonal bipyramidal complex 3, from ligand Lvia the C3-symmetric metalloligand 2. For clarity only two of the six ligand arms forming complex 3 are shown explicitly. Iron spin and oxidation states are depicted by coloured spheres: high-spin iron(II) red, low-spin iron(II) purple and iron(III) orange. | ||
Herein we describe the design and synthesis of a heteroditopic ligand that, in combination with iron(II) tetrafluoroborate salt, generates a metal–organic cage in which the metal ions outline a trigonal bipyramidal structure of approximate D3 symmetry. The complex incorporates six iron atoms in a mixture of spin and oxidation states and includes an [FeIII(μ2-F)6(FeII)3]3+ star motif17–19 at its core.
Reaction of one equivalent of iron(II) tetrafluoroborate salt with three equivalents of L resulted in the formation of a dynamic mixture containing the mononuclear [FeL3]2+ complex 1 (Fig. S6, ESI†). This complex undergoes a gradual and incomplete thermally induced spin crossover (SCO), which is reversible (Fig. S30, ESI†) and consistent with an FeII centre in a pseudo-octahedral FeIIN6 crystal field of moderate strength.20,21 Subsequent addition of one equivalent of tris(2-aminoethyl)amine (TREN) to the reaction mixture resulted in a dramatic change in the 1H NMR resonances consistent with formation of the LS iron(II) trispyridylimine complex 2. In the presence of TREN, iron(II) is preferentially accommodated at the pyridyl imine binding site, rather than the pyridyl benzimidazole site, due to the higher level of preorganisation afforded by the multidentate trispyridylimine. The decreased bond length and reduced lability of the LS FeII–N bonds relative to their HS analogues, also promotes formation of complex 2. The single-crystal X-ray structure of 2 (Fig. S20, ESI†) confirms a facial arrangement of the pyridyl imine ligands which is consistent with the single set of resonances per ligand proton observed in the 1H NMR spectrum.7
Following characterisation of mononuclear complex 2, additional equivalents of iron(II) were added to the reaction mixture as we hypothesised the C3-metalloligands (2) could be brought together using their uncoordinated pyridyl benzimidazole binding sites.
Analysis of crystals grown through diffusion of diethyl ether into an acetonitrile mixture of Fe(BF4)2 and 2 revealed the structure of the multinuclear species (3) to be a mixed oxidation iron complex incorporating an [Fe4F6]3+ star motif at its centre (Fig. 1). High-resolution mass spectral analysis (Fig. S14, ESI†) was consistent with a complex cation containing two equivalents of metalloligand 2 and a mixed valent Fe(II)3Fe(III) core bridged by six fluoride ligands.
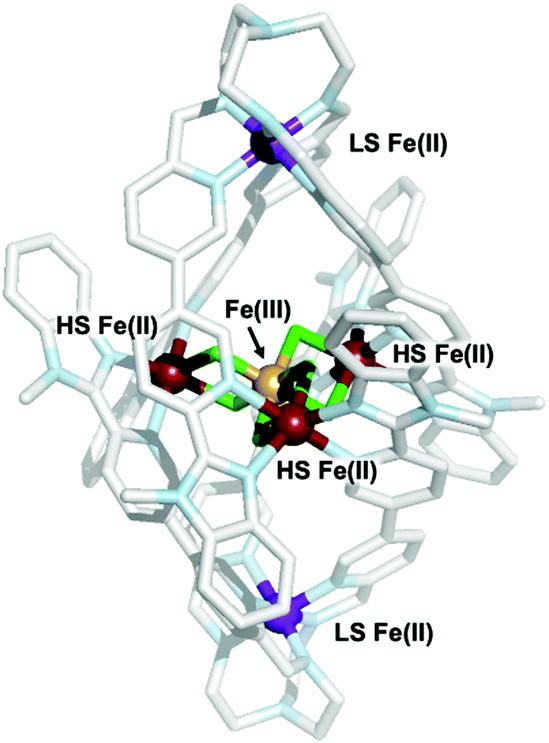 | ||
| Fig. 1 Single-crystal X-ray structure of the cationic portion of complex 3. The different spin and oxidation states of the iron atoms are highlighted; purple: low-spin (LS) iron(II); red: high-spin(HS) iron(II); yellow: high-spin iron(III); green: fluoride ions; pale-blue: nitrogen; light-grey: carbon. | ||
Bond valence sum (BVS) analysis (ESI,† S3) supports assignment of the apical irons in 3 as LS iron(II), while those bound by two bridging fluorides and two pyridyl benzimidazoles moieties were assigned as HS iron(II) sites. The latter connect to the central FeIII ion via two μ2-F− ligands (FeII–F bond lengths 2.089(8)–2.112(6) Å; FeIII–F bond lengths 1.898(8)–1.932(5) Å; average FeII–F–FeIII bridging angle 102.53(3)°). This is the first literature example of a F-bridged mixed valence iron star. The average FeII⋯FeIII and FeII⋯FeII distances of 3.121(4) and 5.407(14) Å, respectively, are shorter than those observed in oxo-bridged iron stars.18 Comparison of the trispyridylimine iron bonds in complexes 2 and 3 confirmed both were LS FeII ions, and no significant change in the FeII–N bond lengths are required to generate the higher nuclearity structure.
1H NMR analysis of the intense purple solution of 3 revealed resonances spanning chemical shift values from −2 to 136 ppm (Fig. S11, ESI†). Diffusion ordered spectroscopy (DOSY) NMR (Fig. S12, ESI†) confirmed that resonances at 23 and 28 ppm were consistent with formation of a structure with a diffusion coefficient of 6.32 × 10−10 m2 s−1, corresponding to a structure with a hydrodynamic radius of 12.6 Å. This value is in agreement with the solid state data which indicates that 3 is 22.4 Å along its maximum dimension. Variable temperature 1H NMR studies (Fig. S13, ESI†) provided no evidence of SCO for complex 3 within the solution state accessible temperature range (−38 to 70 °C).
Formation of 3 could not have been predicted based on previous results and established design criteria.3 In combination with transition metals, linear homotopic bisbidentate ligands featuring either two pyridyl benzimidazoles22 or two pyridyl imine moieties generated with TREN23 generate M4L6 tetrahedra and M2L3 helicates. Furthermore, analysis of self-assembly reactions with six equivalents of p-toluidine and L alongside four equivalents of metal supported formation of a [Fe4L6]8+ tetrahedron (ESI,† S1.3.4).
Central to the formation of 3 are the fluoride bridges which connect the FeIII ion with the three surrounding HS FeII centres. Since no traditional fluorinating agent was added, the tetrafluoroborate counterions are proposed as the source of fluoride. Generation of fluoride from tetrafluoroborate has previously been attributed to Lewis acid assisted abstraction, hydrolysis or the presence of a base,24 all of which are present under the conditions of our reaction. Formation of a M4(μ2-F)6 coordination motif has only previously been reported with a family of complexes with the outer metal atoms being supplied through titanocenes.25
In addition to fluoride generation, we also report in situ oxidation of iron(II).11,21,26 Following formation of 3 no further oxidation was observed and the complex was stable in air, in the solid state over a period of weeks, and in acetonitrile which had a stream of air blown through it for a day. Attempts to synthesize 3via in situ reduction of iron(III) with DMF, following a recent report,10 were unsuccessful and yielded an orange solution of unknown composition.
SQUID measurements for 3 gave χMT = 12.85 cm3 K mol−1 (χM = molar magnetic susceptibility) at room temperature in agreement with the presence of a magnetic FeII3FeIII entity (Fig. 2; χMT = 13.37 cm3 K mol−1 for three S = 2 and one S = 5/2 non-interacting centres, assuming g = 2), along with two non-magnetic (S = 0) LS FeII centres. Upon cooling, χMT decreases slowly until 50 K and then more rapidly to reach 3.67 cm3 K mol−1 at 2 K (Fig. 2B), indicative of weak antiferromagnetic interactions between metal centres coupled with zero-field splitting (ZFS) effects at the lowest temperature. In agreement with this, the M vs. H curves (M = molar magnetization) at 2 and 4 K show no sign of saturation under the 0–7 T applied magnetic fields (inset Fig. 2B), indicative of large magnetic anisotropy. Consistent with the solution state data, no evidence for temperature dependant SCO was observed for 3. Simultaneous fitting of χMT vs. T and M vs. H was performed using PHI.27 The spin Hamiltonian used28 includes the exchange between the peripheral HS FeII with the central HS FeIII (J1) and with its nearest neighbouring HS FeII (J2) (Fig. 2A), and gave g = 1.98(01), D = 9.06(13) cm−1, J1 = −1.58(03) cm−1 and J2 = −0.19(02) cm−1, where g is the g-factor of individual Fe centres and D is the axial ZFS term for HS FeII ions (ESI,† S5). Attempts to model the experimental magnetic data with J2 = 0 gave unsatisfactory results, but further inclusion of an intermolecular interaction term of zJ = −0.011 cm−1 enabled a good fit (Fig. S31 and Table S10, ESI†). Nevertheless, both models give the exchange interaction through μ2-F bridges as weakly antiferromagnetic (J2FeII–FeIII ≅ −1.6 cm−1). As 3 is the first molecular mixed-valence FeII3FeIII system with μ2-F bridges comparison of our coupling constants with precedent is not straightforward. Structurally related [FeIII4(μ-O)6]6+ compounds were reported to display antiferromagnetic coupling.19,29 In contrast, the [FeIII(μ-O)6FeII3]3+ homologue displays weak ferromagnetic exchange via μ2-O (J = 2.77 cm−1),18 though the peripheral FeII centres still couple antiferromagnetically. The FeII–F and FeIII–F bond lengths in 3 are shortened by 0.048 and 0.084 Å, respectively, compared to the equivalent ones in the oxo-bridged homologue, while the FeII⋯FeIII distance is reduced by 0.104 Å. These differences are sufficient to cause variation in magnetic behaviour. Diiron(II) complexes with an [FeII(μ2-F)2FeII]2+ core were found to be either weakly antiferromagnetic (J = −0.26 cm−1)30 or weakly ferromagnetic (J = 0.6 cm−1).31 A triple fluoride-bridged complex [F3FeIII(μ2-F)3FeIIIF3]3− whose Fe–F–Fe bridging angles average to 90.6° also shows weak ferromagnetism (J = 0.24 cm−1).32 While, diiron complexes with a single μ2-F bridging unit manifest a stronger antiferromagnetic exchange (16 < −J < 36 cm−1)33 due to a better magnetic orbital overlap enabled by a wider (151–180°) Fe–F–Fe bridging angle. In agreement with this, the [FFeII(μ2-F)FeIIIF]2+ complex (Fe–F–Fe 166.1°) exhibits stronger antiferromagnetic exchange (J = −10.1 cm−1)30 than 3. Ac susceptibility measurements on 3 detected frequency-dependent tails above 1.8 K that could indicate weak slow magnetic relaxation.
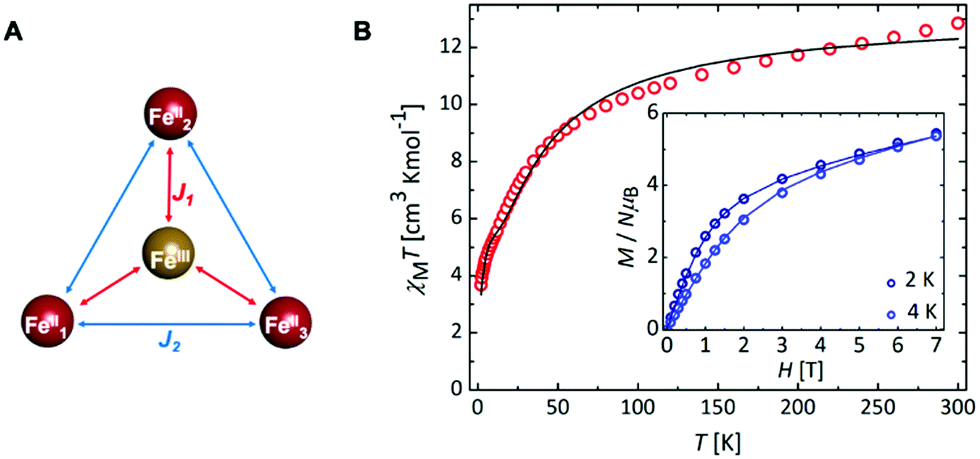 | ||
| Fig. 2 (A) Coupling scheme depicting the magnetic FeII3FeIII unit present in 3, where J1 and J2 represent the magnetic coupling constants; (B) χMT (T) and M (H) (inset) for 3 with the best fit (solid line). | ||
In conclusion, we report a novel one-step synthesis that generates an air-stable complex containing site-specifically incorporated metal ions in three electronic configurations. Isolation of 3 represents a significant advance in construction of multi-metallic architectures, where the goal is to emulate biological systems that control metal spin and oxidation states to direct a myriad of chemical processes. Magnetic measurements support F-mediated antiferromagnetic exchange between the peripheral FeII ions and the central FeIII of the star motif. Future work will focus on identifying reaction conditions that give rise to structurally related complexes and evaluating their magnetic and physical properties. Analysis of mixed oxidation state iron star complexes containing halogens other than fluoride will be invaluable in determining the role of the bridging ligand on the exchange interactions between the peripheral and central metal ions.
This research was supported by a University of Manchester Dame Kathleen Ollerenshaw Fellowship and a Royal Society University Research Fellowship (IAR), the Engineering and Physical Sciences Research Council (grants EP/K039547/1 and EP/R00482X/1), the Leverhulme Trust (RF/2018-545/4), the National EPSRC UK EPR Facility, and Diamond Light Source (beamline I19; cy23480). The authors thank Dr R. W. Adams, C. Bawn, G. Smith, D. Bell and Profs D. Collison and S. Liddle for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. C. O’Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O’Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72 CrossRef CAS PubMed; S. M. Jansze and K. Severin, Acc. Chem. Res., 2018, 51, 2139–2147 CrossRef PubMed; A. J. Metherell and M. D. Ward, Chem. Sci., 2016, 7, 910–915 RSC.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- P. J. Steel, Acc. Chem. Res., 2005, 38, 243–250 CrossRef CAS PubMed; P. J. Stang, Chem. – Eur. J., 1998, 4, 19–27 CrossRef.
- S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2006, 128, 72–73 CrossRef CAS PubMed; W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef PubMed; M. Wang, Y.-R. Zheng, K. Ghosh and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 6282–6283 CrossRef PubMed; Q. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 126, 13728–13731 CrossRef; D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef PubMed.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed; F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H. Yao and F. Li, Inorg. Chem., 2014, 53, 688–690 CrossRef PubMed; S. Sanz, H. M. O’Connor, P. Comar, A. Baldansuren, M. B. Pitak, S. J. Coles, H. Weihe, N. F. Chilton, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Inorg. Chem., 2018, 57, 3500–3506 CrossRef PubMed; S. Sanz, H. M. O’Connor, V. Martí-Centelles, P. Comar, M. B. Pitak, S. J. Coles, G. Lorusso, E. Palacios, M. Evangelisti, A. Baldansuren, N. F. Chilton, H. Weihe, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Chem. Sci., 2017, 8, 5526–5535 RSC; M. Schmittel, V. Kalsani and J. W. Bats, Inorg. Chem., 2005, 44, 4115–4117 CrossRef PubMed.
- M. Hardy, N. Struch, F. Topić, G. Schnakenburg, K. Rissanen and A. Lützen, Inorg. Chem., 2018, 57, 3507–3515 CrossRef CAS PubMed.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170–10178 CrossRef CAS PubMed; E. T. Luis, H. Iranmanesh, K. S. A. Arachchige, W. A. Donald, G. Quach, E. G. Moore and J. E. Beves, Inorg. Chem., 2018, 57, 8476–8486 CrossRef PubMed.
- S. Cardona-Serra, E. Coronado, P. Gaviña, J. Ponce and S. Tatay, Chem. Commun., 2011, 47, 8235–8237 RSC; A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797 CrossRef CAS PubMed.
- A. S. Rathnayake, H. W. L. Fraser, E. K. Brechin, S. J. Dalgarno, J. E. Baumeister, J. White, P. Rungthanaphatsophon, J. R. Walensky, S. P. Kelley, C. L. Barnes and J. L. Atwood, J. Am. Chem. Soc., 2018, 140, 15611–15615 CrossRef CAS PubMed.
- A. S. Rathnayake, H. W. L. Fraser, E. K. Brechin, S. J. Dalgarno, J. E. Baumeister, J. White, P. Rungthanaphatsophon, J. R. Walensky, C. L. Barnes, S. J. Teat and J. L. Atwood, Nat. Commun., 2018, 9, 2119 CrossRef PubMed.
- I. C. Berdiell, T. Hochdörffer, C. Desplanches, R. Kulmaczewski, N. Shahid, J. A. Wolny, S. L. Warriner, O. Cespedes, V. Schünemann, G. Chastanet and M. A. Halcrow, J. Am. Chem. Soc., 2019, 47, 18759–18770 CrossRef PubMed.
- T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC.
- C. Brewer, G. Brewer, C. Luckett, G. S. Marbury, C. Viragh, A. M. Beatty and W. R. Scheidt, Inorg. Chem., 2004, 43, 2402–2415 CrossRef CAS PubMed; R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- T. Lathion, L. Guénée, C. Besnard, A. Bousseksou and C. Piguet, Chem. – Eur. J., 2018, 24, 16873–16888 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed; J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2011, 4, 15 CrossRef PubMed.
- E. Tancini, M. J. Rodriguez-Douton, L. Sorace, A. Barra, R. Sessoli and A. Cornia, Chem. – Eur. J., 2010, 16, 10482–10493 CrossRef CAS PubMed; Y. Zhu, T. Yin, S. Jiang, A. Barra, W. Wernsdorfer, P. Neugebauer, R. Marx, M. Dörfel, B. Wang, Z. Wu, J. van Slageren and S. Gao, Chem. Commun., 2014, 50, 15090–15093 RSC; T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CrossRef PubMed.
- D. Sertphon, P. Harding, K. S. Murray, B. Moubaraki, N. F. Chilton, S. Hill, J. Marbey, H. Adams, C. G. Davies, G. N. L. Jameson and D. J. Harding, Dalton Trans., 2018, 47, 7118–7122 RSC.
- A. Cornia, M. Mannini, R. Sessoli and D. Gatteschi, Eur. J. Inorg. Chem., 2019, 552–568 CrossRef CAS; J. Mayans, M. Font-Bardia and A. Escuer, Dalton Trans., 2018, 47, 8392–8401 RSC.
- B. Brachňková, J. Adamko Kožíšková, J. Kožíšek, E. Melníková, M. Gál, R. Herchel, T. Dubaj and I. Šalitroš, Dalton Trans., 2020, 49, 17786–17795 RSC.
- F. Tuna, M. R. Lees, G. J. Clarkson and M. J. Hannon, Chem. – Eur. J., 2004, 10, 5737–5750 CrossRef CAS PubMed.
- M. J. Burke, G. S. Nichol and P. J. Lusby, J. Am. Chem. Soc., 2016, 138, 9308–9315 CrossRef CAS PubMed.
- W. Meng, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1017–1021 CrossRef CAS PubMed; J. Mosquera, S. Zarra and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 1556–1559 CrossRef PubMed.
- Y. I. Cho, M. L. Ward and M. J. Rose, Dalton Trans., 2016, 45, 13466–13476 RSC; E. Tomat, L. Cuesta, V. M. Lynch and J. L. Sessler, Inorg. Chem., 2007, 46, 6224–6226 CrossRef CAS PubMed; F. Jiang, M. A. Siegler and E. Bouwman, Inorg. Chem. Commun., 2018, 94, 53–56 CrossRef; D. L. Reger, R. P. Watson, J. R. Gardinier, M. D. Smith and P. J. Pellechia, Inorg. Chem., 2006, 45, 10088–10097 CrossRef PubMed.
- F. Liu, A. Künzel, A. Herzog, H. W. Roesky, M. Noltemeyer, R. Fleischer and D. Stalke, Polyhedron, 1997, 16, 61–65 CrossRef CAS; F. Liu, H. Gornitzka, D. Stalke and H. W. Roesky, Angew. Chem., Int. Ed. Engl., 1993, 32, 442–444 CrossRef.
- A. S. Rathnayake, H. W. L. Fraser, E. K. Brechin, S. J. Dalgarno, J. E. Baumeister, P. Rungthanaphatsophon, J. R. Walensky, C. L. Barnes and J. L. Atwood, J. Am. Chem. Soc., 2018, 140, 13022–13027 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
-
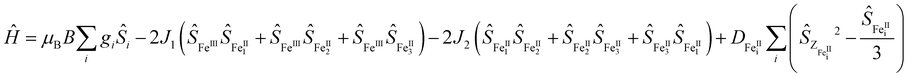 .
. - C. Schlegel, E. Burzurí, F. Luis, F. Moro, M. Manoli, E. K. Brechin, M. Murrie and J. van Slageren, Chem. – Eur. J., 2010, 16, 10178–10185 CrossRef CAS PubMed.
- S. Dammers, T. P. Zimmermann, S. Walleck, A. Stammler, H. Bögge, E. Bill and T. Glaser, Inorg. Chem., 2017, 56, 1779–1782 CrossRef CAS PubMed.
- Z. Yan, H. G. Jang, Y. Chiou, M. P. Hendrich and L. Que, Inorg. Chim. Acta, 1993, 213, 41–48 CrossRef.
- J. M. Dance, J. Mur, J. Darriet, P. Hagenmuller, W. Massa, S. Kummer and D. Babel, J. Solid State Chem., 1986, 63, 446–451 CrossRef CAS.
- D. Sil, A. Kumar and S. P. Rath, Chem. – Eur. J., 2016, 22, 11214–11223 CrossRef CAS PubMed; D. L. Reger, A. E. Pascui, M. D. Smith, J. Jezierska and A. Ozarowski, Inorg. Chem., 2012, 51, 11820–11836 CrossRef PubMed.
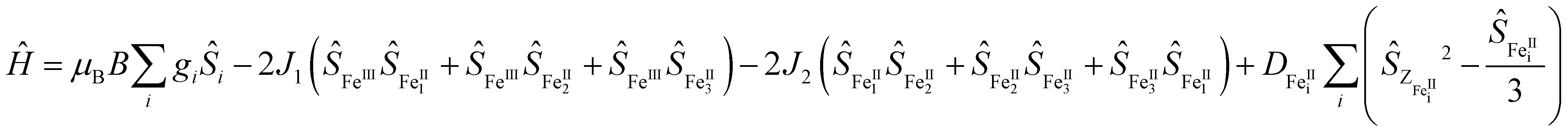
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR, ESI-MS and UV-vis spectra, SQUID data and analysis, and CIFS. Crystallographic have been deposited with the CCDC 1952397–1952402 and 2101118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04413c |
| This journal is © The Royal Society of Chemistry 2021 |