
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn2+-Dependent peptide nucleic acid-based artificial ribonucleases with unprecedented efficiency and specificity†
Olivia
Luige
 a,
Partha Pratim
Bose
a,
Rouven
Stulz
abc,
Peter
Steunenberg‡
a,
Omar
Brun§
a,
Shalini
Andersson
b,
Merita
Murtola
*a and
Roger
Strömberg
*a
a,
Partha Pratim
Bose
a,
Rouven
Stulz
abc,
Peter
Steunenberg‡
a,
Omar
Brun§
a,
Shalini
Andersson
b,
Merita
Murtola
*a and
Roger
Strömberg
*a
aDepartment of Biosciences and Nutrition, Karolinska Institutet, Neo, 141 83 Huddinge, Sweden. E-mail: Roger.Stromberg@ki.se
bOligonucleotide Discovery, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
cDMPK, Research and Early Development, Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
First published on 24th September 2021
Abstract
We present Zn2+-dependent dimethyl-dipyridophenazine PNA conjugates as efficient RNA cleaving artificial enzymes. These PNAzymes display site-specific RNA cleavage with 10 minute half-lives and cleave clinically relevant RNA models.
Modified oligonucleotides (ONs) such as gapmer antisense (ASO) and small interfering RNA (siRNA) technologies are powerful therapeutics used in the treatment of life-threatening diseases.1,2 Both strategies rely on the recruitment of endogenous enzymes (RNase H or RISC complex, respectively), which results in cleavage of the RNA targets.1 The complexes of RNA with these ONs must be recognised as substrates for the respective enzymes, which limits the modifications that can be incorporated.1,3 The chemical design of these ONs is, in turn, linked to issues affecting these technologies such as off-target effects and sequence- and chemistry-dependent toxicity.3,4 The reliance on cellular enzymes can also lead to non-specific toxicity due to the saturation of endogenous RNA processing pathways.5
Oligonucleotide-based artificial (ribo)nucleases (OBANs)6–8 aim to offer an alternative technology for the reduction of RNA levels, independent of endogenous enzymes. These artificial constructs consist of an ON backbone and a covalently linked “molecular scissors” moiety, which together ensure sequence-specific target recognition followed by catalytic cleavage of the RNA target.9–11
Three types of artificial ribonucleases have been investigated in recent years, including Cu2+ or Zn2+ cofactor dependent PNA–neocuproine conjugates (PNAzymes),12–15 metal independent tris(2-aminobenzimidazole)-DNA/LNA mixmer conjugates,16,17 and peptidyl-oligonucleotide conjugates (POCs) that consist of hairpin DNA or 2′-OMe ONs and a catalytic arginine and leucine-rich peptide.18–20 The metal-free systems, although attractive due to their promise of self-reliance, suffer from low rates of RNA cleavage16,20 and the aforementioned POCs require assistance from cellular enzymes (RNase H) to achieve turnover of the substrate.20 Cu2+-Dependent PNAzymes cleave their RNA targets site-specifically with half-lives in the 20–30 minute range and also give turnover.12,14,15 However, Cu2+ PNAzymes are not ideally suited for therapeutic applications, since a sufficient copper ion concentration is required to maintain their activity, while free copper is virtually not available in cells.21 Zn2+ is an attractive cofactor for artificial nucleases due to its presence in various concentrations in intracellular fluids.22–24 Despite the inferior RNA cleavage kinetics displayed by Zn2+-neocuproine PNAzymes13 compared to their Cu2+-dependent counterparts,12,14,15 the great potential of biocompatible artificial ribonucleases warrants further research into Zn2+-dependent PNAzymes.
Herein, we report on Zn2+-dimethyl-dipyridophenazine (dppz) as “molecular scissors” conjugated to PNA, creating novel biocompatible Zn2+ PNAzymes. Initially, 3,6-dimethyl-dipyrido[3,2-a:2′,3′-c]phenazine)-11-carboxylic acid was synthesised and conjugated to a PNA designed to target a leukaemia-related bcr/abl mRNA model (RNA 1, Fig. 1a) (see ESI,† S1 and S2). The placement of “molecular scissors” in a central part of the RNA/PNAzyme complex ensures that the cleavage of the RNA leaves the complex destabilised and prone to dissociation, which in turn facilitates turnover of the substrate.25 However, the formation of a double strand creates a barrier for RNA cleavage since RNA is cleaved significantly more slowly in a double strand than a single strand.26 This is likely due to an energy penalty from disruption of hydrogen bonding required to achieve the conformational change needed for the intramolecular attack of the 2′-OH group on the vicinal phosphodiester. Therefore, partial complementarity is exploited to force the formation of a more cleavage-prone single-stranded bulge region in the RNA, adjacent to the “molecular scissors” moiety.
 | ||
| Fig. 1 Schematic representations of PNAzyme I complexes with (a) RNA 1 and (b) RNA 2 where single stranded RNA bulges of 4 or 3 adenosine nucleotides are formed. Ion-exchange HPLC chromatograms showing the extent of cleavage of (c) RNA 1 (51 ± 2% in 3 h) and (d) RNA 2 (44 ± 2% in 50 min) after incubation at 37 °C, pH 7 and 4 μM RNA/PNAzyme concentration in the presence of 100 μM Zn2+. | ||
In the RNA 1/PNAzyme I complex, a 4-nucleotide AAAA bulge is formed in the RNA (Fig. 1a). Cleavage of RNA 1 was observed in the presence of equimolar Zn2+ PNAzyme I with a half-life of 3 h at pH 7 at 37 °C (Fig. 1c). A significantly higher cleavage rate was observed for RNA 2, where a 3-nucleotide AAA bulge (Fig. 1b) was cleaved with a 1 h half-life at a single site (Fig. 1d). In prior reports, these RNAs were cleaved at multiple sites by the Zn2+-neocuproine PNAzyme with ca. 12 and 24 h half-lives, while 7–8 h cleavage half-lives were achieved for the most rapidly cleaved RNA target sequences.13 As such, the Zn2+ dimethyl-dppz PNAzyme shows unprecedented efficiency and specificity for Zn2+-dependent artificial ribonucleases.
 | ||
| Fig. 2 (a) Schematic representation of PNAzyme I complexes with 3-nucleotide bulge-forming RNAs 2–16 and the extent of RNA cleavage observed for each bulge sequence. (b) RNA 3 degradation curve in the presence of Zn2+ PNAzyme I at pH 7. (c) Representative chromatogram of turnover experiments where the mean cleavage of RNA 13 in 6 experiments was 86 ± 4%. | ||
The RNA/PNAzyme complex that leads to this efficient site-specific RNA cleavage is likely to be governed by potential interactions that the novel “molecular scissors” can have with the RNA nucleobases. Aromatic heterocycles containing large rigid hydrophobic surfaces such as dppz are known intercalators, and thus prone to interact by stacking on to the heterocyclic bases in nucleic acids.27 In fact, intercalative binding between dppz and nucleic acids has been utilised in various applications.28–31 Increased rigidity and interactions with the dimethyl-dppz chelating moiety can result in pre-organisation of the RNA bulge, potentially making the bulge more susceptible to cleavage and possibly also helping to optimise the position of the metal ion to allow for more efficient catalysis. The presence of interactions involving dimethyl-dppz was confirmed by thermal melting analysis, which showed an increase in the RNA/PNA melting temperature from 52 to 56 °C in the presence of the covalently linked dimethyl-dppz moiety (see ESI,† S1). A prominent change was also observed in the CD spectrum, where the presence of dimethyl-dppz was clearly shown to affect the interaction in the complex (see ESI,† S1).
The sequence of the RNA bulge is highly likely to influence the interactions in the vicinity of the cleavage site. The rate of RNA cleavage was indeed improved further when the AAA bulge was replaced with AUA, GUA or UUA bulges (RNA 3, 11, 13, Fig. 2a). The observed half-lives were now as low as 16 minutes, making the Zn2+ dimethyl-dppz PNAzyme presented herein the most efficient oligonucleotide-based artificial ribonuclease reported, showing unprecedented activity and site-specificity for Zn2+-dependent systems13 and displaying rates even higher than the most efficient Cu2+ based systems.12,14,15 The UA internucleosidic phosphate linkage was confirmed as the site of cleavage by ESI-MS analysis in the most rapidly cleaved RNA targets. The central uridine in the bulge provides the 2′-hydroxyl nucleophile for attack on the adjacent phosphodiester bond in the cleavage site, leading to the formation of the 2′,3′-cyclic phosphate in the longer fragment, while the adenosine 5′-hydroxyl is the leaving group in the shorter fragment (ESI-MS data presented in ESI,† S1). The degradation curve for the cleavage of RNA 3 by PNAzyme I is depicted in Fig. 2b and the characteristic cleavage patterns for RNAs 2–16 are shown in ESI,† S3.
The critical importance of the artificial enzyme as well as the metal cofactor was demonstrated, as the RNA remained intact even after 24 h of incubation if either the PNAzyme or Zn2+ ions were absent (see ESI,† S4). The PNAzyme was shown to give turnover of the RNA substrate, as 86 ± 4% of RNA 13 was cleaved in 6 h when the RNA was present in a 33-fold excess compared to the PNAzyme (Fig. 2c).
Zn2+-Dependent PNAzyme I-promoted cleavage of RNA 3 displayed a pH-rate profile with a distinctive bell shape and an optimum at pH 7.4, where the estimated half-life of RNA cleavage was 10 minutes (Fig. 3a). This amounts to a 40–50-fold reduction in the cleavage half-life compared to Zn2+-neocuproine PNAzymes at the same pH.13 The bell-shaped pH-rate profile obtained for the PNAzyme-mediated RNA cleavage is indicative of acid–base catalysis, where one part is deprotonation of the 2′-OH nucleophile and the other part is protonation of the leaving 5′-oxygen. Varying the buffer concentration at the same pH did not affect the rate (see ESI,† S5) which suggests that general base catalysis is not present and that the deprotonation equilibrium of the 2′-OH, i.e. specific base catalysis, is responsible for the increase in rate of cleavage between pH 6.8 and 7.4.32 A further increase in pH from 7.4 to 8.0 leads to a decrease in the rate which fits with the shifting of the equilibrium of the metal-aqua complex (that can protonate the 5′-O leaving group) towards the inactive metal–hydroxide.32 It has been suggested that zinc aquo ions can exert general acid catalysis in this fashion while forming a six-membered ring (O–P–O–Zn2+–O–H) complex that can protonate the leaving group.33 A hypothetical mechanism involving a pre-equilibrium deprotonation followed by general acid catalysis by the metal aquo ion can be envisaged (Fig. 3b). This mechanism could be favoured, since the 5′-O leaving typically is limiting for non-catalysed cleavage, but other options can be considered: (1) deprotonation of the 2′-OH via coordination (direct or mediated by a water molecule) (2) coordination to water and hydroxide, the latter deprotonating 2′-OH and the former replaced by the phosphate to assert Zn2+ Lewis acid catalysis. Steric aspects may here become limiting, but it would fit the pH rate profile as deprotonation of the water prevents phosphate binding and eliminates catalysis.
 | ||
| Fig. 3 (a) pH-Rate profile for RNA 3 cleavage by Zn2+ PNAzyme I. (b) Hypothetical mechanism for the acid–base catalysis of cleavage of RNA by Zn2+ dimethyl-dppz-PNAzymes. | ||
Moreover, further factors, such as interactions of the chelate with nucleobases, positioning of the chelate etc. are likely to be important, but more structural information is needed to elucidate more details of the mechanism.
An important aspect of the PNAzymes is their adaptability to different target sequences, including clinically relevant therapeutic targets. PNAzymes could have diverse applications in disease therapy as well as molecular biology research, but the sensitivity to small structural changes in the RNA/PNAzyme complex increases uncertainty around whether it is possible to design a PNAzyme towards a desired target. In order to demonstrate the possibility to tailor PNAzymes for the cleavage of clinically relevant RNA sequences, we investigated a Plasmodium falciparum (malaria parasite) mRNA model as well as a SARS-CoV-2 genomic RNA model as targets for Zn2+ PNAzymes. We selected suitable targets with partial sequence similarities to our most rapidly cleaved RNA models, particularly the bulge sequence and the bulge closing base pairs. RNA 17 (Fig. 4a) is a model target from the Plasmodium falciparum exported protein 1 (EXP1) transcript which encodes an integral membrane protein essential for nutrient uptake.34,35 RNA 18 (Fig. 4b) is a fragment originating from the genomic RNA of SARS-CoV-2 that encodes a viral protease (betacoronavirus papain-like protease, SCoV2-PLpro) which is essential for processing viral polyproteins required for a functional replicase complex that enables viral spread.36 The cleavage of the Plasmodium falciparum mRNA fragment as well as the SARS-CoV-2 genomic RNA fragment by the corresponding PNAzymes proceeded as efficiently as the most rapidly cleaved RNA models above (Fig. 4c–e). As such, the Zn2+ PNAzymes are proving to be efficient and versatile agents that can be designed to cleave various RNA targets in a sequence and site-specific manner.
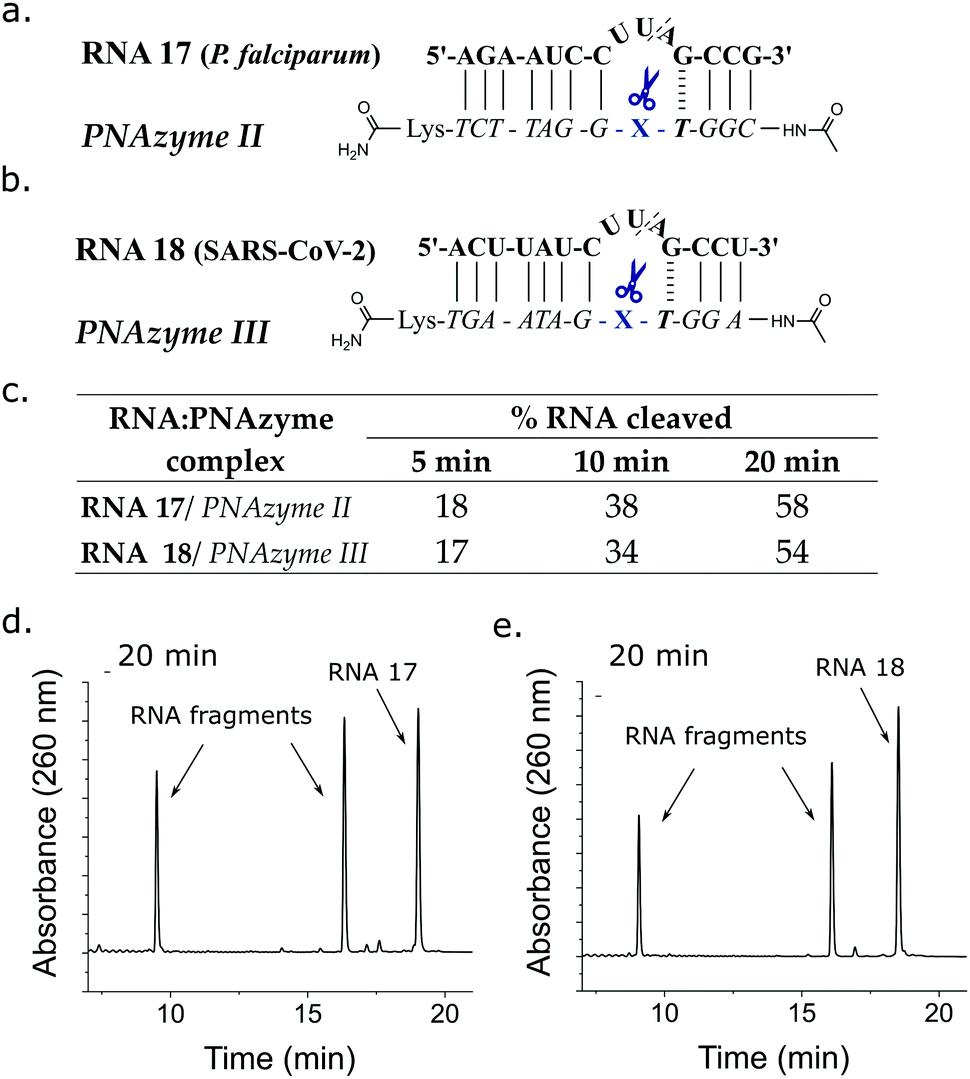 | ||
| Fig. 4 Schematic representations of PNAzymes II and III in complex with their respective clinically relevant RNA targets (a) a Plasmodium falciparum (malaria parasite) mRNA model target RNA 17 and (b) a SARS-CoV-2 genomic RNA model target RNA 18. (c) The extent of cleavage of RNA 17 and 18 by PNAzymes II and III, respectively. Ion-exchange HPLC chromatograms showing the extent of cleavage of (d) RNA 17 (58 ± 2%) and (e) RNA 18 (54 ± 2%) after 20 minutes of incubation at 37 °C, pH 7 and 4 μM RNA/PNAzyme concentration in the presence of 100 μM Zn2+. | ||
PNAzyme reliance on a sufficient local concentration of the metal cofactor is a requirement for efficient activity (see Zn2+ dependence in ESI,† S6, which is similar in the Zn2+ neocuproine systems6,13). The cellular Zn2+ can vary (0.2–0.3 mM on average, but most of it tightly bound),22,24 while some disease conditions are associated with significantly elevated Zn2+ levels.37,38 Malaria is a particularly interesting candidate for future PNAzyme developments, as extreme accumulation of zinc ions has been detected in malaria-infected red blood cells (1 mM average cellular Zn2+), where free Zn2+ is required for parasitic growth.37 We hope that these findings encourage further research into Zn2+ PNAzymes as biocompatible artificial ribonucleases that can be used as tools for molecular biology research or as candidates for therapeutic intervention.
Conceptualisation – M. M. and R. S. Formal analysis – O. L. and R. S. Funding acquisition – R. S. Investigation – O. L with contributions from P. B., R. Stulz, P. S., O. B., M. M. Methodology – O. L., P. S., M. M., R. S. Project administration – O. L., M. M., R. S. Resources – S. A. and R. S. Supervision – S. A., M. M., R. S. Visualisation – O. L. Writing – original draft – O. L. Writing – review and editing – O. L. and R. S. with contributions from all.
The authors would like to extend their gratitude to the Separation Science Laboratory at AstraZeneca Gothenburg for experimental assistance and advice on RNA purification. Eva-Lotta Käsper and Johanna Luige are acknowledged for helpful discussions and advice. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 721613. This material reflects only the authors’ views and the Union is not liable for any use that may be made of the information contained therein.
Conflicts of interest
There are no conflicts to declare.References
- S. T. Crooke, B. F. Baker, R. M. Crooke and X. Liang, Nat. Rev. Drug Discovery, 2021, 20, 427–453 CrossRef CAS PubMed.
- Y. Aoki and M. J. A. Wood, J. Neuromuscul. Dis., 2021, 1–16 CAS.
- A. Khvorova and J. K. Watts, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- D. Grimm, K. L. Streetz, C. L. Jopling, T. A. Storm, K. Pandey, C. R. Davis, P. Marion, F. Salazar and M. A. Kay, Nature, 2006, 441, 537–541 CrossRef CAS PubMed.
- H. Åström, N. H. Williams and R. Strömberg, Org. Biomol. Chem., 2003, 1, 1461–1465 RSC.
- H. Åström and R. Strömberg, Org. Biomol. Chem., 2004, 2, 1901–1907 RSC.
- M. Murtola and R. Strömberg, ARKIVOC, 2009, 84–94 Search PubMed.
- A. Kuzuya and M. Komiyama, Curr. Org. Chem., 2007, 11, 1450–1459 CrossRef CAS.
- T. Niittymäki and H. Lönnberg, Org. Biomol. Chem., 2006, 4, 15–25 RSC.
- A. Ghidini, M. Murtola and R. Strömberg, DNA in Supramolecular Chemistry and Nanotechnology, John Wiley and Sons, Ltd, Hoboken, NJ, USA, 1st edn, 2015, pp. 158–171 Search PubMed.
- O. Luige, M. Murtola, A. Ghidini and R. Strömberg, Molecules, 2019, 24, 672 CrossRef CAS.
- M. Murtola, A. Ghidini, P. Virta and R. Strömberg, Molecules, 2017, 22, 1856 CrossRef PubMed.
- A. Ghidini, M. Murtola and R. Strömberg, Org. Biomol. Chem., 2016, 14, 2768–2773 RSC.
- M. Murtola, M. Wenska and R. Strömberg, J. Am. Chem. Soc., 2010, 132, 8984–8990 CrossRef CAS PubMed.
- F. Zellmann and M. W. Göbel, Molecules, 2020, 25, 1842 CrossRef CAS PubMed.
- F. Zellmann, L. Thomas, U. Scheffer, R. K. Hartmann and M. W. Göbel, Molecules, 2019, 24, 807 CrossRef PubMed.
- Y. Staroseletz, B. Amirloo, A. Williams, A. Lomzov, K. K. Burusco, D. J. Clarke, T. Brown, M. A. Zenkova and E. V. Bichenkova, Nucleic Acids Res., 2020, 48, 10662–10679 CrossRef CAS PubMed.
- M. Gebrezgiabher, W. A. Zalloum, D. J. Clarke, S. M. Miles, A. A. Fedorova, M. A. Zenkova and E. V. Bichenkova, J. Biomol. Struct. Dyn., 2020, 1–20 Search PubMed.
- O. Patutina, D. Chiglintseva, E. Bichenkova, S. Gaponova, N. Mironova, V. Vlassov and M. Zenkova, Molecules, 2020, 25, 2459 CrossRef CAS PubMed.
- Z. N. Baker, P. A. Cobine and S. C. Leary, Metallomics, 2017, 9, 1501–1512 CrossRef CAS PubMed.
- D. Beyersmann, Materwiss. Werksttech., 2002, 33, 764–769 CrossRef CAS.
- R. B. Franklin, J. Ma, J. Zou, Z. Guan, B. I. Kukoyi, P. Feng and L. C. Costello, J. Inorg. Biochem., 2003, 96, 435–442 CrossRef CAS PubMed.
- I. Sekler, S. L. Sensi, M. Hershfinkel and W. F. Silverman, Mol. Med., 2007, 13, 337–343 CAS.
- D. Hüsken, G. Goodall, M. J. J. Blommers, W. Jahnke, J. Hall, R. Häner and H. E. Moser, Biochemistry, 1996, 35, 16591–16600 CrossRef PubMed.
- U. Kaukinen, L. Bielecki, S. Mikkola, R. W. Adamiak and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2, 2001, 1024–1031 RSC.
- A. Mukherjee and W. D. Sasikala, Advances in Protein Chemistry and Structural Biology, Academic Press, Elsevier Inc., 2013, vol. 92, pp. 1–62 Search PubMed.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 RSC.
- A. E. Friedman, J. K. Barton, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- A. J. McConnell, H. Song and J. K. Barton, Inorg. Chem., 2013, 52, 10131–10136 CrossRef CAS PubMed.
- S. T. Li, Z. Y. Ma, X. Liu, J. L. Tian and S. P. Yan, Appl. Organomet. Chem., 2017, 31, 1–14 Search PubMed.
- J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192–200 CrossRef CAS PubMed.
- S. Mikkola, E. Stenman, K. Nurmi, E. Yousefi-Salakdeh, R. Strömberg and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2, 1999, 1619–1626 RSC.
- K. Günther, M. Tümmler, H. H. Arnold, R. Ridley, M. Goman, J. G. Scaife and K. Lingelbach, Mol. Biochem. Parasitol., 1991, 46, 149–157 CrossRef.
- P. Mesén-Ramírez, B. Bergmann, T. T. Tran, M. Garten, J. Stäcker, I. Naranjo-Prado, K. Höhn, J. Zimmerberg and T. Spielmann, PLoS Biol., 2019, 17, 1–33 CrossRef PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K. P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- R. G. Marvin, J. L. Wolford, M. J. Kidd, S. Murphy, J. Ward, E. L. Que, M. L. Mayer, J. E. Penner-Hahn, K. Haldar and T. V. O’Halloran, Chem. Biol., 2012, 19, 731–741 CrossRef CAS PubMed.
- T. J. A. Craddock, J. A. Tuszynski, D. Chopra, N. Casey, L. E. Goldstein, S. R. Hameroff and R. E. Tanzi, PLoS One, 2012, 7, e33552 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HPLC chromatograms and MS data for the presented RNA cleavage as well as control experiments, Zn2+ dependence study and a full description of the experimental work available. See DOI: 10.1039/d1cc04383h |
| ‡ Current address: TNO Delft, Leeghwaterstraat 44. NL-2628 CA Delft, Netherlands. |
| § Current address: Carrer can Vinyalets, 11, 08130 Santa Perpètua de Mogoda, Spain. |
| This journal is © The Royal Society of Chemistry 2021 |